

Abstract
The metabolic instability of an antitubercular small molecule CD117 was addressed through iterative alteration of a key sulfide substituent and interrogation of the effect on growth inhibition of cultured Mycobacterium tuberculosis. This process was informed by studies of the intramycobacterial metabolism of CD117 and its inactive carboxylic acid derivative. Isoxazole 4e and thiazole 4m demonstrated significant gains in mouse liver microsomal stability with slight losses in whole-cell activity. This work illustrates the challenges of antitubercular hit evolution, requiring a balance of chemical and biological insights.
Keywords: tuberculosis, thienopyrimidine, metabolomics, prodrug
Tuberculosis (TB) represents a global health pandemic, on a per annum basis infecting 9.0 million people and leading to the death of 1.5 million.1 The causative agent – Mycobacterium tuberculosis – poses significant challenges to the discovery and development of novel therapeutic treatments due to drug resistance, mycobacterial persistence, and latency.2 In particular, the ability of M. tuberculosis to mutate genes directly or indirectly related to the mode of action of TB drugs has led to multi drug-, extensively drug-, and totally drug-resistant strains of M. tuberculosis and severely if not completely diminished the effectiveness of approved therapies.3
We have been focused on the issue of resistance to the front-line drug isoniazid (INH). INH is arguably the most important component of TB front-line therapy and targets predominantly the enoyl acyl-carrier protein reductase (InhA).4 INH is activated by the M. tuberculosis catalase-peroxidase KatG to form a probable isonicotinoyl radical which reacts with NAD+/NADH to inhibit InhA. Given this requirement for activation of the prodrug INH, it is not surprising that the majority of clinical INH-resistant mutants harbor mutations in katG.5 Thus, we and other have focused on the development of InhA inhibitors that do not require KatG activation and reports have highlighted a variety of chemotypes include diaryl ether phenols,6-8 pyrrolidine carboxamides,9 pyrazoles,10 proline amides,11 and thienopyrimidines.12
Our own work on the thienopyrimidine class of InhA inhibitors led to a hit – CD117 (Table 1) – that exhibited in vitro bactericidal activity against drug-sensitive and multidrug-resistant strains of M. tuberculosis (e.g, MIC = minimum inhibitory concentration for growth inhibition of bacteria = 0.39 μg/mL versus the H37Rv lab strain which is used in all experiments reported herein) and lack of significant relative cytotoxicity to Vero cells (CC50 = amount of compound inhibiting 50% of cultured cell growth = 47 μg/mL; SI = selectivity index = CC50/MIC = 120). Preliminary mechanistic studies evidenced a complex mechanism of action where InhA inhibition is accompanied by modulation of at least one additional target within mycobacterial fatty acid biosynthesis (FAS).12 Clearly, InhA does not represent the dominant target of CD117 given an IC50 (concentration of compound required to inhibit 50% of the enzymatic activity) of 34 ± 6 μM and an MIC of 0.39 μg/mL (1.0 μM). Unfortunate for mechanistic studies but promising for the drug discovery value of CD117, we were unable to obtain CD117-resistant mutants when plating 10 and 20 fold the MIC of CD117 on 1 × 109 CFU of M. tuberculosis.
Table 1.
CD117, its carboxylic acid derivative, and ester analogs compared by their whole-cell activity versus M. tuberculosis H37Rv
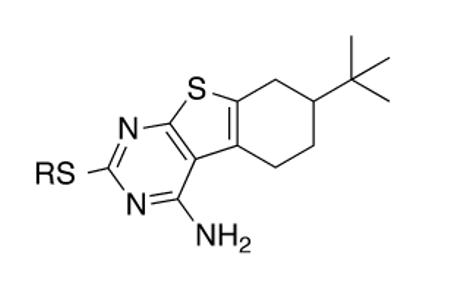
| Cmpd | R | MIC (μg/mL) |
|---|---|---|
| CD117 | CH2CO2CH2CH3 | 0.39 |
| 1a | CH2CO2H | >200 |
| 1b | CH2CO2(CH2)2CH3 | 1.5 |
| 1c | CH2CO2CH(CH3)2 | 3.1 |
| 1d | CH2CO2(CH2)3CH3 | 1.5 |
| 1e | CH2CO2CH(CH3)CH2CH3 | 1.5 |
| 1f | CH2CO2CH2(c-C6H12) | 0.78 |
| 1g | CH2CO2CH2Ph | 1.5 |
| 1h | CH2CO2C(CH3)2Ph | 25 |
| 1i | CH2CO2C(CH3)3 | >200 |
| 2a |
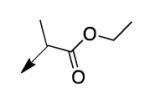
|
6.2 |
| 2b |
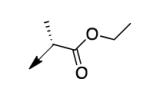
|
3.1 |
| 2c |
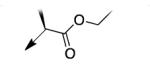
|
12.5 |
| 2d |
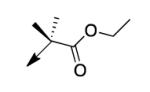
|
>200 |
| 2e |
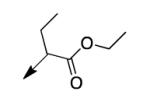
|
>200 |
| 2f |
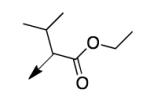
|
200 |
| 2g |
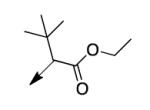
|
>200 |
Given our early lack of mechanistic information to guide a target-based approach, we initially proposed to pursue the evolution of CD117 as a chemical tool and/or drug discovery hit through a medicinal chemistry-guided optimization of the thienopyrimidine structure with primary feedback from whole-cell growth inhibition of M. tuberculosis (MIC) and in vitro metabolism assays. This report will discuss the initial profiling of CD117 that informed the medicinal chemistry approach, the pursuit of this strategy and how it was altered by metabolomics studies to provide evolved thienopyrimidine antituberculars.
The promising in vitro efficacy profile previously reported for CD11712 spurred us to explore its potential for assessment in an acute mouse model of infection. CD117 was not available commercially in gram quantities and, thus, we designed a modular and scalable synthesis (Figure 1). Commercial 4-t-Bu-cyclohexanone subjected to Gewald conditions afforded an intermediate aminonitrile in 65% yield.13 The aminonitrile reacted quantitatively with benzoylisothiocyanate to deliver an intermediate benzoylthiourea that upon exposure to sodium hydroxide in ethanol with heating14 provided a presumed intermediate thiolate that was alkylated with ethyl bromoacetate to deliver CD117 in 57% yield and with an overall yield of 29% over the four steps.
Figure 1.

Synthetic scheme to afford CD117 and its S-substituted analogs
This procedure afforded multi-gram quantities of CD117 and facilitated in vivo studies in Balb/c mice. Formulated in PEG200, CD117 was administered to the mice in groups of 3 at a single dose of 100, 300, or 500 mg/kg. After two days, the mice did not exhibit any overt signs of toxicity. Thus, 300 and 500 mg/kg single doses were administered to three groups of mice (2 by oral gavage and 1 by intraperitoneal injection) in a serum bactericidal assessment. After 2 h, the mice were exsanguinated and the serum was added to M. tuberculosis H37Rv cultures. The cultures were incubated for 3 days and then plated for colony-forming units. Sera obtained from CD117-treated mice did not show a reduction in bacterial counts as compared to the no-drug control (Supplementary Data; Figure S1). LC-MS analysis (data now shown) failed to quantify CD117 in the sera, lending doubt to the compound’s stability in mouse blood. Confirmatory of this concern were mouse liver microsomal stability studies demonstrating rapid hydrolysis (t1/2 < 1 min in the presence or absence of NADPH) of CD117 to a compound whose mass (via LCMS) was consistent with the carboxylic acid analog of CD117. Synthesis of this proposed metabolite – compound 1a – was achieved by lithium hydroxide-mediated hydrolysis of CD117. The MIC of this compound was >200 μg/mL (Table 1), supporting the hypothesis that the absence of activity in the mouse serum bactericidal assay was due to rapid metabolism of CD117 to the corresponding whole-cell inactive 1a. It is important, however, to note that CD117 was stable in the growth media for Middlebrook 7H9 media supplemented with albumin, dextrose, and saline (Supplementary Data; Figure S2).
Efforts to address the metabolic stability of CD117, thus, commenced by focusing on the preparation of a range of esters with the hope that a larger/bulkier ester substituent would at least maintain whole-cell activity while improving metabolic stability. The synthetic route to CD117 was adapted through modification of the final electrophile to include various alkyl bromoacetates, which were purchased or prepared via Fischer esterification of the commercial carboxylic acid. Esters 1b – 1i were assayed versus M. tuberculosis and a clear trend was observable: the whole-cell efficacy decreased with increasing bulk of the ester substituent. Most prominent was the lack of significant activity of α,α-dimethylphenyl ester 1h and t-butyl ester 1i, while smaller ester substituents (1b – 1g) afforded compounds with MIC values within 4 – 8 fold of that for CD117. Disappointingly, the mouse liver microsomal stability of the i-Pr ester 1c (MIC = 3.1 μg/mL) and the t-butyl ester 1i were both negligible (0.1% remaining after 1 h in the presence of NADPH; 1.6% and 0.10%, respectively, remaining after 1 h in the absence of NADPH).
A second strategy was to potentially preclude metabolism of the ethyl ester of CD117 via steric blockade through mono- or di-substitution at the α-carbonyl position. The requisite analogs of CD117 (2a – 2f) were prepared via ethyl bromoacetates with substitution at the αcarbonyl position. Interestingly, mono-methyl substitution at the α-position (compound 2a with a racemic mixture of epimers) exhibited an MIC = 6.2 μg/mL with the (R)-epimer in 2b being slightly more active (MIC = 3.1 μg/mL) than the (S)-epimer in 2c (MIC = 12.5 μg/mL). However, the dimethyl analog 2d was inactive (MIC > 200 μg/mL) as were the mono- ethyl (2e; MIC >200 μg/mL), i-Pr (e; MIC = 200 μg/mL) and the t-butyl (f; MIC > 200 μg/mL) derivatives. Unfortunately, 2a did not exhibit an improvement in mouse liver microsomal stability over CD117 (0% remaining after 1 h in presence of NADPH, 0.32% remaining after 1 h in absence of NADPH). These results, coupled with those for the alkyl ester variants, cast doubt on the likelihood of finding an ester analog with both significant whole-cell activity and mouse liver microsomal stability.
We retreated to investigate the essentiality of the ester moiety for whole-cell efficacy. Simple alkyl groups (methyl 3a, ethyl 3b, ethoxyethyl 3c, and benzyl 3d), prepared via an alkyl halide input in the CD117 synthesis), led to significant losses in whole-cell activity. Combined with knowledge of the MIC of >200 μg/mL for the corresponding ketone 3e, prepared using 1-bromo-2-butanone as the electrophile, these structure-activity relationship data (SAR) demonstrate the importance of the ester moiety of CD117 for whole-cell activity. Given the previously reported MIC of 12 μg/mL for the parent amide 3f,12 a small range of amides (3g – 3j) was then prepared, via a bromoacetamide input – synthesized from the corresponding amine and bromoacetyl bromide (Supplementary Data). Simple substitutions (methyl, phenyl, or benzyl) to provide secondary or tertiary amides (3g – 3j) led to MIC values of >200 μg/mL.
The compiled SAR thus far supported a prodrug hypothesis: the ethyl ester of CD117 favors permeability of the waxy cell wall of M. tuberculosis15 while offering sufficient chemical lability to be hydrolyzed within the cell to afford the carboxylic acid 1a that is not cell-permeable due to its hydrophilicity/charge. Given the advances in M. tuberculosis metabolomics16-18 building on early work by the laboratories of Rabinowitz and Botstein,19-23 we were aware of the potential to semi-quantitatively assess this hypothesis. Following published procedures,16 we demonstrated that both CD117 and its carboxylic acid analog 1a permeate the M. tuberculosis cell wall (Supplementary Data; Figure S3) in a dose-dependent fashion. It is notable that CD117 is partially hydrolyzed within M. tuberculosis to afford 1a (Supplementary Data; Figure S4).
The metabolomics data focusing on the fate of CD117 (data and analysis with a focus on M. tuberculosis metabolite pools will be published elsewhere) demonstrated that the lack of whole-cell efficacy of acid 1a is not due to an inability to accumulate within M. tuberculosis. This observation was contrary to our ester prodrug hypothesis. In fact, 1a was found to accumulate within CD117-treated M. tuberculosis, suggesting CD117 may suffer partial hydrolysis within the intramycobacterial environment. Our chemocentric evolution of CD117, therefore, shifted to ester isosteres that would maintain the antitubercular whole-cell efficacy of CD117 but be stable with regard to hydrolysis (both within M. tuberculosis and the mouse).24 Specifically, we sought to replace the ethyl ester with various heterocycles that would mimic aspects of the moiety (hydrogen bonding and/or hydrophobic), while likely being more metabolically stable. The synthesis of analogs 4a – 4r relied on shifting the electrophile input to the requisite halomethyl heterocycle. 2-pyridyl (4a) displayed very modest activity (MIC = 25 μg/mL) while the 3- (4b) and 4-analogs (4c) were essentially whole-cell inactive (MIC > 200 μg/mL). Isoxazole 4d demonstrated an MIC = 25 μg/mL and interestingly its 5-methyl analog 4e had an MIC = 3.1 μg/mL while the 5-phenyl 4f and benzoxazole 4g were essentially inactive. Linkage of the isoxazole to the sulfide through the 2-position led to a loss of activity with either a 5-methyl (4h; MIC = 50 μg/mL) or 5-methoxy substituent (4i; MIC > 200 μg/mL). While the oxazoles 4j – 4l did not approach the whole-cell activity of 4e, thiazole 4m had an MIC = 6.1 μg/mL. Close analogs of 4m, such as 4n and 4o, were much less efficacious (MIC = 50 μg/mL). Also, imidazole 4p and pyrazoles 4q and 4r featured higher MIC values.
With our identification of two CD117 analogs with MIC values within 6–12 fold of the original hit compound, we assessed their mouse liver microsomal stability. Isoxazole 4e remained in the amounts of 13.6% and 98.3% after 1 h in the presence and absence of NADPH, respectively. Thiazole 4m was present to the extents of 37.8% and 99.8% with and without NADPH, respectively, after 1 h. These significant important improvements in mouse metabolic stability within the CD117 chemical series will spur further profiling of 4e, 4m, and their close analogs for key physiochemical and Absorption-Distribution-Metabolism-Excretion properties before seeking to evaluate them in a mouse model of infection. We have begun by determining the Vero cell CC50 for 4e and 4m to be 31 (SI = 10) and 12 μg/mL (SI = 1.9), respectively.
In conclusion, we report an optimization strategy for our antitubercular hit compound CD117 that relied on a chemocentric approach enlightened by a significant application of M. tuberculosis metabolomics to inform us of the metabolic fate of the initial hit within the bacteria. We are exploring further use of a metabolomics platform in the evolution of antitubercular small molecules, as the effects of mycobacterial metabolism are crucial to the design of more potent compounds. Finally, it is critical to mention that this hit evolution process proceeded without consideration of the M. tuberculosis biological target/s of CD117. It may be presumptuous to assume that small alterations to the structure of these thienopyrimidines do not alter their target profiles. Given what we know about the mechanism of action of and the potential for target synergy being critical to bactericidal activity, we assert that assaying for both whole-cell growth inhibition and modulation of M. tuberculosis target/s (e.g., InhA) and fatty acid biosynthetic pathways (e.g., FAS-I25 and FAS-II26), may represent a more informed and efficient process to optimize antituberculars. This will be conducted with regard to CD117, 4e, and 4m while also leveraging our demonstrated pharmacophore27,28 and machine-learning methods.29
Supplementary Material
Table 2.
CD117 alkyl, ketone, and amide analogs compared by their whole-cell activity versus M. tuberculosis H37Rv
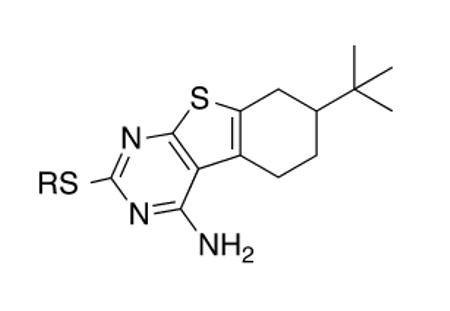
| Cmpd | R | MIC (μg/mL) |
|---|---|---|
| 3a | CH3 | 25 |
| 3b | CH2CH3 | 200 |
| 3c | (CH2)2OCH2CH3 | >100 |
| 3d | CH2C6H5 | >200 |
| 3e |
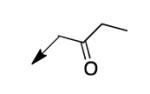
|
>200 |
| 3f |
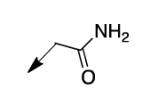
|
12 |
| 3g |
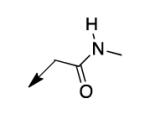
|
>200 |
| 3h |
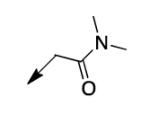
|
>200 |
| 3i |
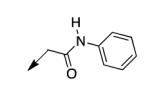
|
>200 |
| 3j |
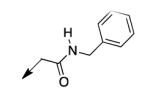
|
>200 |
Table 3.
CD117 heterocyclic analogs compared by their whole-cell activity versus M. tuberculosis H37Rv
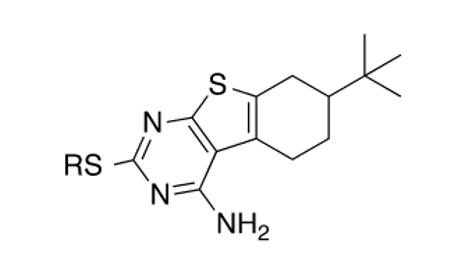
| Cmpd | R | MIC (μg/mL) |
|---|---|---|
| 4a |
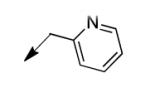
|
25 |
| 4b |
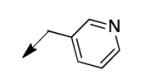
|
>200 |
| 4c |
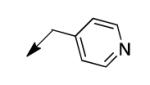
|
>200 |
| 4d |
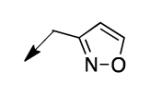
|
25 |
| 4e |
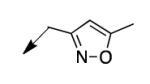
|
3.1 |
| 4f |
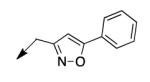
|
>200 |
| 4g |
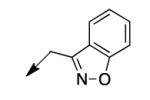
|
>200 |
| 4h |
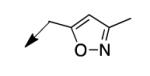
|
50 |
| 4i |
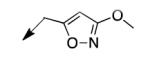
|
>200 |
| 4j |
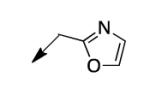
|
25 |
| 4k |
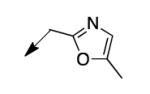
|
12.5 |
| 4l |
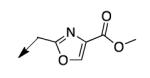
|
>200 |
| 4m |
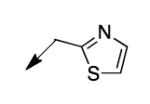
|
6.2 |
| 4n |
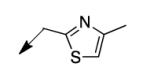
|
50 |
| 4o |
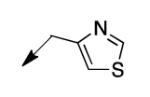
|
50 |
| 4p |
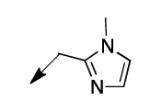
|
25 |
| 4q |
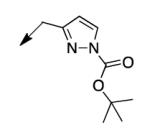
|
>200 |
| 4r |
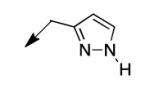
|
100 |
Acknowledgements
J.S.F. and S.E. acknowledge support from award number R44TR000942-02 “Biocomputation across distributed private datasets to enhance drug discovery” from the National Institutes of Health and National Center for Advancing Translational Sciences. J.S.F. also acknowledges support from the New Jersey Health Foundation. W.R.J. acknowledges generous support from the National Institutes of Health Grant AI26170. K.Y.R. acknowledges support from the Bill & Melinda Gates Foundation TB Drug Accelerator and Burroughs Wellcome Career Award in the Biomedical Sciences.
Footnotes
Supplementary data (Experimentals for synthesized compounds and biological assays and referenced supplementary figures) associated with this article may be found in the online version.
This paper is dedicated to the memory of Professor Harry H. Wasserman.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- (1).Global Tuberculosis Report. World Health Organization; 2014. [Google Scholar]
- (2).Sacchettini JC, Rubin EJ, Freundlich JS. Nat. Rev. Microbiol. 2008;6:41. doi: 10.1038/nrmicro1816. [DOI] [PubMed] [Google Scholar]
- (3).Dheda K, Gumbo T, Gandhi NR, Murray M, Theron G, Udwadia Z, Migliori GB, Warren R. Lancet. Respir. Med. 2014;2:321. doi: 10.1016/S2213-2600(14)70031-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Vilchèze C, Jacobs WR., Jr. Annu. Rev. Microbiol. 2007;61:35. doi: 10.1146/annurev.micro.61.111606.122346. [DOI] [PubMed] [Google Scholar]
- (5).Hazbon MH, Brimacombe M, Bobadilla del Valle M, Cavatore M, Guerrero MI, Varma-Basil M, Billman-Jacobe H, Lavender C, Fyfe J, Garcia-Garcia L, Leon CI, Bose M, Chaves F, Murray M, Eisenach KD, Sifuentes-Osornio J, Cave MD, Ponce de Leon A, Alland D. Antimicrob. Agents Chemother. 2006;50:2640. doi: 10.1128/AAC.00112-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Freundlich JS, Wang F, Vilchèze C, Gulten G, Langley R, Schiehser GA, Jacobus DP, Jacobs WRJ, Sacchettini JC. ChemMedChem. 2009;4:241. doi: 10.1002/cmdc.200800261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Pan P, Tonge PJ. Curr. Top. Med. Chem. 2012;12:672. doi: 10.2174/156802612799984535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sullivan TJ, Truglio JJ, Boyne ME, Novichenok P, Zhang X, Stratton CF, Li H-J, Kaur T, Amin A, Johnson F, Slayden RA, Kisker C, Tonge PJ. ACS Chem. Biol. 2006;1:43. doi: 10.1021/cb0500042. [DOI] [PubMed] [Google Scholar]
- (9).He X, Alian A, Stroud R, Ortiz de Montellano PR. J. Med. Chem. 2006;49:6308. doi: 10.1021/jm060715y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kuo MR, Morbidoni HR, Alland D, Sneddon SF, Gourlie BB, Staveski MM, Leonard M, Gregory JS, Janjigian AD, Yee C, Musser JM, Kreiswirth B, Iwamoto H, Perozzo R, Jacobs WR, Jr., Sacchettini JC, Fidock DA. J. Biol. Chem. 2003;278:20851. doi: 10.1074/jbc.M211968200. [DOI] [PubMed] [Google Scholar]
- (11).Encinas L, O’Keefe H, Neu M, Remuinan MJ, Patel AM, Guardia A, Davie CP, Perez-Macias N, Yang H, Convery MA, Messer JA, Perez-Herran E, Centrella PA, Alvarez-Gomez D, Clark MA, Huss S, O’Donovan GK, Ortega-Muro F, McDowell W, Castaneda P, Arico-Muendel CC, Pajk S, Rullas J, Angulo-Barturen I, Alvarez-Ruiz E, Mendoza-Losana A, Ballell Pages L, Castro-Pichel J, Evindar G. J. Med. Chem. 2014;57:1276. doi: 10.1021/jm401326j. [DOI] [PubMed] [Google Scholar]
- (12).Vilcheze C, Baughn AD, Tufariello J, Leung LW, Kuo M, Basler CF, Alland D, Sacchettini JC, Freundlich JS, Jacobs WR., Jr Antimicrob. Agents Chemother. 2011;55:3889. doi: 10.1128/AAC.00266-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Rosowsky A, Chaykovsky M, Chen KK, Lin M, Modest EJ. J. Med. Chem. 1973;16:185. doi: 10.1021/jm00261a002. [DOI] [PubMed] [Google Scholar]
- (14).Hacker HG, de la Haye A, Sterz K, Schnakenburg G, Wiese M, Gutschow M. Bioorg. Med. Chem. Lett. 2009;19:6102. doi: 10.1016/j.bmcl.2009.09.023. [DOI] [PubMed] [Google Scholar]
- (15).Kieser KJ, Rubin EJ. Nat. Rev. Microbiol. 2014;12:550. doi: 10.1038/nrmicro3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Chakraborty S, Gruber T, Barry CE, 3rd, Boshoff HI, Rhee KY. Science. 2013;339:88. doi: 10.1126/science.1228980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).de Carvalho LP, Darby CM, Rhee KY, Nathan C. ACS Med. Chem. Lett. 2011;2:849. doi: 10.1021/ml200157f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).de Carvalho LP, Fischer SM, Marrero J, Nathan C, Ehrt S, Rhee KY. Chem. Biol. 2010;17:1122. doi: 10.1016/j.chembiol.2010.08.009. [DOI] [PubMed] [Google Scholar]
- (19).Brauer MJ, Yuan J, Bennett BD, Lu W, Kimball E, Botstein D, Rabinowitz JD. Proc. Natl. Acad. Sci. 2006;103:19302. doi: 10.1073/pnas.0609508103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Bajad SU, Lu W, Kimball E, Yuan J, Peterson C, Rabinowitz JD. J. Chromatogr. A. 2006;1125:76. doi: 10.1016/j.chroma.2006.05.019. [DOI] [PubMed] [Google Scholar]
- (21).Lu W, Kimball E, Rabinowitz JD. J. Am. Soc. Mass Spectrom. 2006;17:37. doi: 10.1016/j.jasms.2005.09.001. [DOI] [PubMed] [Google Scholar]
- (22).Munger J, Bajad SU, Coller HA, Shenk T, Rabinowitz JD. PLoS Pathog. 2006;2:e132. doi: 10.1371/journal.ppat.0020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Yuan J, Rabinowitz JD. J. Am. Chem. Soc. 2007;129:9294. doi: 10.1021/ja072997c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Meanwell NA. J. Med. Chem. 2011;54:2529. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- (25).Zimhony O, Cox JS, Welch JT, Vilcheze C, Jacobs WR., Jr. Nature Med. 2000;6:1043. doi: 10.1038/79558. [DOI] [PubMed] [Google Scholar]
- (26).Bhatt A, Molle V, Besra GS, Jacobs WR, Jr., Kremer L. Mol. Microbiol. 2007;64:1442. doi: 10.1111/j.1365-2958.2007.05761.x. [DOI] [PubMed] [Google Scholar]
- (27).Lamichhane G, Freundlich JS, Ekins S, Wickramaratne N, Nolan ST, Bishai WR. MBio. 2011;2:e00301. doi: 10.1128/mBio.00301-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Sarker M, Talcott C, Madrid P, Chopra S, Bunin BA, Lamichhane G, Freundlich JS, Ekins S. Pharm. Res. 2012;29:2115. doi: 10.1007/s11095-012-0741-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ekins S, Reynolds RC, Kim H, Koo M-S, Ekonomidis M, Talaue M, Paget SD, Woolhiser LK, Lenaerts A, Bunin BA, Connell N, Freundlich JS. Chem. Biol. 2013;20:370. doi: 10.1016/j.chembiol.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.