

ABSTRACT
Centronuclear myopathy (CNM) is a congenital myopathy that is histopathologically characterized by centrally located nuclei, central aggregation of oxidative activity, and type I fiber predominance and hypotrophy. Here, we obtained commercially available mice overexpressing phospholamban (PlnOE), a well-known inhibitor of sarco(endo)plasmic reticulum Ca2+-ATPases (SERCAs), in their slow-twitch type I skeletal muscle fibers to determine the effects on SERCA function. As expected with a 6- to 7-fold overexpression of phospholamban, SERCA dysfunction was evident in PlnOE muscles, with marked reductions in rates of Ca2+ uptake, maximal ATPase activity and the apparent affinity of SERCA for Ca2+. However, our most significant discovery was that the soleus and gluteus minimus muscles from the PlnOE mice displayed overt signs of myopathy: they histopathologically resembled human CNM, with centrally located nuclei, central aggregation of oxidative activity, type I fiber predominance and hypotrophy, progressive fibrosis and muscle weakness. This phenotype is associated with significant upregulation of muscle sarcolipin and dynamin 2, increased Ca2+-activated proteolysis, oxidative stress and protein nitrosylation. Moreover, in our assessment of muscle biopsies from three human CNM patients, we found a significant 53% reduction in SERCA activity and increases in both total and monomeric PLN content compared with five healthy subjects, thereby justifying future studies with more CNM patients. Altogether, our results suggest that the commercially available PlnOE mouse phenotypically resembles human CNM and could be used as a model to test potential mechanisms and therapeutic strategies. To date, there is no cure for CNM and our results suggest that targeting SERCA function, which has already been shown to be an effective therapeutic target for murine muscular dystrophy and human cardiomyopathy, might represent a novel therapeutic strategy to combat CNM.
KEY WORDS: SERCA, Dynamin 2, Skeletal muscle, Calcium regulation, Congenital myopathy
Summary: Phospholamban overexpression in mouse slow-twitch muscle impairs SERCA function and causes histopathological features associated with human centronuclear myopathy.
INTRODUCTION
The sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) pumps catalyze the active transport of Ca2+ into the sarcoplasmic reticulum (SR) and play a crucial role in muscle relaxation and the maintenance of resting intracellular Ca2+ [Ca2+]i, which ranges from 30 to 100 nM in skeletal muscle (Tupling, 2009; Schiaffino and Reggiani, 2011). Phospholamban (PLN) is a small, 52-residue SR protein capable of physically interacting with and inhibiting SERCA activity by reducing its apparent affinity for Ca2+ (Morita et al., 2008). Mouse (Song et al., 2004) and rabbit (Pattison et al., 2008) models overexpressing PLN in slow-twitch (type I) muscle fibers (PlnOE) have been generated by attaching a Pln transgene to the β-myosin heavy chain (β-MHC) promoter, which preferentially directs high levels of expression of type I skeletal-muscle-specific transgenes (Rindt et al., 1993, 1995; Knotts et al., 1994). In the PlnOE rabbit, obvious signs of muscular dystrophy, including central nucleation, severe muscle wasting, fibrosis, fatty infiltration and muscle weakness, were observed in soleus muscles, which are rich in type I fibers (Pattison et al., 2008). In contrast, PlnOE mice exhibited soleus muscle atrophy, but no other signs of myopathy were visualized under the light microscope (Song et al., 2004).
We purchased these commercially available PlnOE mice and their wild-type (WT) littermates (000067-MU, FVB/N background, Mutant Mouse Regional Resource Centre, Columbia, MO) to assess SERCA function because the effect of PLN overexpression on SERCA activity and Ca2+ transport in skeletal muscle remains uncharacterized. Unlike the study by Song et al. (2004), we discovered that the postural soleus and gluteus minimus muscles from PlnOE mice exhibited overt signs of myopathy. Collectively, our data suggest that the PlnOE mouse is a novel myopathy model resembling human centronuclear myopathy (CNM), with additional dystrophic features and potential core-like lesions.
RESULTS
SERCA function is impaired in PlnOE muscle
PlnOE mice were generated in order to examine the functional importance of PLN in skeletal muscle; however, in the only study published using these mice (Song et al., 2004), no measurements of SERCA function were reported because of technical limitations. In order to better define the functional role of PLN in skeletal muscle we assessed SERCA function in these mice, focusing our analyses on the postural muscles – soleus and gluteus minimus – since these muscles normally contain approximately 55-65% and 27% type I fibers, respectively. However, these type I fiber levels increase in response to overexpression of PLN (discussed below). Western blotting of whole homogenates demonstrated clear overexpression of PLN in these muscles because only 2.5 μg of total protein was loaded for PlnOE mice compared with the 25 μg required for WT mice (Fig. 1A and supplementary material Fig. S3A). Specifically, in the PlnOE soleus relative to WT (n=7-9 per genotype), monomeric and pentameric PLN levels, normalized to actin, were 6.3-fold (P=0.00001) and 80-fold (P=0.0007) higher, respectively. In the PlnOE gluteus minimus muscles relative to WT (n=5-6 per genotype), monomeric and pentameric PLN levels were 6.7-fold (P=0.0001) and 23-fold (P=0.0001) higher, respectively. In addition to the analysis in homogenates, we also performed single-fiber western blot analyses confirming overexpression of PLN specific to type I fibers in this model (supplementary material Fig. S1). In contrast, PLN was barely detectable in some WT type I and type IIA fibers (supplementary material Fig. S1). This corresponds well with the fact that PLN expression in rodent skeletal muscle is far lower than that in human skeletal muscle, where PLN can be found in all type I and type IIA fibers in the vastus lateralis (Fajardo et al., 2013).
RESOURCE IMPACT.
Background
Phospholamban (PLN) is a well-known inhibitor of the sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) pumps in muscle that maintain low levels of cytosolic Ca2+ and that play a crucial role in muscle contraction. The importance of PLN regulation of SERCA function in cardiac muscle health and disease is well established but whether PLN plays a similar role in skeletal muscle disease remains unknown. Centronuclear myopathy (CNM) is a congenital myopathy characterized by muscle weakness, centrally located nuclei, type I fiber predominance and central aggregation of oxidative activity in the skeletal muscles. To date, there is no curative treatment for CNM and the exact mechanism leading to these skeletal muscle defects remains unknown. Thus, animal models that accurately recapitulate these histological abnormalities are required.
Results
In this study, the authors characterize a mouse model (PlnOE) in which PLN is overexpressed in type I skeletal muscle fibers. They show that SERCA function is greatly impaired in this model and that the soleus and gluteus minimus muscles, which normally contain many type I fibers, from PlnOE mice exhibit phenotypes that resemble human CNM including centrally located nuclei, type I fiber predominance, central aggregation of oxidative activity and weakness. These muscles also present with progressive atrophy, fibrosis and potential core formations. Finally, the authors report that SERCA function was on average 53% lower in muscle biopsies from three patients with CNM compared with biopsies from five healthy individuals, whereas PLN expression seemed to be elevated in biopsies from patients with CNM compared with healthy controls.
Implications and future directions
These results identify a novel CNM mouse model that can be used for the investigation of the mechanisms underlying CNM and for the development of therapeutic strategies. The findings also suggest that studies to assess the role of PLN and SERCA dysfunction in human and animal CNM and myopathy in general might be worthwhile. To date, 25-30% of CNM cases remain genetically unresolved but several mutations in the human gene encoding PLN are known to lead to SERCA inhibition and cardiac complications. Given that many clinical cases of CNM present with cardiomyopathy, future studies should determine whether skeletal muscle defects are also present in patients harboring PLN mutations. Finally, these findings add to the notion that aberrant Ca2+ regulation is central to many cardiac and skeletal muscle diseases and that targeting SERCA function might represent a viable therapeutic strategy for these diseases.
Fig. 1.

SERCA function in soleus muscles in PlnOE mice at 4-6 months of age. (A) Western blotting for PLN in WT and PlnOE mice from soleus muscle homogenates. For WT mice, 25 μg of total protein was loaded, whereas only 2.5 μg was required for PlnOE mice to detect PLN protein. (B) Ca2+-ATPase activity–pCa curves in WT (n=5) and PlnOE mice (n=6) in the presence of the Ca2+ ionophore. (C) Ca2+ uptake assessed in soleus muscles from WT (n=4) and PlnOE mice (n=5). (D-F) Western blotting for SERCA1a (D), SERCA2a (E) and SLN (F) in soleus muscle from WT and PlnOE mice (n=6 per genotype). Actin was used as a loading control and all values are expressed relative to WT. *P≤0.05 versus WT. All values are presented as means±s.e. PLN (p), PLN pentamer; PLN (m), PLN monomer.
In agreement with the known effects of PLN overexpression on SERCA function in HEK 293 cells and transgenic hearts (MacLennan and Kranias, 2003; Bhupathy et al., 2007), we observed a reduction in SERCA's apparent affinity for Ca2+, as indicated by a higher KCa value and a corresponding rightward shift in the activity–pCa curve in soleus homogenates prepared from PlnOE mice compared with WT (Table 1 and Fig. 1B). Furthermore, maximal rates of SERCA activity (−27%) and rates of Ca2+ uptake (−74%) were reduced in the PlnOE soleus compared with WT (Fig. 1B and C and Table 1). The SERCA dysfunction in PlnOE soleus cannot be explained by altered SERCA expression since western blot analysis showed that SERCA1a content is similar in WT and PlnOE muscles (Fig. 1D), whereas a significant 3-fold increase in SERCA2a expression was observed in the soleus of PlnOE mice (Fig. 1E). Sarcolipin (SLN), a small SR protein that is structurally and functionally homologous with PLN (Asahi et al., 2002, 2003), was also found to be upregulated 9-fold in the soleus of PlnOE mice compared with WT (Fig. 1F). As SLN can inhibit SERCA pumps alone or in combination with PLN (Asahi et al., 2003), these results suggest that SLN may also contribute to the impaired SERCA function we observed in PlnOE muscles. Indeed, the forced overexpression of SLN acutely in rat soleus reduces SR Ca2+ transport (−31%) and causes severe contractile dysfunction (Tupling et al., 2002).
Table 1.
SERCA activity in mouse soleus muscles from WT and PlnOE mice at 4-6 months of age

Soleus muscle atrophy and Ca2+-activated proteolysis in PlnOE mice
Similar to previously described results (Song et al., 2004), we also observed atrophy of soleus muscles in PlnOE mice (Fig. 2A and supplementary material Table S1). Interestingly, when breeding male and female PlnOE mice, soleus muscles weighing ≤1 mg were observed, presumably from homozygous PlnOE mice (+/+), because these muscles were no longer seen after breeding male PlnOE mice with female WT mice (Fig. 2A). Owing to this limitation of the tissue, we restricted all of our analyses in the present study to heterozygous PlnOE mice (+/−). In the study published by Song et al. (2004), a 35-40% reduction in soleus:body-weight ratio was observed in PlnOE mice at 10-12 weeks of age. In our hands, assessment of soleus:body-weight ratio in PlnOE mice at 1 month, 4-6 months and 10-12 months of age indicates a 21%, 43% and 41% reduction, respectively, when compared with WT mice (supplementary material Table S1). When we include age as a factor, absolute soleus weight and the soleus:body-weight ratios were significantly different at 4-6 months and 10-12 months, but not at 1 month. To elucidate the mechanisms behind the diminished soleus muscle mass, Song and colleagues reported an approximate 2-fold increase in calpain expression (Song et al., 2004). Calpains are a class of proteolytic enzymes that are activated by high levels of [Ca2+]i (Murphy et al., 2006), and our results show that calpain activity was 1.5-fold higher in soleus from PlnOE mice compared with WT (Fig. 2B), which probably results from reduced SERCA function and greater [Ca2+]i in PlnOE mice. We also observed a significant 1.5- to 2-fold increase in other proteolytic pathways, such as caspase-3, cathepsin-B/L activity and protein ubiquitylation (Fig. 2C-E), which suggests that the soleus muscle atrophy in PlnOE mice is due to a general overall enhancement of proteolytic activity in those muscles.
Fig. 2.

Soleus muscles of PlnOE mice are atrophied at 4-6 months of age. (A) Representative images of soleus muscles extracted from WT and PlnOE mice. +/−, heterozygous; +/+, homozygous. (B) Calpain activity in soleus homogenates from WT (n=4) and PlnOE mice (n=5). (C) Caspase-3 activity in soleus muscle homogenates from WT (n=4) and PlnOE mice (n=7). (D) Cathepsin-B/L activity in soleus homogenates from WT and PlnOE mice (WT, n=4; PlnOE, n=6). Calpain activity, caspase-3 activity, and cathepsin-L activity are in arbitrary units normalized to mg protein and are presented relative to WT. (E) Total protein extracts from soleus muscles (15 μg) were immunoblotted with anti-ubiquitin (Ub) antibody. Ponceau stain was used as a loading control. The sum of optical densities from detectable ubiquitylated proteins (p-Ub) as well as the optical density of monomeric ubiquitin (m-Ub, 10 kDa) was measured and compared between genotypes (n=4 per group). Values were normalized to the sum of optical densities of bands visualized through Ponceau stain. *P≤0.05 versus WT. All values are presented as means±s.e.
Central nuclei, progressive fibrosis and oxidative stress in PlnOE mice
As mentioned earlier, previous analyses of PlnOE mice revealed no signs of myopathy under the light microscope (Song et al., 2004). Consequently, we were surprised that our histological analyses of the soleus muscles from PlnOE mice showed obvious signs of pathology that were repeatedly observed (Fig. 3). In fact, the myopathy was at least partially consistent with the dystrophic-like phenotype observed in PlnOE rabbits (Pattison et al., 2008), including greater central nucleation of fibers, fiber splitting, atrophy and progressive fibrosis, which became evident at 4-6 months of age (Fig. 3A-D). Corresponding well with the dystrophic phenotype, we also observed greater whole muscle oxidative stress, as indicated by DCF (2′,7′-dichlorofluorescein) fluorescence and protein nitrosylation – effects that may be explained by Ca2+ dysregulation (Altamirano et al., 2012). Furthermore, we found elevated plasma CK in 4- to 6-month-old PlnOE mice (Fig. 3E-G); however, the relatively small increase in plasma CK suggests that the dystrophic-like phenotype in PlnOE mice is a direct result of impaired SERCA activity and elevated [Ca2+]i, and is less likely to be caused by sarcolemmal damage per se. A similar finding has been reported in mice overexpressing transient receptor potential canonical 3, which led to greater Ca2+ influx, muscular dystrophy and only slightly elevated plasma CK as a result of a generally intact sarcolemmal membrane (Millay et al., 2009).
Fig. 3.

Dystrophic features in soleus muscles from PlnOE mice. (A) H&E-stained sections of the soleus muscles from WT and PlnOE mice at 1 month, 4-6 months and 10-12 months of age. (B) Percentage of fibers containing central nuclei in the soleus at 1 month, 4-6 months and 10-12 months of age (n=3 per genotype at each age with 300-600 fibers counted per mouse). (C) Van-Geison-stained sections of the soleus muscles from WT and PlnOE mice at 1 month, 4-6 months and 10-12 months of age. (D) Quantification of fibrotic area in the soleus at 1 month, 4-6 months and 10-12 months of age (n=3-4 per group at each age). ImageJ software was used to quantify fibrotic area. (E) Reactive oxygen species generation in whole soleus determined using DCF assay at 4-6 months of age (WT, n=4; PlnOE, n=5). (F) Total protein extracts from soleus muscles (15 μg) were immunoblotted with anti-nitrotyrosine antibody. The sum of optical densities from detectable nitrosylated proteins (50-100 kDa) was measured and compared between genotypes at 4-6 months of age (n=6 per group). Values were normalized to the sum of optical densities of bands visualized through Ponceau stain. (G) Plasma CK levels in WT (n=5) and PlnOE mice (n=9) at 4-6 months of age. Scale bars: 50 μm (A,C). *P≤0.05 versus WT using two-way ANOVA and Tukey's post hoc analysis for % central nuclei and % fibrosis; Student's t-test for DCF, protein nitrosylation and plasma CK. All values are presented as means±s.e.
CNM in the PlnOE mouse
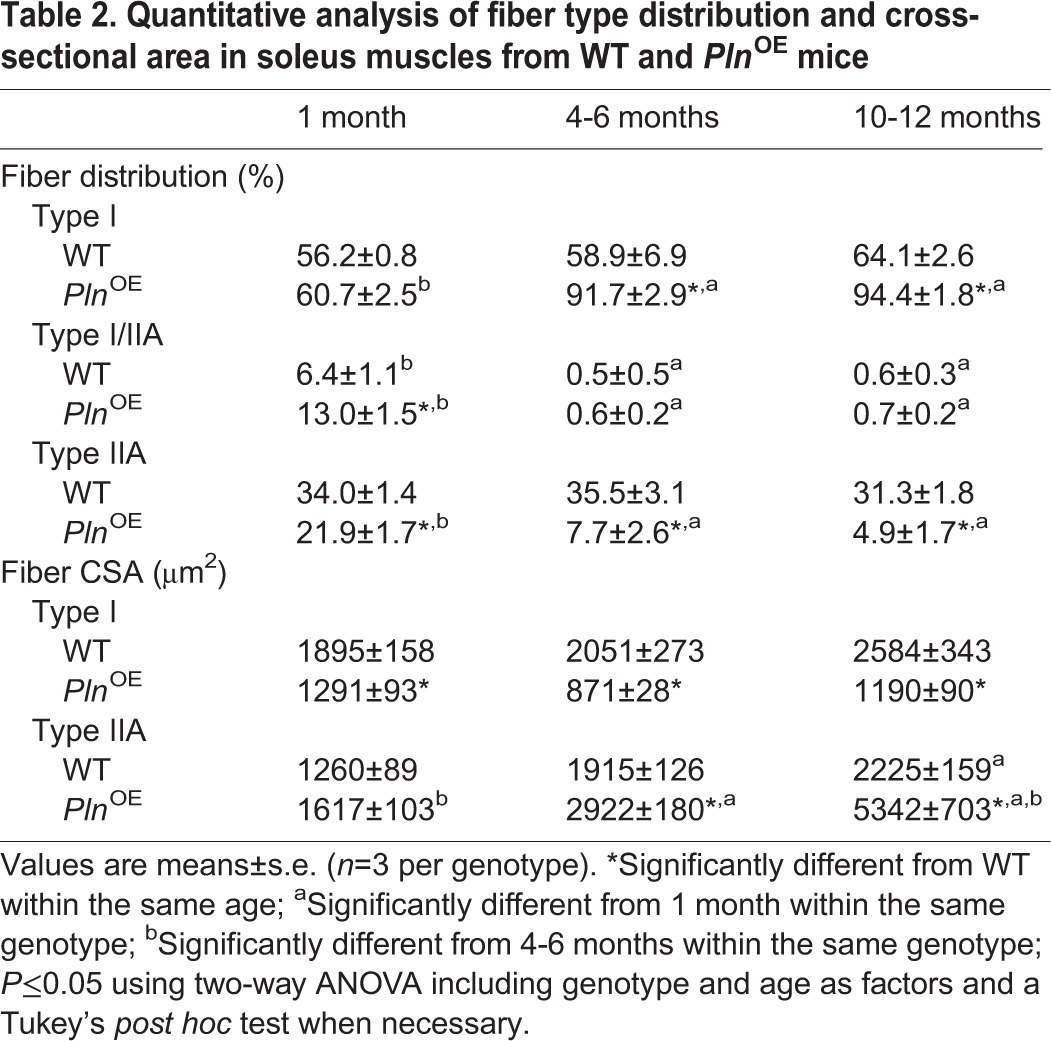
Although central nucleation might be an active marker in muscle regeneration and a common feature in muscular dystrophy (Charge and Rudnicki, 2004; Dubowitz et al., 2013; Folker and Baylies, 2013), we observed only a small proportion of fibers that were positive for embryonic MHC (supplementary material Fig. S2), which is normally expressed transiently during muscle regeneration (Sartore et al., 1982; DiMario et al., 1991; Grady et al., 1997). These results, combined with the small elevation in plasma CK, suggest that very little degeneration–regeneration cycling is occurring in PlnOE muscle. Therefore, the PlnOE mouse does not closely resemble dystrophic myopathy; however, central nucleation in the face of low regeneration, progressive fibrosis, and moderate elevations in CK levels is also found in CNM (Jungbluth et al., 2008). CNM is a congenital myopathy that is diagnosed by the existence of three histopathological features found in muscle biopsies, which include: (1) central nucleation, (2) central aggregation of oxidative activity, and (3) predominance of type I fibers and hypotrophy (Jungbluth et al., 2008; Romero, 2010). Both central aggregation of oxidative activity and type I fiber hypotrophy were evident in soleus sections as early as 1 month of age in PlnOE mice (Fig. 4A and B and Table 2). Type I fiber predominance was established at 4-6 months of age, whereas at 1 month, fibers were still transitioning towards the type I fiber distribution as the PlnOE soleus had lower type IIA and greater type I/IIA content compared with WT (Fig. 4A and Table 2). The type I fiber predominance found from 4-6 months is in agreement with previous findings (Song et al., 2004), and the increase in the slow-twitch SERCA2a isoform in PlnOE soleus (Fig. 1E) is also consistent with type I fiber predominance. Taken together, these results indicate that the PlnOE mouse model is a phenocopy of the histological features found in human CNM. Importantly, the soleus muscle was not the only muscle affected by PLN overexpression because the postural gluteus minimus muscle, which normally comprises roughly 27% type I fibers in WT mice, also displayed signs of impaired SERCA function, CNM and endomysial fibrosis (supplementary material Fig. S3, Tables S2,S3). Despite a similar level of monomeric PLN overexpression, the relative impairment in SERCA function in the gluteus minimus muscles compared with the PlnOE soleus was lower, with only a 20% reduction in the rates of Ca2+ uptake (supplementary material Fig. S3C). This might be explained by the fact that the gluteus minimus muscles have a greater number of type II fibers that do not overexpress PLN, which could mask the inhibition of SERCA pumps occurring in the type I fibers. In any event, both the PlnOE soleus and gluteus minimus muscles resembled the histopathological features associated with CNM.
Fig. 4.

Centronuclear myopathy in the soleus muscles from PlnOE mice. (A) Representative images of soleus muscles showing type I fiber predominance and hypotrophy. MHC immunofluorescence stained sections of the soleus muscles at 1 month, 4-6 months and 10-12 months of age. Cross sections were stained with antibodies against MHC to identify type I (blue), type IIA (green), type IIB (red) and type IIX (unstained) fibers. (B) Representative images of soleus muscles showing central accumulation of oxidative activity. Succinate dehydrogenase (SDH)-stained sections display central aggregation of oxidative activity in the PlnOE mice at 1 month, 4-6 months and 10-12 months of age. (C) NADH-TR-stained cross sections demonstrating radiating SR strands in soleus muscles from PlnOE mice (arrows). Asterisks represent the same fiber across cryosections. Scale bars: 50 μm.
Table 2.
Quantitative analysis of fiber type distribution and cross-sectional area in soleus muscles from WT and PlnOE mice
To date, CNMs are known to be genetically heterogeneous (Dowling et al., 2014; Jungbluth and Gautel, 2014) and have been attributed to X-linked recessive mutations in the MTM1 gene encoding myotubularin (Laporte et al., 1996), autosomal-dominant mutations in the DNM2 gene encoding dynamin-2 (Bitoun et al., 2005) and the BIN1 gene encoding amphiphysin-2 (Bohm et al., 2014), and autosomal-recessive mutations in BIN1 (Nicot et al., 2007), RYR1 encoding the SR ryanodine receptor (Jungbluth et al., 2007; Wilmshurst et al., 2010) and TTN encoding titin (Ceyhan-Birsoy et al., 2013). Many different structural abnormalities have been identified that aid in distinguishing between the various human CNMs (Romero, 2010; Jungbluth and Gautel, 2014). For example, the autosomal-dominant DNM2-related CNMs present with radiating SR strands and marked contrast of the diameters between type I and type II fibers (Romero, 2010). In addition to these features, increases in connective tissue and the presence of core formations can also be observed in DNM2-related CNMs (Jungbluth and Gautel, 2014). Since we observed radiating SR strands (Fig. 4C), type II fiber hypertrophy (Table 2 and supplementary material Table S3), progressive fibrosis (Fig. 3C and D) and potential core-like formations (Fig. 5) in the PlnOE mice, our results suggest that the CNM phenotype found in these mice resembles autosomal-dominant DNM2-related CNM. However, because increases in endomysial connective tissue and the presence of core formations may also be observed in RYR1-related and TTN-related CNM (Jungbluth et al., 2007; Jungbluth and Gautel, 2014), the PlnOE mouse also shares a resemblance to these forms of CNM. We use the term ‘potential core-like lesions’ because future studies with electron microscopy are required to verify the presence and characterize the structure of cores in the PlnOE mouse.
Fig. 5.
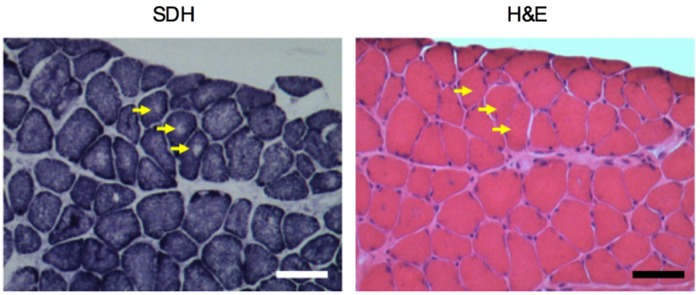
Potential core-like lesions in PlnOE mouse at 4-6 months of age. SDH-stained sections from the soleus muscle show areas devoid of oxidative staining representing potential core-like lesions (yellow arrows). Corresponding H&E-stained serial section shows that the lack of oxidative staining with SDH is not due to the presence of a vacuole or an artifact in the muscle fiber.
Muscle function in the PlnOE mouse
General muscle weakness is common in cases of CNM and our analyses of contractile function in isolated intact soleus muscle, as well as whole-body endurance during treadmill exercise, demonstrate that muscle function is impaired in PlnOE mice (supplementary material Fig. S4A-E). Specifically, we found that PlnOE mice generate lower normalized force at both submaximal and maximal stimulation frequencies (30-100 Hz) (supplementary material Fig. S4D) and reached exhaustion much sooner than WT mice during a treadmill exercise test (supplementary material Fig. S4E). In agreement with previous findings (Song et al., 2004), we found that normalized twitch force generation of soleus muscle was not different between PlnOE and WT mice, whereas the twitch kinetics were significantly slower in PlnOE compared with WT (supplementary material Fig. S4A-C).
PLN and SERCA dysfunction in human CNM
Thus far, it is evident that PLN overexpression in mouse skeletal muscle leads to CNM; however, whether PLN plays a primary pathogenic role in CNM remains unknown. In support of this notion, PLN protein was found to be upregulated in the tibialis anterior muscles from mice lacking microRNA 133a-1 and 133a-2, where CNM was observed (Liu et al., 2011). In addition, we obtained muscle biopsies from three patients with CNM and when compared with biopsies from five healthy controls, we found that the maximal rate of SERCA activity was significantly reduced by 53% (supplementary material Fig. S5A) and that monomeric PLN (P=0.12) and total PLN (P=0.08) content were greater (supplementary material Fig. S5B). Therefore, we provide the first indication of a potential role for SERCA dysfunction and elevated PLN in human CNM; however, caution should be taken when interpreting these results. Recently, we established through single-fiber western blotting and immunohistochemical analyses that PLN expression follows SERCA2a and MHC I expression, whereas SLN follows SERCA1a and MHC II expression in healthy human vastus lateralis (Fajardo et al., 2013). Since we observed a trend for higher MHC I (supplementary material Fig. S5C, P=0.10) and SERCA2a (supplementary material Fig. S5D, P=0.10) expression, and significant reductions in both SERCA1a (supplementary material Fig. S5E) and SLN (supplementary material Fig. S5F) in CNM muscles compared with healthy controls, it is possible that these differences, including upregulated PLN, are due mostly to the expected type I fiber predominance in CNM patients. Furthermore, in addition to the low sample size used in the present study, the CNM cases obtained were of notable age variability and were genetically unrelated, with one patient harboring a DNM2 mutation (R369W) and the other two patients remaining genetically unresolved. Importantly, despite these limitations, we were still able to show a 53% reduction in SERCA activity and the expression patterns of the SERCA isoforms, SLN and PLN are consistent with the expected type I fiber predominance often found in CNM.
Dynamin 2 levels in CNM
As a result of the dominant inheritance of the DNM2 mutation, one theory is that DNM2-related CNM may be caused by a toxic gain-of-function in dynamin 2 (Cowling et al., 2014; Dowling et al., 2014). In support of this, overexpression of WT dynamin 2 in skeletal muscle results in features associated with CNM (Cowling et al., 2011; Liu et al., 2011). In addition, upregulation of dynamin 2 was observed in skeletal muscles from mtm1–/y mice, which accurately models human X-linked myotubular myopathy (XLCNM), and heterozygous knockdown of dynamin 2 in the mtm1–/y mouse improves muscle pathology and function (Cowling et al., 2014). We analyzed dynamin 2 protein content in PlnOE muscles and found a 5-fold (supplementary material Fig. S6A) and 3-fold (supplementary material Fig. S6B) higher level in the soleus and gluteus minimus muscles, respectively, compared with WT. Whether dynamin 2 plays a pathological role in the CNM found in the PlnOE mouse and the exact mechanism leading to its augmented expression requires further investigation.
Elevated levels of dynamin 2 are thought to also play a pathological role in human CNM because results from Laporte's laboratory have shown an approximate 1.5-fold increase in dynamin 2 expression in muscle lysates from three XLCNM patients compared with two healthy controls (Liu et al., 2011; Cowling et al., 2014). To confirm whether elevated dynamin 2 contributes to uman CNM pathology, we assessed the dynamin 2 protein content in the three CNM muscle biopsies obtained here; however, in contrast to what was previously found in human CNM (Liu et al., 2011; Cowling et al., 2014), we found a 35% reduction in dynamin 2 compared with healthy subjects (supplementary material Fig. S6C). Notwithstanding the aforementioned sample size and other limitations with our human CNM cases, these findings may suggest that reducing dynamin 2 as a therapeutic strategy may not be appropriate for all forms of human CNM as previously suggested (Demonbreun and McNally, 2014) and may be limited to XLCNM.
DISCUSSION
To date, most studies concerning the physiological significance of PLN inhibition of SERCA have focused solely on its role in cardiac muscle health and disease (for a review, see MacLennan and Kranias, 2003). To our knowledge, our current study is the first to demonstrate an important role for PLN and SERCA dysfunction in skeletal muscle health and CNM pathology. Specifically, we obtained commercially available PlnOE mice to determine PLN's effect on SERCA function in skeletal muscle and uncovered a mouse model that phenotypically resembles the histopathological features associated with human CNM. In addition, our results with three human CNM patients suggest that SERCA dysfunction, possibly through increased PLN expression, contributes to CNM pathology. Although it is possible that the predominance of type I fibers in human CNM may explain the elevations in PLN expression, it is important to consider our results from the PlnOE mouse, which indicate that overexpression of PLN specifically in type I fibers can cause type I fiber predominance and CNM. Thus, future studies with more CNM patients that are similar in age and genetically related are required to better investigate whether PLN plays a primary pathological role in human CNM.
We cannot fully explain why we repeatedly observed myopathy in the PlnOE mouse whereas Song and co-workers did not report such a finding (Song et al., 2004). Comparisons are difficult to make because SERCA function was not assessed and histology results were not shown in their study. Strain differences seem unlikely to account for the discrepant results since both transgenic lines were generated on an FVB/N background; however, it is possible that genetic drift and the level of PLN overexpression may be significant factors. Nevertheless, several results are in agreement between the two studies, including type I fiber predominance, atrophy, increased proteolytic markers and slower force kinetics in soleus muscle.
It has been estimated that 25-30% of human CNM cases remain genetically unresolved (Romero, 2010; Dowling et al., 2014) and our results raise the possibility that mutations in Pln leading to increased SERCA inhibition could be involved in those cases. Although Pln mutations either resulting in elevated PLN expression (Minamisawa et al., 2003) or reduced PLN phosphorylation (Schmitt et al., 2003; Haghighi et al., 2006) have been causally linked to human cardiomyopathy, it is unknown whether these mutations can affect skeletal muscle health. For example, deletion of arginine 14 (R14del) in the coding region of human PLN results in increased SERCA inhibition through increased monomeric PLN and reduced PLN phosphorylation at Ser16 by PKA (Haghighi et al., 2006). Interestingly, one patient with an R14del mutation in PLN developed late-onset mild dilated cardiomyopathy, but was initially evaluated in a muscle dystrophy clinic for a 25-year history of slowly progressive muscle weakness (DeWitt et al., 2006). Since there were no significant abnormalities in the staining patterns of sarcolemmal proteins, muscular dystrophy was discounted; however, to our knowledge, the existence of CNM was not tested. Recent reports of human cases of CNM coexisting with cardiomyopathy (Ceyhan-Birsoy et al., 2013; Agrawal et al., 2014; Gal et al., 2015) further highlight the importance of assessing the existence of CNM in skeletal muscles from patients with cardiomyopathy arising from PLN mutations.
Currently, the mechanisms leading to CNM are not completely understood; however, several mechanisms have been suggested, including abnormalities of triad structure and function (Al-Qusairi et al., 2009; Al-Qusairi and Laporte, 2011; Bohm et al., 2014; Dowling et al., 2014; Gibbs et al., 2014). As a result of these triad defects, aberrant Ca2+ handling and excitation–contraction (EC) coupling are also thought to be important for CNM pathogenesis. Indeed, the identification of mutations in RYR1 leading to CNM (Jungbluth et al., 2007; Wilmshurst et al., 2010) further supports the notion that triad dysfunction, Ca2+ dysregulation and EC coupling are key pathogenic drivers of CNM (Dowling et al., 2014). Since the SERCA pumps regulate SR Ca2+ load and, thus, contractility, the SERCA pumps are also crucial for Ca2+ regulation and EC coupling. Thus, our results with the PlnOE mice add further support to the hypothesis that defects in triad function, Ca2+ handling and EC coupling are important for CNM pathogenesis and extend the hypothesis by adding SERCA dysfunction as a potential pathogenic mechanism. Since abnormalities in triad structure are often found in human and animal CNM, future studies using electron microscopy should examine the triad structure in the PlnOE mouse to determine whether structural abnormalities also contribute to the Ca2+ dysregulation seen in these mice. Furthermore, although areas devoid of oxidative (SDH) staining that cannot be explained by artifacts or the presence of vacuoles may represent core formations in the PlnOE mouse, future studies with electron microscopy will confirm and characterize these potential core-like lesions.
One other interesting question raised by our study pertains to the role of SLN in CNM. Similar to PLN, SLN is a SERCA pump inhibitor (Asahi et al., 2002, 2003; Gorski et al., 2013), which suggests that the 7- to 9-fold upregulation in SLN protein found in the PlnOE soleus and gluteus minimus muscles may contribute to the SERCA dysfunction, Ca2+ dysregulation and CNM pathology. Interestingly, this response in SLN expression is consistent with other mouse models of myopathy (Nakagawa et al., 2005; Ottenheijm et al., 2008; Al-Qusairi et al., 2009; Liu et al., 2011; Calvo et al., 2012; Schneider et al., 2013), but the role of SLN remains unknown. Our finding that SLN content was reduced in human CNM reveals a potential species difference and suggests that this SERCA regulator may have minimal contribution to the SERCA dysfunction and overall CNM pathology. To test whether or not SLN actively contributes to the CNM phenotype found in the PlnOE mice, future studies targeting the Sln gene are required. Furthermore, a novel micropeptide, myoregulin (MLN), was found to be structurally and functionally homologous with both PLN and SLN (Anderson et al., 2015). When new antibodies targeting MLN protein expression become available, it will be interesting to determine its response and potential involvement in murine and human CNM and myopathy in general.
In summary, the commercially available PlnOE mouse histopathologically resembles human CNM. To date, there is no cure for CNM and many unresolved questions remain, including the mechanisms leading to skeletal muscle defects in patients. Mechanistically, our results from the PlnOE mouse are consistent with the hypothesis that triad dysfunction, aberrant Ca2+ handling and EC coupling are important for CNM pathogenesis; but whether it is higher cytosolic Ca2+ and/or altered Ca2+ dynamics that lead to CNM histopathology, and how the muscle translates this dysregulated Ca2+ into the CNM phenotype remains unknown and future studies using the PlnOE mouse will prove valuable. Furthermore, treatment strategies aimed at improving SERCA function have already shown promise in murine muscular dystrophy (Goonasekera et al., 2011; Gehrig et al., 2012; Mazala et al., 2015) and human cardiomyopathy (Horowitz et al., 2011; Jessup et al., 2011; Zsebo et al., 2014). Thus, future studies that improve SERCA function through various modes, including alterations in PLN content and/or inhibitory function, in the PlnOE mouse and other animal models of CNM, could lead to the development of viable therapeutic strategies for CNM patients.
MATERIALS AND METHODS
PlnOE mice
PlnOE mice were resuscitated from cryopreserved embryos by the mmRRC (000067-MU, FVB/N background) to generate a breeding colony with WT FVB/N mice in our facility. The Pln transgene was attached to the β-MHC promoter so that these mice overexpress PLN in their slow-twitch type I skeletal muscle fibers. Animals were housed in an environmentally controlled room with a standard 12 h:12 h light:dark cycle and allowed access to food and water ad libitum. Experiments were performed on littermate heterozygous PlnOE and WT males that were between the ages of 1 and 12 months. Reintroduction of homozygous PlnOE mice was avoided by breeding heterozygous male PlnOE mice with female WT mice. Analysis of 287 newborn mice showed that WT and heterozygous PlnOE mice were born at the expected mendelian frequency with a ratio of 137:150. All animal procedures were reviewed and approved by the Animal Care Committee of the University of Waterloo and are consistent with the guidelines established by the Canadian Council on Animal Care.
Human subjects
Five untrained university students were used as healthy controls. All subjects were fully informed of all experimental procedures and all associated risks before written consent was obtained. Written approval for the research was granted by the Human Research Ethics Committee at the University of Waterloo. Three patients with CNM from McMaster University Medical Centre were used in this study. One female (55 years old) patient had CNM due to a missense mutation in dynamin 2 (DNM2, 1105C→T, R369W) whereas the other two male CNM patients (17 and 44 years old) remain genetically unresolved. Of the two unresolved cases, one underwent 163-gene next-generation sequencing, which included MTM1, DNM2, RYR1, CACNA1S and TTN. The other unresolved patient was screened for MTM1 and DNM2; however, he did not return to complete a screen for additional genes. Written approval for the use of material from previously consented and diagnosed patients was granted by the Chair of the Human Research Ethics Committee at McMaster University. All muscle tissue samples (∼100 mg) were obtained from the vastus lateralis using the needle biopsy technique under suction (Tarnopolsky et al., 2011). These samples were homogenized and then frozen at −80°C prior to being used for western blotting and SERCA activity assays.
SERCA activity and Ca2+ uptake
Ca2+-dependent SERCA activity was assessed in homogenates prepared from mouse (WT and PlnOE) soleus and gluteus minimus muscles and human vastus lateralis muscles over Ca2+ concentrations ranging from pCa 7.5 to 4.5 in the presence of the Ca2+ ionophore A23187 (Sigma C7522) using a spectrophotometric plate reader assay that has been described previously (Duhamel et al., 2007). SERCA activity–pCa curves were generated with GraphPad Prism™ by non-linear regression curve fitting using an equation for a general cooperative model for substrate activation. Ca2+ uptake was measured in homogenates in the presence of the precipitating anion, oxalate, using the fluorescent dye Indo-1 and a spectrofluorometer equipped with dual-emission monochromators (Tupling and Green, 2002). Rates of Ca2+ uptake assessed at a free cytosolic Ca2+ concentration of 1500 nM are reported.
Antibodies
Primary antibodies against SERCA2a (2A7-A1), PLN (2D12), dynamin 2 (PA5-19800) and RyR (MA3-925) were obtained from Pierce Antibodies. The primary antibody for SERCA1a (A52) was a kind gift from Dr David MacLennan (University of Toronto) (Zubrzycka-Gaarn et al., 1984). The primary antibody directed against SLN was generated by Lampire Biological Laboratories (Fajardo et al., 2013). Anti-ubiquitin (P4D1) and anti-nitrotyrosine (189542) antibodies were obtained from Cell Signaling Technology and Cayman Chemicals, respectively. The primary antibody against α-actin (A4700) was obtained from Sigma-Aldrich. The primary antibodies against MHCI (BA-F8), MHCIIa (SC-71), MHCIIb (BF-F3), embryonic MHC (BF-F6) (Schiaffino et al., 1989; Lucas et al., 2000) and dystrophin (3B7) (Nguyen thi et al., 1990) were obtained from Developmental Studies Hybridoma Bank. Secondary antibodies for western blotting, goat anti-mouse IgG (peroxidase conjugated) and goat anti-rabbit IgG (peroxidase conjugated) were obtained from Santa Cruz Biotechnology. Secondary antibodies for immunofluorescence staining, Alexa Fluor 350 anti-mouse IgG2b, Alexa Fluor 488 anti-mouse IgG1 and Alexa Fluor 555 anti-mouse IgM, were obtained from Molecular Probes.
Western blot analysis
Single-fiber western blots were performed as previously described (Fajardo et al., 2013) to determine fiber-type specificity of PLN overexpression. Briefly, single fibers extracted from soleus muscles of WT and PlnOE mice were placed into 1× solubilizing buffer. Solubilized proteins were then separated using Tricine-based SDS-PAGE (6-13% layered gel). Separated proteins were then transferred onto 0.2 μm polyvinylidene difluoride (PVDF) membranes. Membranes were cut at the 75 kDa band (Western C Precision Plus™, Bio-Rad, CA, USA) and were immunoprobed with PLN (<75 kDa strip) and with MHCI (>75 kDa strip). Following this, membranes were immunoprobed with horseradish-peroxidase-conjugated secondary antibodies and antigen–antibody complexes were detected by Luminata Forte™ (Millipore, MA, USA) for PLN, and ECL Western Blot Substrate (BioVision, MA, USA) for MHCI. After detection of MHCI, the membrane was stripped and re-probed with MHCIIa and antigen–antibody complexes were detected using ECL Western Blot Substrate.
Similarly, western blot analysis was performed to determine expression levels of SLN, PLN, SERCA isofroms, dynamin 2, and RyR as well as levels of protein ubiquitylation (Ub) and nitrosylation (Ny) in mouse (WT and PlnOE) and human CNM muscles after homogenates were placed into 1× solubilizing buffer. Solubilized proteins from tissue homogenates, were separated using Tricine based SDS-PAGE (13% total acrylamide for PLN and SLN) or a standard SDS-PAGE (7.5% total acrylamide for SERCA isoforms and dynamin 2 and a 7.5-15% layered gel for Ub and Ny). Separated proteins were then transferred onto 0.2 μm PVDF membranes (PLN, SERCAs, Ub, Ny, RyR) or nitrocellulose membranes (SLN). Membranes were then immunoprobed with their corresponding primary antibodies. Following this, membranes were immunoprobed with horseradish-peroxidase-conjugated secondary antibodies and signals were detected by SuperSignal West Femto™ substrate (Pierce, Thermo Fisher Scientific Inc.) for SLN; Luminata Forte™ for SERCA2a, PLN, Ub, Ny and RyR; and ECL Western Blot Substrate for SERCA1a and dynamin 2. Quantification of optical densities was performed using GeneTools (Syngene, MD, USA) and values were normalized to total protein or α-actin.
Histological, histochemical and immunofluorescence staining
Soleus and gluteus minimus muscles from WT and PlnOE mice were removed and embedded in OCT compound (Tissue-Tek), frozen in liquid nitrogen-cooled isopentane, stored at −80°C, and cut into 10-μm-thick cryosections with a cryostat (Thermo Electronic) maintained at −20°C. Histological staining included H&E and Van Gieson and histochemical staining included succinate dehydrogenase (SDH) and NADH-TR activity. Images were acquired with a brightfield Nikon microscope linked to a PixeLink digital camera and quantified with Image-Pro PLUS analysis and ImageJ software. Immunofluorescence analysis of MHC expression was previously described (McMillan and Quadrilatero, 2011; Bloemberg and Quadrilatero, 2012; Fajardo et al., 2013) and performed with primary antibodies against MHCI, MHCIIa and MHCIIb. Additional immunofluorescence analysis of embryonic MHC expression was conducted with primary antibodies against embryonic MHC along with primary antibodies against dystrophin. Slides were visualized with an Axio Observer Z1 fluorescent microscope equipped with standard red, green, blue filters, an AxioCam HRm camera, and AxioVision software (Carl Zeiss). Quantification of fibers and cross-sectional area (CSA) was performed using ImageJ software.
Proteolytic activity and DCF assays
Calpain, caspase-3 and cathepsin-B/L activity were determined in soleus muscle homogenates using the substrates, Suc-LLVY-AMC (Enzo-Life Sciences), Ac-DEVD-AMC (Alexis Biochemicals) and z-FR-AFC (Enzo Life Sciences), respectively (McMillan and Quadrilatero, 2011; Bloemberg et al., 2014). These fluorogenic substrates are weakly fluorescent but yield highly fluorescent products following proteolytic cleavage by their respective proteases. Fluorescence was measured using a SPECTRAmax Gemini XS microplate spectrofluorometer (Molecular Devices, Sunnyvale, CA) with excitation and emission wavelengths: 360 nm and 440 nm, respectively for caspase-3; 380 nm and 460 nm, respectively for calpain; 400 nm and 505 nm, respectively for cathepsin. Calpain activity was taken as the difference in fluorescence from homogenate incubated with and without calpain inhibitor. Calpain, caspase-3 and cathepsin activities were normalized to total protein content and expressed as fluorescence intensity in arbitrary units per mg of protein.
Generation of reactive oxygen species in soleus muscle homogenates from WT and PlnOE mice was determined as previously described (McMillan and Quadrilatero, 2011) using 2′,7′-dichlorodihydrofluorescein-diacetate (DCFH-DA, Invitrogen, Carlsbad, CA) at 37°C. Cellular esterases hydrolyze DCFH-DA to the non-fluorescent DCFH, which can then be oxidized by a variety of ROS to form highly fluorescent DCF. Fluorescence was measured using a SPECTRAmax Gemini XS microplate spectrofluorometer with excitation and emission wavelengths of 490 nm and 525 nm, respectively. Fluorescence intensity was normalized to total protein content and expressed as arbitrary units per mg protein.
Plasma CK analysis
WT and PlnOE mice were anesthetized using somnotol (0.65 mg/kg body weight) and blood from the left ventricle was drawn into a heparinized syringe. Blood was centrifuged at 5000 g for 8 min and the plasma was decanted and stored at −80°C until analysis. CK activity was measured using a kinetic fluorometric assay as previously described (Szasz et al., 1976).
Electrical stimulation and muscle contractility measurements
Experiments were performed on adult (4-6 month) WT and PlnOE mice. Mice were killed by cervical dislocation, and the intact soleus muscles were removed and placed into a bath with oxygenated Tyrode solution (95% O2, 5% CO2) containing 121 mM NaCl2, 5 mM KCl, 24 mM NaHCO3, 1.8 mM CaCl2, 0.4 mM NaH2PO4, 5.5 mM glucose, 0.1 mM EDTA and 0.5 mM MgCl2, pH 7.3 (Lännergren et al., 2000), and was maintained at 25°C. Muscles were situated between flanking platinum electrodes driven by a biphasic stimulator (Model 710B, Aurora Scientific) and electrically evoked muscle force was assessed across a range of stimulation frequencies from 1 to 100 Hz. Data were analyzed using Dynamic Muscle Control Data Acquisition software (Aurora Scientific). Specifically, peak isometric force amplitude (mN) and the maximal rates of force development (+dF/dt) and relaxation (−dF/dt) were determined during a twitch and across the range of stimulation frequencies. Peak isometric force was then normalized to muscle weight (mN/g).
Treadmill exercise
WT and PlnOE mice were assessed for muscle performance by running them to exhaustion on an enclosed motorized treadmill. All animals exercised at a running speed of 8 m/min for 10 min followed by 10 min at 16 m/min and then allowed to reach exhaustion at 24 m/min at a 5° incline.
Statistics
All values are presented as means±s.e. Statistical significance was set to P≤0.05. Comparisons between WT and PlnOE mice were performed using Students t-test; however, a two-way repeated-measures ANOVA was used for force–frequency analysis, and when age was included as a factor. Post hoc testing was done using Tukey's HSD. Comparisons between healthy controls and human CNM patients were performed using Student's t-test.
Supplementary Material
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
A.R.T., V.A.F. and E.B. conceived the study idea. V.A.F. coordinated western blot experiments and with K.T. performed the western blot analyses. E.B. and C.B. performed the histological and histochemical analysis. E.B., C.B. and E.M. performed the immunofluorescence staining experiments. V.A.F., D.G. and A.H. performed SERCA ATPase activity. V.A.F. and C.V. performed Ca2+-uptake measures. E.M. performed proteolytic activity and oxidative stress assays. V.A.F. and E.B. performed plasma creatine kinase analysis. B.J.W. and I.C.S. conducted soleus contractile analysis. V.A.F. conducted treadmill running. J.Q. and A.R.T. contributed reagents, materials and analysis tools. M.A.T. provided the muscle biopsies from CNM patients. A.R.T. and V.A.F. wrote the manuscript that was reviewed by all authors.
Funding
This work was supported by research grants from the Canadian Institutes of Health Research (CIHR) [grant numbers MOP 86618 and MOP 47296 to A.R.T.]. I.C.S. and C.V. were supported by postgraduate scholarship doctoral awards from the Natural Sciences and Engineering Research Council of Canada. V.A.F. was supported by a doctoral award from CIHR.
Supplementary material
Supplementary material available online at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.020859/-/DC1
References
- Agrawal P. B., Pierson C. R., Joshi M., Liu X., Ravenscroft G., Moghadaszadeh B., Talabere T., Viola M., Swanson L. C., Haliloğlu G. et al. (2014). SPEG interacts with myotubularin, and its deficiency causes centronuclear myopathy with dilated cardiomyopathy. Am. J. Hum. Genet. 95, 218-226. 10.1016/j.ajhg.2014.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Qusairi L. and Laporte J. (2011). T-tubule biogenesis and triad formation in skeletal muscle and implication in human diseases. Skelet. Muscle 1, 26 10.1186/2044-5040-1-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Qusairi L., Weiss N., Toussaint A., Berbey C., Messaddeq N., Kretz C., Sanoudou D., Beggs A. H., Allard B., Mandel J.-L. et al. (2009). T-tubule disorganization and defective excitation-contraction coupling in muscle fibers lacking myotubularin lipid phosphatase. Proc. Natl. Acad. Sci. USA 106, 18763-18768. 10.1073/pnas.0900705106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altamirano F., Lopez J. R., Henriquez C., Molinski T., Allen P. D. and Jaimovich E. (2012). Increased resting intracellular calcium modulates NF-kappaB-dependent inducible nitric-oxide synthase gene expression in dystrophic mdx skeletal myotubes. J. Biol. Chem. 287, 20876-20887. 10.1074/jbc.M112.344929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson D. M., Anderson K. M., Chang C.-L., Makarewich C. A., Nelson B. R., McAnally J. R., Kasaragod P., Shelton J. M., Liou J., Bassel-Duby R. et al. (2015). A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 160, 595-606. 10.1016/j.cell.2015.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahi M., Kurzydlowski K., Tada M. and MacLennan D. H. (2002). Sarcolipin inhibits polymerization of phospholamban to induce superinhibition of sarco(endo)plasmic reticulum Ca2+-ATPases (SERCAs). J. Biol. Chem. 277, 26725-26728. 10.1074/jbc.C200269200 [DOI] [PubMed] [Google Scholar]
- Asahi M., Sugita Y., Kurzydlowski K., De Leon S., Tada M., Toyoshima C. and MacLennan D. H. (2003). Sarcolipin regulates sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) by binding to transmembrane helices alone or in association with phospholamban. Proc. Natl. Acad. Sci. USA 100, 5040-5045. 10.1073/pnas.0330962100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhupathy P., Babu G. J. and Periasamy M. (2007). Sarcolipin and phospholamban as regulators of cardiac sarcoplasmic reticulum Ca2+ ATPase. J. Mol. Cell. Cardiol. 42, 903-911. 10.1016/j.yjmcc.2007.03.738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitoun M., Maugenre S., Jeannet P.-Y., Lacène E., Ferrer X., Laforêt P., Martin J.-J., Laporte J., Lochmüller H., Beggs A. H. et al. (2005). Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat. Genet. 37, 1207-1209. 10.1038/ng1657 [DOI] [PubMed] [Google Scholar]
- Bloemberg D. and Quadrilatero J. (2012). Rapid determination of myosin heavy chain expression in rat, mouse, and human skeletal muscle using multicolor immunofluorescence analysis. PLoS ONE 7, e35273 10.1371/journal.pone.0035273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloemberg D., McDonald E., Dulay D. and Quadrilatero J. (2014). Autophagy is altered in skeletal and cardiac muscle of spontaneously hypertensive rats. Acta Physiol 210, 381-391 10.1111/apha.12178. [DOI] [PubMed] [Google Scholar]
- Bohm J., Biancalana V., Malfatti E., Dondaine N., Koch C., Vasli N., Kress W., Strittmatter M., Taratuto A. L., Gonorazky H. et al. (2014). Adult-onset autosomal dominant centronuclear myopathy due to BIN1 mutations. Brain 137, 3160-3170. 10.1093/brain/awu272 [DOI] [PubMed] [Google Scholar]
- Calvo A. C., Manzano R., Atencia-Cibreiro G., Oliván S., Muñoz M. J., Zaragoza P., Cordero-Vázquez P., Esteban-Pérez J., García-Redondo A. and Osta R. (2012). Genetic biomarkers for ALS disease in transgenic SOD1(G93A) mice. PLoS ONE 7, e32632 10.1371/journal.pone.0032632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceyhan-Birsoy O., Agrawal P. B., Hidalgo C., Schmitz-Abe K., DeChene E. T., Swanson L. C., Soemedi R., Vasli N., Iannaccone S. T., Shieh P. B. et al. (2013). Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy. Neurology 81, 1205-1214. 10.1212/WNL.0b013e3182a6ca62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charge S. B. P. and Rudnicki M. A. (2004). Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 84, 209-238. 10.1152/physrev.00019.2003 [DOI] [PubMed] [Google Scholar]
- Cowling B. S., Toussaint A., Amoasii L., Koebel P., Ferry A., Davignon L., Nishino I., Mandel J.-L. and Laporte J. (2011). Increased expression of wild-type or a centronuclear myopathy mutant of dynamin 2 in skeletal muscle of adult mice leads to structural defects and muscle weakness. Am. J. Pathol. 178, 2224-2235. 10.1016/j.ajpath.2011.01.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowling B. S., Chevremont T., Prokic I., Kretz C., Ferry A., Coirault C., Koutsopoulos O., Laugel V., Romero N. B. and Laporte J. (2014). Reducing dynamin 2 expression rescues X-linked centronuclear myopathy. J. Clin. Invest. 124, 1350-1363. 10.1172/JCI71206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demonbreun A. R. and McNally E. M. (2014). Dynamin 2 the rescue for centronuclear myopathy. J. Clin. Invest. 124, 976-978. 10.1172/JCI74434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitt M. M., MacLeod H. M., Soliven B. and McNally E. M. (2006). Phospholamban R14 deletion results in late-onset, mild, hereditary dilated cardiomyopathy. J. Am. Coll. Cardiol. 48, 1396-1398. 10.1016/j.jacc.2006.07.016 [DOI] [PubMed] [Google Scholar]
- DiMario J. X., Uzman A. and Strohman R. C. (1991). Fiber regeneration is not persistent in dystrophic (MDX) mouse skeletal muscle. Dev. Biol. 148, 314-321. 10.1016/0012-1606(91)90340-9 [DOI] [PubMed] [Google Scholar]
- Dowling J. J., Lawlor M. W. and Dirksen R. T. (2014). Triadopathies: an emerging class of skeletal muscle diseases. Neurotherapeutics 11, 773-785. 10.1007/s13311-014-0300-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubowitz V., Sewry C. and Oldfors A. (2013). Muscle Biopsy: A Practical Approach. Oxford: Saunders. [Google Scholar]
- Duhamel T. A., Green H. J., Stewart R. D., Foley K. P., Smith I. C. and Ouyang J. (2007). Muscle metabolic, SR Ca(2+) -cycling responses to prolonged cycling, with and without glucose supplementation. J. Appl. Physiol. 103, 1986-1998. 10.1152/japplphysiol.01440.2006 [DOI] [PubMed] [Google Scholar]
- Fajardo V. A., Bombardier E., Vigna C., Devji T., Bloemberg D., Gamu D., Gramolini A. O., Quadrilatero J. and Tupling A. R. (2013). Co-expression of SERCA isoforms, phospholamban and sarcolipin in human skeletal muscle fibers. PLoS ONE 8, e84304 10.1371/journal.pone.0084304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folker E. S. and Baylies M. K. (2013). Nuclear positioning in muscle development and disease. Front. Physiol. 4, 363 10.3389/fphys.2013.00363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal A., Inczedy-Farkas G., Pal E., Remenyi V., Bereznai B., Geller L., Szelid Z., Merkely B. and Molnar M. J. (2015). The coexistence of dynamin 2 mutation and multiple mitochondrial DNA (mtDNA) deletions in the background of severe cardiomyopathy and centronuclear myopathy. Clin. Neuropathol 34, 89-95. 10.5414/NP300789 [DOI] [PubMed] [Google Scholar]
- Gehrig S. M., van der Poel C., Sayer T. A., Schertzer J. D., Henstridge D. C., Church J. E., Lamon S., Russell A. P., Davies K. E., Febbraio M. A. et al. (2012). Hsp72 preserves muscle function and slows progression of severe muscular dystrophy. Nature 484, 394-398. 10.1038/nature10980 [DOI] [PubMed] [Google Scholar]
- Gibbs E. M., Davidson A. E., Telfer W. R., Feldman E. L. and Dowling J. J. (2014). The myopathy-causing mutation DNM2-S619L leads to defective tubulation in vitro and in developing zebrafish. Dis. Model. Mech. 7, 157-161. 10.1242/dmm.012286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goonasekera S. A., Lam C. K., Millay D. P., Sargent M. A., Hajjar R. J., Kranias E. G. and Molkentin J. D. (2011). Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. J. Clin. Invest. 121, 1044-1052. 10.1172/JCI43844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski P. A., Glaves J. P., Vangheluwe P. and Young H. S. (2013). Sarco(endo)plasmic reticulum calcium ATPase (SERCA) inhibition by sarcolipin is encoded in its luminal tail. J. Biol. Chem. 288, 8456-8467. 10.1074/jbc.M112.446161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady R. M., Teng H., Nichol M. C., Cunningham J. C., Wilkinson R. S. and Sanes J. R. (1997). Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell 90, 729-738. 10.1016/S0092-8674(00)80533-4 [DOI] [PubMed] [Google Scholar]
- Haghighi K., Kolokathis F., Gramolini A. O., Waggoner J. R., Pater L., Lynch R. A., Fan G.-C., Tsiapras D., Parekh R. R., Dorn G. W. II. et al. (2006). A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc. Natl. Acad. Sci. USA 103, 1388-1393. 10.1073/pnas.0510519103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz J. D., Rosenson R. S., McMurray J. J. V., Marx N. and Remme W. J. (2011). Clinical Trials Update AHA Congress 2010. Cardiovasc. Drugs Ther. 25, 69-76. 10.1007/s10557-011-6285-9 [DOI] [PubMed] [Google Scholar]
- Jessup M., Greenberg B., Mancini D., Cappola T., Pauly D. F., Jaski B., Yaroshinsky A., Zsebo K. M., Dittrich H., Hajjar R. J. et al. (2011). Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation 124, 304-313. 10.1161/CIRCULATIONAHA.111.022889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungbluth H. and Gautel M. (2014). Pathogenic mechanisms in centronuclear myopathies. Front. Aging Neurosci. 6, 339 10.3389/fnagi.2014.00339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungbluth H., Zhou H., Sewry C. A., Robb S., Treves S., Bitoun M., Guicheney P., Buj-Bello A., Bönnemann C. and Muntoni F. (2007). Centronuclear myopathy due to a de novo dominant mutation in the skeletal muscle ryanodine receptor (RYR1) gene. Neuromuscul. Disord. 17, 338-345. 10.1016/j.nmd.2007.01.016 [DOI] [PubMed] [Google Scholar]
- Jungbluth H., Wallgren-Pettersson C. and Laporte J. (2008). Centronuclear (myotubular) myopathy. Orphanet. J. Rare Dis. 3, 26 10.1186/1750-1172-3-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knotts S., Rindt H., Neumann J. and Robbins J. (1994). In vivo regulation of the mouse beta myosin heavy chain gene. J. Biol. Chem. 269, 31275-31282. [PubMed] [Google Scholar]
- Lännergren J., Bruton J. D. and Westerblad H. (2000). Vacuole formation in fatigued skeletal muscle fibres from frog and mouse: effects of extracellular lactate. J. Physiol. 526, 597-611. 10.1111/j.1469-7793.2000.00597.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte J., Hu L. J., Kretz C., Mandel J.-L., Kioschis P., Coy J. F., Klauck S. M., Poustka A. and Dahl N. (1996). A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat. Genet. 13, 175-182. 10.1038/ng0696-175 [DOI] [PubMed] [Google Scholar]
- Liu N., Bezprozvannaya S., Shelton J. M., Frisard M. I., Hulver M. W., McMillan R. P., Wu Y., Voelker K. A., Grange R. W., Richardson J. A. et al. (2011). Mice lacking microRNA 133a develop dynamin 2-dependent centronuclear myopathy. J. Clin. Invest. 121, 3258-3268. 10.1172/JCI46267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas C. A., Kang L. H. D. and Hoh J. F. Y. (2000). Monospecific antibodies against the three mammalian fast limb myosin heavy chains. Biochem. Biophys. Res. Commun. 272, 303-308. 10.1006/bbrc.2000.2768 [DOI] [PubMed] [Google Scholar]
- MacLennan D. H. and Kranias E. G. (2003). Phospholamban: a crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 4, 566-577. 10.1038/nrm1151 [DOI] [PubMed] [Google Scholar]
- Mazala D. A. G., Pratt S. J. P., Chen D., Molkentin J. D., Lovering R. M. and Chin E. R. (2015). SERCA1 overexpression minimizes skeletal muscle damage in dystrophic mouse models. Am. J. Physiol. Cell Physiol. 308, C699-C709. 10.1152/ajpcell.00341.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan E. M. and Quadrilatero J. (2011). Differential apoptosis-related protein expression, mitochondrial properties, proteolytic enzyme activity, and DNA fragmentation between skeletal muscles. Am. J. Physiol. Regul. Integr. Comp. Physiol. 300, R531-R543. 10.1152/ajpregu.00488.2010 [DOI] [PubMed] [Google Scholar]
- Millay D. P., Goonasekera S. A., Sargent M. A., Maillet M., Aronow B. J. and Molkentin J. D. (2009). Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc. Natl. Acad. Sci. USA 106, 19023-19028. 10.1073/pnas.0906591106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamisawa S., Sato Y., Tatsuguchi Y., Fujino T., Imamura S.-i., Uetsuka Y., Nakazawa M. and Matsuoka R. (2003). Mutation of the phospholamban promoter associated with hypertrophic cardiomyopathy. Biochem. Biophys. Res. Commun. 304, 1-4. 10.1016/S0006-291X(03)00526-6 [DOI] [PubMed] [Google Scholar]
- Morita T., Hussain D., Asahi M., Tsuda T., Kurzydlowski K., Toyoshima C. and MacLennan D. H. (2008). Interaction sites among phospholamban, sarcolipin, and the sarco(endo)plasmic reticulum Ca(2+)-ATPase. Biochem. Biophys. Res. Commun. 369, 188-194. 10.1016/j.bbrc.2007.11.098 [DOI] [PubMed] [Google Scholar]
- Murphy R. M., Verburg E. and Lamb G. D. (2006). Ca2+ activation of diffusible and bound pools of μ-calpain in rat skeletal muscle. J. Physiol. 576, 595-612. 10.1113/jphysiol.2006.114090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa O., Arnold M., Nakagawa M., Hamada H., Shelton J. M., Kusano H., Harris T. M., Childs G., Campbell K. P., Richardson J. A. et al. (2005). Centronuclear myopathy in mice lacking a novel muscle-specific protein kinase transcriptionally regulated by MEF2. Genes Dev. 19, 2066-2077. 10.1101/gad.1338705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen thi M., Cartwright A. J., Morris G. E., Love D. R., Bloomfield J. F. and Davies K. E. (1990). Monoclonal antibodies against defined regions of the muscular dystrophy protein, dystrophin. FEBS Lett. 262, 237-240. 10.1016/0014-5793(90)80199-S [DOI] [PubMed] [Google Scholar]
- Nicot A.-S., Toussaint A., Tosch V., Kretz C., Wallgren-Pettersson C., Iwarsson E., Kingston H., Garnier J.-M., Biancalana V., Oldfors A. et al. (2007). Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat. Genet. 39, 1134-1139. 10.1038/ng2086 [DOI] [PubMed] [Google Scholar]
- Ottenheijm C. A. C., Fong C., Vangheluwe P., Wuytack F., Babu G. J., Periasamy M., Witt C. C., Labeit S. and Granzier H. (2008). Sarcoplasmic reticulum calcium uptake and speed of relaxation are depressed in nebulin-free skeletal muscle. FASEB J. 22, 2912-2919. 10.1096/fj.07-104372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattison J. S., Waggoner J. R., James J., Martin L., Gulick J., Osinska H., Klevitsky R., Kranias E. G. and Robbins J. (2008). Phospholamban overexpression in transgenic rabbits. Transgenic Res. 17, 157-170. 10.1007/s11248-007-9139-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rindt H., Gulick J., Knotts S., Neumann J. and Robbins J. (1993). In vivo analysis of the murine beta-myosin heavy chain gene promoter. J. Biol. Chem. 268, 5332-5338. [PubMed] [Google Scholar]
- Rindt H., Knotts S. and Robbins J. (1995). Segregation of cardiac and skeletal muscle-specific regulatory elements of the beta-myosin heavy chain gene. Proc. Natl. Acad. Sci. USA 92, 1540-1544. 10.1073/pnas.92.5.1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero N. B. (2010). Centronuclear myopathies: a widening concept. Neuromuscul. Disord. 20, 223-228. 10.1016/j.nmd.2010.01.014 [DOI] [PubMed] [Google Scholar]
- Sartore S., Gorza L. and Schiaffino S. (1982). Fetal myosin heavy chains in regenerating muscle. Nature 298, 294-296. 10.1038/298294a0 [DOI] [PubMed] [Google Scholar]
- Schiaffino S. and Reggiani C. (2011). Fiber types in mammalian skeletal muscles. Physiol. Rev. 91, 1447-1531. 10.1152/physrev.00031.2010 [DOI] [PubMed] [Google Scholar]
- Schiaffino S., Gorza L., Sartore S., Saggin L., Ausoni S., Vianello M., Gundersen K. and Lømo T. (1989). Three myosin heavy chain isoforms in type 2 skeletal muscle fibres. J. Muscle Res. Cell Motil. 10, 197-205. 10.1007/BF01739810 [DOI] [PubMed] [Google Scholar]
- Schmitt J. P., Kamisago M., Asahi M., Li G. H., Ahmad F., Mende U., Kranias E. G., MacLennan D. H., Seidman J. G. and Seidman C. E. (2003). Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 299, 1410-1413. 10.1126/science.1081578 [DOI] [PubMed] [Google Scholar]
- Schneider J. S., Shanmugam M., Gonzalez J. P., Lopez H., Gordan R., Fraidenraich D. and Babu G. J. (2013). Increased sarcolipin expression and decreased sarco(endo)plasmic reticulum Ca2+ uptake in skeletal muscles of mouse models of Duchenne muscular dystrophy. J. Muscle Res. Cell Motil. 34, 349-356. 10.1007/s10974-013-9350-0 [DOI] [PubMed] [Google Scholar]
- Song Q., Young K. B., Chu G., Gulick J., Gerst M., Grupp I. L., Robbins J. and Kranias E. G. (2004). Overexpression of phospholamban in slow-twitch skeletal muscle is associated with depressed contractile function and muscle remodeling. FASEB J. 18, 974-976. 10.1096/fj.03-1058fje [DOI] [PubMed] [Google Scholar]
- Szasz G., Gruber W. and Bernt E. (1976). Creatine kinase in serum: 1. Determination of optimum reaction conditions. Clin. Chem. 22, 650-656. [PubMed] [Google Scholar]
- Tarnopolsky M. A., Pearce E., Smith K. and Lach B. (2011). Suction-modified Bergstrom muscle biopsy technique: experience with 13,500 procedures. Muscle Nerve 43, 716-725. 10.1002/mus.21945 [DOI] [PubMed] [Google Scholar]
- Tupling A. R. (2009). Excitation-contraction coupling. In Encyclopedia of Neuroscience (ed. Binder M., Hirokawa N. and Windhorst U.), pp. 1479-1483. Berlin; Heidelberg: Springer. [Google Scholar]
- Tupling R. and Green H. (2002). Silver ions induce Ca2+ release from the SR in vitro by acting on the Ca2+ release channel and the Ca2+ pump. J. Appl. Physiol. 92, 1603-1610. 10.1152/japplphysiol.00756.2001 [DOI] [PubMed] [Google Scholar]
- Tupling A. R., Asahi M. and MacLennan D. H. (2002). Sarcolipin overexpression in rat slow twitch muscle inhibits sarcoplasmic reticulum Ca2+ uptake and impairs contractile function. J. Biol. Chem. 277, 44740-44746. 10.1074/jbc.M206171200 [DOI] [PubMed] [Google Scholar]
- Wilmshurst J. M., Lillis S., Zhou H., Pillay K., Henderson H., Kress W., Müller C. R., Ndondo A., Cloke V., Cullup T. et al. (2010). RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann. Neurol. 68, 717-726. 10.1002/ana.22119 [DOI] [PubMed] [Google Scholar]
- Zsebo K., Yaroshinsky A., Rudy J. J., Wagner K., Greenberg B., Jessup M. and Hajjar R. J. (2014). Long-term effects of AAV1/SERCA2a gene transfer in patients with severe heart failure: analysis of recurrent cardiovascular events and mortality. Circ. Res. 114, 101-108. 10.1161/CIRCRESAHA.113.302421 [DOI] [PubMed] [Google Scholar]
- Zubrzycka-Gaarn E., MacDonald G., Phillips L., Jorgensen A. O. and MacLennan D. H. (1984). Monoclonal antibodies to the Ca2++Mg2+-dependent ATPase of sarcoplasmic reticulum identify polymorphic forms of the enzyme and indicate the presence in the enzyme of a classical high-affinity Ca2+ binding site. J. Bioenerg. Biomembr. 16, 441-464. 10.1007/BF00743238 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.