

Summary
There are two main mechanisms by which cells become multidrug resistant (MDR): by increasing drug efflux pumps on the cell membrane and by increasing anti-apoptotic pathways. The use of nanotechnology to develop nanodelivery systems has allowed researchers to overcome limitations of antineoplastic drugs by increasing the solubility of the drug and decreasing the toxicity to healthy tissues. By encapsulating drugs into nanoparticles that bypass the efflux pumps, drug efflux is reduced, hence increasing the intracellular concentration of the drug. siRNA has the ability to disrupt cellular pathways by knocking down genes, opening the door to down regulating anti-apoptotic pathways.
The use of nanocarriers to deliver siRNA, prevents both renal clearance and RNase degradation by protecting siRNA chains, increasing their half life in blood. It has been suggested that co-delivering drugs and siRNA together in the same delivery system would be more effective in overcoming resistance of cancer cells than co-treatment of cancer cells with delivery systems carrying either siRNA or drugs. In this study we discuss the progress of nanoscale co-delivery systems in overcoming multidrug cancer resistance.
Keywords: Co-delivery, siRNA, Cancer therapy, Multi drug resistance (MDR), Nanoparticles, Doxorubicin
Introduction
Chemotherapeutical agents present many limitations that hinder the effectiveness of chemotherapy: poor solubility in aqueous solutions (making them difficult to administer) [1], non-specific distribution throughout the body (which causes insufficient penetration to tumors) [2], toxicity to healthy tissues [3] (which limits the dose and frequency of the treatment) and cancer cell resistance [4].
There are two main mechanisms by which cells become multidrug resistant (MDR): by increasing drug efflux pumps on the cell membrane and by increasing anti-apoptotic pathways [5]. An increase in efflux pumps causes a decrease in intracellular concentration of the drug, compromising the efficiency of the treatment [6]. The onset of nanotechnology has fostered the ability to provide solutions to these limitations, by encapsulating drugs in hydrophilic nanocarriers, which increases the solubility of the drug, decreases the toxicity to healthy tissues, and bypasses the effect of the efflux pumps [7,8].
Although the initial limitations of antineoplastic drugs have been mitigated through the use of nanocarriers [9,10], new limitations have arisen. Cells have developed strategies to avoid death, increasing their resistance to chemotherapy, by the activation of anti-apoptotic pathways [11]. Small interference RNA (siRNA) has the ability to disrupt cellular pathways by knocking down genes, opening the door for new treatments of diseases caused by aberrant gene expression [12,13]. However, siRNA chains exhibit a short half-life in blood if injected intravenously due to intravascular degradation by the catalytic activity of Ribonuclease (RNase) enzymes present in the bloodstream [13]. Rapid systemic clearance of siRNA through the renal system, low selectivity for the desired tissue, and poor cellular uptake have been reported for intravenous injection of siRNA in chemotherapeutic studies, decreasing the effectiveness of the therapy.
The use of nanocarriers for siRNA encapsulation can prevent both renal clearance and RNase degradation, effectively increasing its half-life in blood [14,15]. Promising results have been shown in which resistant cancer cells were sensitized to chemotherapy by knocking down one of the multidrug resistance mechanisms [16]. While some types of cancer cells were completely sensitized to antineoplastic drugs by siRNA therapy, others maintained their resistance to the treatment, compensating the loss of one type of resistance mechanism by increasing other multidrug resistance mechanisms.
Even though co-treatment of cancer cells with nano delivery systems carrying either siRNA or drugs proved to be important in decreasing resistance of cancer cells [17—20], it has been proposed that co-delivering drugs and siRNA together in the same delivery system would be more effective in overcoming resistance of cancer cells [21]. In this study we discuss the progress of nanoscale co-delivery systems in overcoming multidrug cancer resistance.
Mechanisms of cancer drug resistance
The treatment of cancer cells has evolved from general drugs targeting DNA, such as doxorubicin or cisplatin, to more specific molecules that target overexpressed proteins or upregulated pathways present in cancer cells [22]. The specificity of DNA binding chemotherapy slightly directed toward cancer cells rather than healthy cells, has improved cancer treatment. However, due to the high toxicity of most chemotherapeutical agents and the severity of the secondary effects, cancer treatment with chemotherapy requires convalescence time to allow patients to recover from treatment to treatment [23]. Since tumors have heterogeneous populations of cancer cells [24], only sensitive cancer cells within the tumor will die due to chemotherapy, remaining intact only those that are drug resistant remain intact. This allows for a population of almost exclusively non-resistant cancer cells to become a resistant population, making each chemotherapy session less effective (Fig. 1). The lack of effectiveness of chemotherapy to treat cancer is due to several factors, but mostly due to low concentration of drug reaching its target and/or poor effectiveness in killing cancer cells, even when the target its reached [25].
Figure 1.
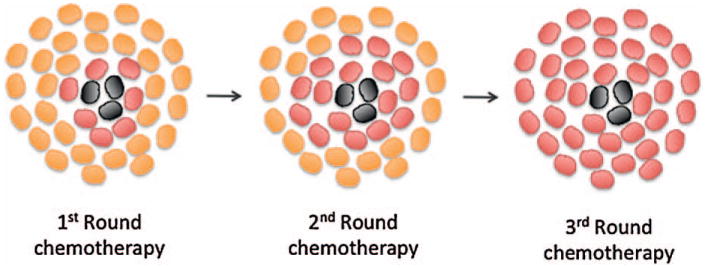
Acquired resistance of a cancer cell population after intervals of chemotherapy. After the first round of chemotherapy, cell population decreases significantly due to the death of sensitive cancer cells. Recovery time between chemotherapy sessions allows for resistant cells to grow and take over the entire population.
The vast majority of tumors (85%) in cancer patients are solid tumors [26]. The first line of treatment for solid tumors is surgery, when possible, followed by radiation and chemotherapy. Drugs that are injected systemically need to reach the cells within the tumor. Because tumors are highly irrigated with abnormal vasculature that leave gaps between the endothelial cells, but poorly irrigated with lymphatic vessels, drugs can easily leak from the blood vessels to the interstitial space within the tumor minimizing clearance through the lymphatic system. This is known as the enhance permeability and retention effect (EPR) [27]. Drugs that reach the tumor must be taken up by cancer cells [28].
Transport across the cell membrane depends on the type of molecule. Alterations in the transport of drugs across the membrane and/or efflux of internalized drugs from the intracellular compartment to the extracellular matrix is one of the major causes of cancer resistance [29]. Drugs that reach their target within the cell start a cascade of reactions that lead cells to enter programmed cell death, known as apoptosis [30]. A wide variety of cells, from many types of cancers, present different strategies to prevent apoptosis induced by chemotherapy, i.e. by upregulating anti-apoptosis pathways, altering cell cycle checkpoints, increasing repair mechanism, etc. [5]. Here we describe the two major mechanism of cancer resistance (Fig. 2).
Figure 2.
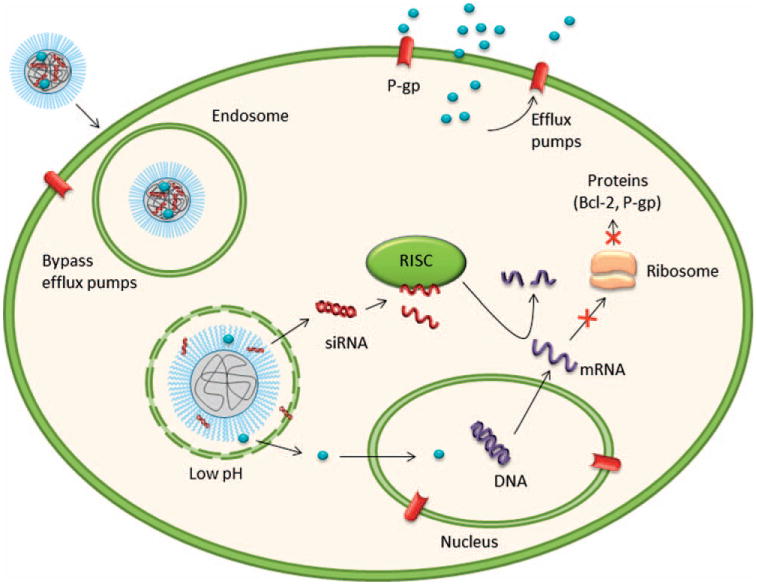
Mechanism of drug resistance and sensitization of cancer cells by co-delivering siRNA and an antineoplastic agent. Drugs encapsulated in nanoparticles evade the efflux pump, by endosomal internalization. Once in the endosome, specifically designed nanoparticles, release siRNA and drug to the cytosol.
Alterations in the membrane transporters or efflux pumps
Cancer cells can develop resistance to specific drugs, or type of drugs. For example, several types of cancer cells present resistance to folate, such as methotrexate [31]. Loss of cell surface receptors or transporters for the drug, increased metabolism of the drug, or alteration of the drug's target are some of the strategies that cells use to avoid cell death [32—34]. By combining several different types of chemotherapy agents, treatments can overcome this type of resistance. However, a wide variety of cancer cells are resistant to a multiple of drugs. This phenomenon is known as multidrug resistance (MDR) [33].
The primary protein known to be involved in MDR is P-gp, an ATP-dependent transporter of the ATP-binding cassette family (ABC), and encoded by the gene MDR1 in humans [6]. P-gp can be present in the cell membrane as well as in the nuclear membrane. P-gp binds to neutral and positively charged molecules. A great amount of antineoplastic drugs are either neutral or positive at physiological pH, hence act as a substrate for P-gp, which pumps the drug across the membrane. This decreases the concentration of drug inside the cell and the nucleus by removing the drug to the extracellular matrix or the cytoplasm, respectively. This mechanism of self-defense is widely known as efflux pump related cell resistance. In healthy cells P-gp is involved not only in the efflux of undesirable molecules but also in the transport of beneficial molecules and nutrients across the cell membrane and intracellular membranes in the cell [35]. P-gp is expressed in many cancers including, but not limited to, small and large intestine, liver, pancreas, kidney, ovary, and testicle. Other important ABC are the multidrug resistant protein 1 (MRP1) and 2 (MRP2) [36]. MRP1 has been found to be expressed in a great variety of cancers. MRP1 binds to negatively charged molecules or molecules that have been modified by the cell through glycosylation, sulfonation, or other post-translational modifications. Other ABC transporters with implications in cancer resistance have been reported [37].
Activation of anti-apoptotic pathways
Among non-drug pump related mechanisms, the most important is the activation of anti-apoptotic pathways; a defense mechanism that rescues cells from cell death [38]. Apoptosis is the most common type of programmed cell death and is an essential part of the cell cycle. Apoptosis is activated by a series of cascade signals in which many proteins are involved. Bcl-2 is a protein of the Bcl-2 family, encoded by the gene BCL-2, which has a major role in preventing apoptosis in healthy cells [11]. Bcl-2 overexpression prevents cells from entering apoptosis. It is correlated with cancer cell survival and cancer cell resistance. Other members of the Bcl-2 family, such as Mcl-1, a protein encoded by the gene MCL-1, have been identified as inhibitors of apoptosis [39].
Another protein involved in cancer resistance is the protein Plk-1, encoded by the Plk-1 gene. Plk-1 is a protooncogene overexpressed in some types of cancer cells, such as breast and colon [40,41]. The loss of Plk-1 has been associated with activation of apoptosis [42]. c-Myc is a gene encoding for a transcription factor that is constitutively expressed in cancer cells. c-Myc overexpression has been linked to breast [38], ovarian [43], and lung [44] cancer resistance.
Sensitization strategies
Considerable efforts have been made recently to suppress multidrug resistance; both in efflux pump and non-efflux pump related multidrug resistance. Sensitization strategies include targeting membrane transporters [45—47], inhibition of cell survival pathways [48,49], altering transcription factors [19], and silencing anti-apoptotic pathways [17,50].
Efflux pump related sensitization strategies
Several sensitization strategies have been tested by using selective inhibitors of the ABC transporters, such as verapamil [46]. PLGA nanoparticles were synthesized to simultaneously deliver verapamil and vincristine, a potent chemotherapeutical agent, to cancer cells to reverse the cell's resistance to the latter [47]. Results have shown the resistance to certain chemotherapy agents by inhibition of efflux transporters can only be achieved for certain cancer cell types. A strategy to increase the success of sensitization is by using a broader inhibitor of the transporter, such as depharantine [51]. This, however, resulted in an increase in toxicity in vivo [5]. These results seem to indicate that sensitization of cancer cells by inhibitors of ABC transporters is not only cell specific but also drug specific.
Nanoparticles are not affected by efflux pumps and they can be used as carriers for drugs that are affected by efflux [52]. Encapsulation of drugs in nanoparticles that are specifically designed to release their cargo in the cytoplasm has been proved to increase drug concentration inside cells [52]. Classic chemotherapeutical drugs such as doxorubicin, cisplatin, and paclitaxel, must reach DNA preserved within the nucleus to decrease cell viability. The use of nanoparticles to bypass transporters present in the surface of cell membranes has increased drug concentration in the cytoplasm, but drugs also must cross the nuclear membrane. The presence of efflux pumps in the nuclear membrane decreases the intranuclear concentration of the drug, compromising the efficacy of the treatment. The use of siRNA against genes encoding for efflux pump proteins has been employed as a promising strategy to sensitize cells to cancer drugs. For example, Yadav et al. [20] synthesized poly(ethylene oxide)-modified poly(beta aminoester) (PEO-PbAE) to deliver siRNA against P-gp in Paclitaxel resistant SKOV3 human ovarian adenocarcinoma cells. The cell membrane P-gp protein was targeted by using monoclonal antibodies to increase sensitization of human ovarian carcinoma cells A2780/AD to doxorubicin [53].
Non-efflux pump related sensitization strategies
A wide variety of sensitization strategies have been used by inhibition of cell survival pathways, altering transcription factors, and silencing anti-apoptotic pathways. Among these strategies, some involved the use of nanocarriers to deliver cargo to either block or inhibit pathways or silence genes. For example, poly(ethylene glycol) lipoplexes were used to deliver siRNA to silence Bcl-2 genes (siBcl-2), which sensitized cancer cells to 5-fluoracil [18]. Nanogels were used to deliver siRNA against the gene encoding for Epidermal Growth Factor Receptor (EGFR) to sensitize SKOV3 human ovarian adenocarcinoma cells to docetaxel [54]. Liposomes were used to sensitize temozolomide resistant glioblastoma mutiforme (GM) cancer cells by delivering siRNA against the gene encoding for MGMT, a DNA repair protein, that is overexpressed in GM cancer cells [17]. An inhibitor of the nuclear factor kappa B and down-regulator of ABC transporters, curcumin, was delivered with paclitaxel using nanoemulsions to sensitize paclitaxel resistant SKOV3 human ovarian adenocarcinoma cells [48].
Nanocarriers to co-deliver siRNA and small drugs
Nanocarriers have been successfully used as platforms for delivery of cargo to sensitize cells to chemotherapy. A promising sensitization strategy is using siRNA to silence genes encoding for proteins involved in cancer resistance. Co-treatment of resistant cancer cells by using siRNA and drugs, administered separately, has been shown to increase efficacy of cancer treatment. However, co-delivery of siRNA and drugs would be more efficient in overcoming cancer resistance to chemotherapy [21]. Several different co-delivery systems have been synthesized but all can be grouped into three categories: polymer based, lipid based, and inorganic based.
Polymer based nanoparticles
Polymer based nanoparticles have been used as delivery systems for a variety of drugs, proteins, and nucleotides [9,10,55,56]. Due to their tunability, biocompatibility, and high transfection rate of DNA and siRNA, polymeric nanoparticles are the preferred nanosystem to co-deliver a drug and siRNA (Table 1). Poly(ethylene amine) (PEI) is a cationic polymer and it has been widely used as a major component for non-viric delivery carriers of siRNA due to its high siRNA complexation and its proton sponge effect for endosomal escape of the cargo to the cytosol [57]. Since high molecular weight PEI nanocarriers are toxic Cao et al. [58] incorporated polycaprolactone (PCL) biodegradable structures containing disulfide or ester covalent linkages between low molecular weight PEI chains. Phosphate species negatively charged in the siRNA were complexed onto the positively charged nitrogen species of the nanoparticles by electrostatic interactions. They studied cytotoxicity of siRNA conjugated PEI-PCL polyplexes at different nitrogen (N)/phosphate (P) ratios and found an optimum N/P ratio of 30, which decreased viability down to 65%. To improve cyto toxicity, the carrier surface was modified with PEG chains, which has been proved to provide stability and reduce toxicity of nanoparticles.
Table 1.
Nanosystems to codeliver siRNA and drugs to overcome multidrug resistance in cancer therapy.
| System | Type of nanoparticle | SiRNA | Drug | Size and Zeta potential |
Cell line | Targeting | Reference |
|---|---|---|---|---|---|---|---|
| FA-PEG-PGA coated onto PEI-PCL | Cationic biodegradable polymeric | Bcl-2 electrostatic complexed | DOX encapsulated | 150nm, −5mV | Bel-7402 hepatoma | FA | [58] |
| FA-PEG-PGA coated onto PEI-PCL | Cationic biodegradable polymeric | Bcl-2 electrostatic complexed | DOX encapsulated | 150nm, −5mV | C6 glioma | FA | [59] |
| PEI-SA | Cationic polymeric | VEGF complexed | DOX encapsulated | 303nm, 64mV | HUH-7 hepatocarcinoma | [60] | |
| Octasilsesquioxane-p(L-Lys) | Cationic biodegradable Polymeric | Cye3 complexed | DOX conjugated biodegradable disulfide spacer. | U87 Glioblastoma | RGD | [65] | |
| Dendritic polyamine-β-CD | Cationic biodegradable polymeric | EGFR complexed | Erlotinib or SAHA encapsulated | 200—400nm | U78 glioblastoma | mAb-EGFR | [49] |
| PLGA-PEI-Biotin | Cationic biodegradable | P-gp complexed | PAC encapsulated | 237nm, −12.2mV | JC breast adenocarcinoma | Biotin | [62] |
| mPEG-PCL-PPEEA | Cationic biodegradable polymeric | Plk1 complexed | PAC encapsulated | 50nm | MDA-MB-435 breast carcinoma | [40] | |
| P(MDS-co-CES) | Cationic biodegradable polymeric | Bcl-2 complexed | PAC encapsulated | 160nm, 44mV | MDA-MB-231 breast carcinoma | [61] | |
| PEO-b-PCL | Biodegradable polymeric | MDR-1 complexed | DOX conjugated pH-sensitive hydrazone linkage | 103nm, 4mV | MDA-MB-435 breast carcinoma | RGD TAT | [63] |
| PDMAEMA—PCL—PDMAEMA | Cationic biodegradable polymeric | VEGF GFP | PAC encapsulated | 95nm, 35mV | PC3 prostate adenocarcinoma | [64] | |
| mono-Pal-MTO di-Pal-MTO |
Cationic mono-di lipidic | Mcl-1 complexed | MTO conjugated | 210nm | KB nasopharynx carcinoma | [74] | |
| LPD LPD-II | Cationic liposome Anionic liposome |
VEGF c-Myc complexed |
Dox Dox complexed |
Cationic liposome 135nm, 35mV Anionic liposome 62nm, −19mV. |
HT-1080 fibrosarcoma | AA | [75] |
| LPD | Cationic liposome-polycation-DNA | c-Myc complexed | DOX complexed | 197nm, 30mV. | HT-1080 fibrosarcoma | NGR | [76] |
| PDGL | Cationic liposome | Mcl1 complexed | PD0325901 encapsulated | 230nm, 16mV | KB nasopharynx carcinoma | [77] | |
| DOTAP | Cationic liposome | MRP1+Bcl2 complexed | DOX encapsulated | 500nm, 4mV | H69AR lung carcinoma | [81] | |
| EDOPC cSLN | Cationic liposome | MCL-1 complexed | PAC encapsulated | 183nm, 44mV | KB nasopharynx carcinoma | [79] | |
| pTLOL | Cationic liposome | Mcl-1 complexed | SAHA encapsulated | 230nm, 19mV | KB nasopharynx carcinoma | [78] | |
| Pyridilthiol-mesoporous silica nanoparticles | Porous silica nanoparticle | MRP1 Bcl2 conjugated through disulfide bonds | DOX CIS encapsulated | 200nm | A549 lung carcinoma | LHRH | [21] |
| PAMA -Silica Mesoporous | Cationic porous silica | Bcl-2 complexed | DOX | 200nm | A2780/AD ovarian carcinoma | [88] | |
| PEI coated Mesoporous silica | Cationic porous silica | P-gp Bcl-2 complexed | DOX encapsulated | 247nm, 31mV | KB-V1 squamous carcinoma | [89] | |
| QD — B-CD — l-Arg or l-His | Cationic quantum dot | MDR1 complexed | DOX encapsulated | 11nm, 8mV | HeLa/Dox cervix adenocarcinoma | [90] |
It has also been reported that PEG can hinder the complexation of siRNA, hence Cao et al. [58] proposed a hierarchical assembly strategy, in which PEGylation is performed after complexation of siRNA to the nanocarrier. Doxorubicin (DOX) was loaded to PEI-PCL micelles using a chloroform/water mixture under sonication. The treatment of hepatic cancer cells with doxorubicin causes an increase in the production of BCl-2 protein as a defense mechanism that leads to resistance of cancer cells to the drug. They studied the ability of the nanocarriers to diminish the upregulation of BCl-2 protein caused by the administration of doxorubicin. Bcl-2 siRNA — doxorubicin loaded nanocarriers were capable of decreasing the overexpression of Bcl-2 protein induced by doxuribicin. The ability of the co-delivery system in reducing cell viability was measured using MTT assay. Nanocarriers loaded with BCl-2 siRNA and doxorubicin were incubated for 96 h with hepatic cancer cells. Cell viability was reduced down to 40% at the highest concentration of doxorubicin (1 μM) when using Bcl-2 siRNA; however for scrambled siRNA cell viability was reduced down to 60%. When using Folate Receptor targeted nanocarriers loaded with Bcl-2 siRNA cell viability was reduced down to 5% at the same doxorubicin concentration. This indicates the synergistic effect a co-delivery system has on cell metabolism, and the importance of developing targeted co-delivery systems. Further investigations in vivo using the same system were carried out [59]. By using Western Blott and Tunel assay, they studied the effect of different treatments on apoptotic response of C6 glioma cells. They concluded that the synthesis of Bcl-2 protein is doxorubicin dose dependant. They also noticed that at high doxorubicin concentration (15μg/mL), the protein Bax is inhibited and there is a higher presence of cleaved caspase 3.
Another strategy to reduce cytotoxicity of high molecular weight polyethylenimine (PEI) is by grafting stearic acid (SA) to PEI through carbodiimide conjugation using EDC reaction [60]. PEI-SA micelles were formed using the oil in water (o/w) solvent evaporation method, obtaining small (≈51 nm) and cationic (≈64 mV) micelles. These micelles contain both a hydrophobic core that can encapsulate a hydrophobic drug and a hydrophilic cationic shell capable of complexing siRNA. Doxorubicin was encapsulated into the micelles by mild agitation. siRNA against the vascular endothelial growth factor (VEGF) was complexed onto the nanoparticle surface. VEGF is a growth factor over secreted by tumors to force the formation of new blood vessels by stimulating the growth and division of endothelial cells to provide oxygen-rich blood to tumor cells. This process is known as angiogenesis and it has been proven to be necessary for the in the tumor to survive. By blocking the formation of new blood vessels irrigating the tumor with oxygen, tumor growth can be stopped. PEI-SA/DOX reduced the volume of the tumor down to 13% relative to the control. When using PEI-SA/DOX/viVEGF the tumor was reduced down to 56.7%.
Another strategy to obtain biodegradable nanoparticles is by introducing the biodegradable polymer poly(ε-caprolactone) into the formulation [40]. Micelleplexes were synthesized using tri-block copolymers of poly(ethylene glycol)-b-poly(ε-caprolactone)-b-poly(2-aminoethylethylene phosphate) (mPEG-b-PCL-b-PPEE). Paclitaxel (PAC) was encapsulated through hydrophobic—hydrophobic interactions. siRNA to silence Plk-1, a serine/threonine protein kinase overexpressed in some tumors, was complexed on the positive surface of the micelle. They studied MDA-MB-435 cell viability in vitro and in vivo when co-delivering paclitaxel and siPlk-1. They concluded that the use of a co-delivery system requires one thousand fold less paclitaxel required for monotherapy. Micelleplexes carrying paclitaxel were administered along with micelleplexes complexed to siPlk-1to mice bearing a MDA-MB-435 tumor. Micelleplexes carrying paclitaxel and complexed to siPlk-1 showed a great decreases in tumor volume when compared to the control. These results clearly indicate that it is necessary to co-deliver both the drug and the siRNA in the same system.
A different strategy to achieve biodegradability is by incorporating hydrophilic cholesterol into an hydrophobic cationic polymer [61]. Poly [(N-methyldietheneamine sebacate)-co- [(cholesteryl oxocarbonylamido ethyl) methyl bis(ethylene) ammonium bromide] sebacate] (P(MDS-co-CES)) was self-assembled into a cationic biodegradable nanoparticle. The hydrophobic drug Paclitaxel was added into the solution at the moment of self-assembly in order to be encapsulated in the nanoparticle through hydrophobic—hydrophobic interactions. siBcl-2 was complexed onto the nanoparticle surface via electrostatic interaction. Synergistic effects between siBcl-2 and the encapsulated drug were studied in breast adenocarcinoma MDA-MB-231 cells. Cell viability decreased from 78% to 59% and from 58% to 39% in the presence of siRNA at paclitaxel concentrations of 100 and 400 nM, respectively. As the cytotoxicity of the siRNA was only 8%, there was indeed a synergistic effect associated with the co-delivery of paclitaxel and siRNA, possibly because the suppression of the anti-apoptotic activity of Bcl-2 by the siRNA made cells more sensitive to paclitaxel.
The biodegradable polymer poly(d,l-lactide-co-glycolide) (PLGA) was mixed with polyethyleneimine (PEI) to form micelles using a water-in-oil (W/O) emulsion [62]. Paclitaxel was encapsulated during the emulsion. siP-gp was complexed through electrostatic interactions to the nanoparticle and increased paclitaxel uptake by resistant breast cancer cells. P-gp silencing increased intracellular paclitaxel accumulation in vitro and it enhanced in vivo activity of paclitaxel, which translated in a reduction of tumor growth.
Another strategy to complex negative siRNA onto a nanoparticle is by grafting a positive polymer chain onto a biodegradable neutral nanoparticle after it is formed [63]. Poly(ethylene oxide)-block-poly(ε-caprolactone) (PEO-b-PCL) polymer was used as a backbone to ensemble a biodegradable polymer that could further be functionalized with different moieties. Polyamine was attached to the PCL block to allow complexation of the siRNA through electrostatic interactions. A pH-sensitive hydrazone link was conjugated to other PCL blocks to covalently conjugate doxorubicin. The cell penetrating peptide TAT and the integrin Rvβ3-specific ligand (RGD4C) were attached to the PEO block to facilitate cell internalization and uptake. The functionalized polymers self-assembled into micelles of ≈103 nm and ≈4.23 mV.
Previous studies from the same group [63], in which they delivered doxorubicin using nanoparticles, showed that doxorubicin released in the cytoplasm was pumped out of the cells and failed to accumulate in the nucleus of P-gp- overexpressing DOX-resistant cells. It has been demonstrated that nanocarriers bypass the P-gp pump efflux system expressed on the surface of the cell membrane [8], increasing the concentration of the drug in the cytoplasm. However, once the drug is released inside the cytoplasm, it still has to reach its site of action, the nucleus. By developing nanoparticles that release the drug in the cytoplasm and block the efflux pump present in the nuclear membrane, Xiong and Lavasanifar [63] increased the drug concentration in the nucleus of MDA-MB-435/LCC6MDR1-resistant cells. This increase translated in a greater decrease in cell viability.
A promising polymer used to deliver siRNA is the poly (2-(N,N-dimethyl aminoethyl) methacrylate), designated as P(DMAEMA), due to its high efficiency in complexing and transfecting siRNA. However, P(DMEAMEA) is highly toxic to cells, limiting its use for biological applications. One strategy to decrease its toxicity is by adding a biodegradable polymer in the backbone. To do so, Zhu et al. [64], synthesized the block co-polymer 2-(N,N-dimethyl aminoethyl) methacrylate-b-poly(ε-caprolactone)-b-2-(N,N-dimethyl aminoethyl) methacrylate (PDMAEMA—PCL—PDMAEMA) by free radical reversible addition-fragmentation chain transfer (RAFT) polymerization and assembled it into biodegradable cationic micelles. Nile red, a hydrophobic molecule used as a drug model, was encapsulated using hydrophobic—hydrophobic interactions after the micelle was formed. siRNA to silence Green Fluorescence Protein (GFP) was complexed onto the nanoparticle surface. Co-delivery of siGFP and Nile red into PC3 human prostate cancer cells was successfully achieved in vitro, according to confocal microscopy studies. Paclitaxel and siVEGF were both successfully incorporated into the micelle, which opens the possibility to use a biodegradable PDMAEMA carrier to co-deliver siRNA and hydrophobic drugs.
A different approach to co-deliver siRNA and a drug is by using a three-dimensional octasilsesquioxane cage as a backbone to grow poly(l-lysine) chains to create a nanoglobular system [65]. Two different moieties were conjugated to the p(l-Lys) chains to confer poly functionality to the nanoparticle. A biodegradable disulfide spacer was used to conjugate doxorubicin. A PEG chain was used as a linker to attach RGD, a cyclic peptide that binds specifically to αvβ3 integrin, conferring the nanoparticle with specificity toward cancer cells that overexpress αvβ3 integrin. U78 glioblastoma cancer cells treated with free doxorubicin reduced cell viability down to 50% at 6.50 μg/mL of doxorubicin. Cells treated with conjugated doxorubicin, reduced viability down to 50% at 0.7 μg/mL of doxorubicin, probably due to the ability of these nanoparticles to bypass the efflux effect. Co-localization studies showed that the siRNA was found mostly in the cytoplasm and not in endosomes and/or lysosomes.
Due to the heterogeneity of cells in tumors, a promising strategy to overcome cancer resistance of a heterogeneous population of cells is to target multiple signaling elements. The synthesis of a carrier capable of co-delivering multiple therapeutic agents is required to achieve a cooperative effect. Kim et al. [49] developed a low toxicity, high transfection efficiency, and high solubility system capable of encapsulating hydrophobic drugs, complexing with siRNA, and attaching to proteins. To do so, they synthesized a dendritic polyamine, which was further conjugated to a β-cyclodextrin to confer solubility to hydrophobic drugs [66]. In this case, erlotinib and suberoylanilide hydroxamic acid (SAHA) were encapsulated via hydrophobic—hydrophobic interactions with β-cyclodextrin. Furthermore, the presence of β-cyclodextrin decreased the cytotoxicity of the dendritic polyamines. The free amines in the dendritic polyamine served to complex siRNA and to conjugate an antibody to target Epidermal Growth Factor Receptor. The final DexAM nanoparticle had significantly less amines available on its surface compared to commercially available transfection systems such as polyethyleneimine (PEI), lipofectamine 2000 (LF) and X-tremeGENE (Xgene). Cytotoxicity of these compounds has been attributed to the presence of positive charge due to the amine groups, which are necessary for siRNA complexation and for endosomal escape once the nanoparticle is internalized in the cell. Cell viability of U87 glioblastoma cells was determined after incubation with DexMA, PEI, LF and Xgene. Cell viability was decrease down to 90% for DexMA, 60% for PEI, and 25% for LF and Xgene, showing that DexMA is a highly biocompatible system compared to other commonly used transfection systems. U78 glioblastoma cells were treated with EGFR targeted DexMA-siEGFR conjugated with and without erlotinib and SAHA. Targeting EGFR with nanoparticles has proven to be effective in increasing internalization of nanoparticles and in improving treatment of EGFR overexpressing cancer cells [67]. EGFR overexpression is correlated with cell survival and response to therapy. Erlotinib and SAHA have been shown to enhance the efficacy of other EGFR antagonists [68]. EGFR targeted DexMA-siEGFR without erlotinib reduced cell viability down to 50%; when using erlotinib conjugated nanoparticles cell viability was reduced down to 30%. A similar trend was observed when comparing nanoparticles with and without SAHA.
Lipid based nanoparticles
Lipidic nanoparticles have been widely used for different biomedical and pharmaceutical applications [69—72]. Liposomes as drug delivery systems gain a lot of attention after Doxil®, a PEGylated liposome, was FDA approved to deliver doxorubicin [73].
An interesting approach to incorporate an antineoplastic drug within a lipidic nanoparticle is by covalently attaching a positively charged drug to a lipidic chain to create an amphiphilic molecule that can be further used to self-assemble into a multilayer cationic nanoparticle. To do so, Chang et al. [74] covalently conjugated mitoxantrone (MTO) to either one or two chains of palmitoleyl to create monopalmitoleys-MTO (mono-Pal-MTO) and dipalmitoleys-MTO (di-Pal-MTO), respectively. The positive charge of the MTO confers cationic properties to the nanoparticle, and allows complexation of siRNA by electrostatic interaction within the layers of the nanoparticle (Fig. 2). They studied the ability of this multilayer system to co-deliver MTO and siRNA against Mcl-1, a Bcl-2 related gene, into human epithelial carcinoma KB cells. After 24h of incubation, they observed a 68% reduction when using pal-MTO and 81% when using siRNA-pal-MTO compared to controls. Efficacy of the co-delivery treatment in reducing tumor growth in vivo in mice bearing KB cell tumors was studied. A reduction in tumor volume after 20 days of starting the treatment down to 53.9% when using pal-MTO and 83.4% when using siRNA-pal-MTO compared to controls was observed (Fig. 3).
Figure 3.
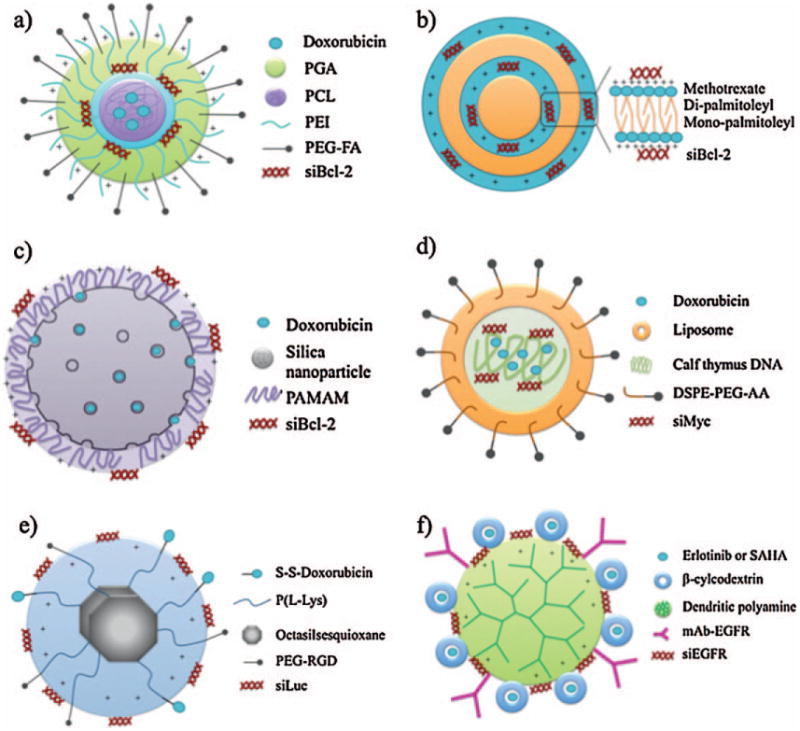
Schematic representation of various types of nanoparticles to co-deliver siRNA and a chemotherapeutical agent. (a) Biodegradable nanoparticle [59], (b) cationic mono-, di-lipidic nanoparticle [74], (c) mesoporous silica nanoparticle [88], (d) liposome [75], (e) octasilesquioxane nanoparticle [65], (f) dendritic nanoparticle [49].
The possibility of functionalizing lipids with molecules leads to a great variety of applications. For example, cationic liposome-DNA (LPD) was synthesized using the guanidine containing cationic lipid N,N-distearyl-N-methyl-N-2-(N-arginyl) aminoethyl ammonium chloride (DSAA) [75]. Doxorubicin was complexed to the negatively charged DNA and encapsulated within the liposome as cargo. siVEGF was complexed onto the liposome surface. Anionic liposome-DNA (LPD-II) was synthesized using the anionic lipid 2-di-(9Z-octadecenoyl)-sn-glycero-3-phosphate (DOPA) and cholesterol. Doxorubicin was complexed to the DNA and encapsulated within the liposome as cargo. The entrapment efficiency of doxorubicine was low (10%) for LPD nanopar ticles compared with LPD-II nanoparticles (90%). Leakage of doxorubicin from LPD nanoparticles was reported, most likely due to the competition of cationic lipids for the negatively charged DNA, displacing the positively charge doxorubicin. Small interference molecules siVEGF and siMyc, respectively, were complexed onto the liposome surface. Transfection efficiency of siRNA in vitro was high for LPD and low for LPD-II. Cytotoxicity studies showed that anionic liposomes LPD-II were nontoxic at every concentration studied. However, cationic liposomes LPD were cytotoxic by increasing interleucine-2 (IL-12) and decreasing white blood cells and platelets. Both systems were capable of inhibiting tumor growth significantly when compared to the untreated group after systemic intravenous injection in mice.
PEGylated LPD [76] were decorated with NGR (aspargine—glycine—arginine) moiety to target amino peptidase N (CD13) expressed in tumor cells and tumor vascular endothelium. Delivery and efficiency of siMyc in mice by LPD-PEG-NGR was studied in CD13 expressing cells HT-1080 and CD13 nonexpressing HT-29 cells. siRNA was efficiently delivered to the cytoplasm and c-Myc was down regulated in HT-1080 cells but not in HT-29 cells. When co-delivering siMyc and doxorubicin using LPD-PEG-NGR, an accumulation of siRNA and DOX in tumor tissue was observed which translated in an enhanced therapeutic effect.
As new lipids are synthesized, new liposomic formulations become available. In this case, cationic liposomes (DGL) were synthesized using a new N′,N″ -dioleylglutamide (DG) cationic lipid [40], 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), and cholesterol [77]. The mitogen-activated protein kinase (MEK) inhibitor PD0325901 was encapsulated through hydrophobic—hydrophobic interactions in the liposome. The inhibition of MEK results in inhibition of phosphorylation and inactivation of the MAPK/ERK signaling pathway that in cancer is known to be involved in proliferation and resistance to apoptosis. The myeloid cell leukemia sequence 1 (Mcl-1) gene has been reported to be overexpressed in tumor cells that presented resistance to chemotherapeutical agents. Small interference RNA against Mcl-1, was complexed to the cationic liposomes. PD0325901 containing liposomes (PDGL) and siMcdl-1 were incubated with KB tumor cells to study the ability of this carrier to co-deliver both cargos. PDGL liposomes were capable of co-delivering ERK inhibitor and siMcl-1 in KB tumor cells. Western Blot shows inhibition of both phosphorylated ERK1/2 protein and MCl-1 protein when treated with PDGL-siMcl-1, indicating not only the feasibility of co-delivery but the therapeutic effect of down regulate both proteins. Cytotoxicity of KB tumor cells was studied when treated with free PD0325901. At a low concentration (0.72 μg/mL) of PD0325901 cell viability was not affected by the MEK inhibitor. However, when the same concentration was delivered using the cationic liposome with siMcl-1, cell viability was reduced down to 10%. Co-delivery of siMcl-1 and the drug using cationic liposomes (PDGL-siMcl-1) in BALB/c mice injected with KB cells resulted in suppression of tumor size by 79% when compared to the control. Suppression of the tumor growth was greater when using PDGL-siMcl-1 compared to any other co-treatment studied.
The results of tumor growth suppression using liposomes as a co-delivery system are promising and encouraging. However, due to the lack of systemic delivery systems currently available, Shim et al. [78] developed new cationic liposomes using oligolysine based lipids and studied their suitability as systemic co-delivery systems. Different formulations using varying amounts of lysine, DOPE, cholesterol and PEG were synthesized. The final multilayer cationic liposomes were systematically studied to determine their ability to complex siRNA and their cytotoxicity when cultured with cancer cells.
In order to increase the transfection efficiency of liposomes, Yu et al. [79] synthesized 1,2-dioleoyl-sn-glycero-3-ethylphosphocholine (EDOPC)-based cationic lipid nanoparticles (cSLN), which have been shown to have higher transfection efficiency than formulations using other lipids [80]. The structure of liposomes allows the encapsulation of poorly soluble drugs, such as paclitaxel (PAC), in the core of the cSLN matrix without altering the chemical structure of the drug. This type of encapsulation allows a gradual release of the drug, instead of a burst release. The cationic nature of the lipids that form the liposome allow for negative moieties, such as siMcl-1, to complex onto the liposome surface through electrostatic interactions. KB cancer cells treated with free paclitaxel and free siMcl-1, show a small decrease in cell viability down to approximately 90%. Cells treated with cSLN liposomes carrying siMcl-1 decreased viability down to 58%. When cells were treated with cSLN liposomes carrying both siMCl-1 and paclitaxel, viability was decreased down to 38%. Mice bearing a KB tumor were treated with free paclitaxel and cSLN liposomes carrying siMcl-1. Tumor volume was reduced 48% compared to the control. When mice were treated with cSLN liposomes carrying both paclitaxel and siMcl-1, the volume was reduced 88% compared to the control.
Saad et al. [81] studied the possibility of co-delivering doxorubicin and two different siRNA: siBcl-2 and siMRP1. They synthesized a liposome using 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), encapsulated doxorubicin via hydrophobic—hydrophobic interactions, and complexed siRNA through electrostatic interactions. They observed that the mere use of liposome-siMRP1 caused a decrease in small cell lung carcinoma (SCLC) cancer cells. A decrease in an effective efflux mechanism caused cell death, probably due to the fact that MRP1 proteins are involved not only in the efflux of drugs from the cell but also in detoxifying cells of their own metabolic products. An accumulation of undesired products could cause cells to enter apoptosis. When doxorubicin was added to the liposome-siMRP1, cell viability was significantly decreased. As reported before, liposomes-siBcl-2 reduce cell viability to a certain extent by promoting cells to enter apoptosis. However, liposomes-siBcl-2 carrying doxorubicin decreased cell viability to a greater extent. Liposomes-siBcl-2-siMRP1 carrying doxorubicin decreased cell viability down to 5%, the most effective treatment. By blocking two resistance mechanisms at the same time while administering doxorubicin, in a single carrier, higher levels of effectiveness can be achieved. However, due to the non-specificity of this system to target cancer cells, adverse effects of doxorubicin toward healthy cells when used in vivo may be significant.
Inorganic based nanoparticles
Increasing use of inorganic based nanoparticles in biological applications has been observed due to their tunable specific properties [82—84]. Silica based nanoparticles have been use for several applications, such as delivery systems to deliver either drugs or siRNA [85,86]. Its high surface area to volume ratio and large pore volume make them ideal for loading large amounts of drugs and conjugation/complexation of other components on the surface [86,87].
The use of silica nanoparticles for simultaneous co-delivery of a hydrophobic drug (doxorubicin) and siRNA (siBcl-2) was explored by Chen et al. [88]. Silica macroporous nanoparticles were conjugated with 3-iso-cyanatopropyltriethoxysilane to obtain an isocyanatopropyl (ICP)-modified surface that could be further functionalized with a polyamidoamine dendrimer (PAMA) to confer nanoparticles with positive surface charge. Doxorubicin was encapsulated within the silica pores and siRNA was complexed to PAMA. They successfully delivered doxorubicin and siRNA into A2780/AD ovarian cancer cells. Cell viability of cells treated with a low concentration of doxorubicin (0.01 μM) encapsulated within silica particles complexed to siBcl-2 decreased down to 50% compared to cells treated with the same concentration of free doxorubicin. These encouraging results showed that at a nontoxic concentration of doxorubicin, cell viability can be reduced down to 50% when doxorubicin is encapsulated and co-delivered with siBcl-2.
To confer good particle dispersion and biocompatibility to mesoporous silica nanoparticles, Meng et al. [89] coated them with phosponates groups. This also allowed for adsorption of molecules such as PEI for complexing siRNA. To improve hydrophilicity of PEI coated nanoparticles, they were treated with a solution of bovine serum albumin (BSA) before being transferred to the culture media. Silica particles were coated with various molecular weight PEI, and toxicity, cell uptake, and siRNA complexation were studied. Particles coated with high molecular weight PEI (25 kD) presented greater cell uptake and better knock down efficiency (90%) but also greater toxicity, compared with lower molecular weight PEI (1.8 and 10 kD). PEI 10 kD nanoparticles were nontoxic when the concentration was kept below 100 μg/mL and presented the same cellular uptake as PEI 25 kD nanoparticles when cultured with KB-V1 resistant cancer cells at various concentrations for 24 h. Doxorubicin was encapsulated in the mesoporous silica pores through electrostatic complexation to negatively charged silica. Doxorubicin was released intracellularly due to the lysosomal low pH due to the sponge proton effect of PEI that disrupts the lysosome, compared to the physiological 7.4 pH.
When using PEI 10kD coated nanoparticles loaded with doxorubicin (PEI-DOX), more doxorubicin was found inside cells compared to cells cultured with free doxorubicin. However, no presence of doxorubicin was found in the nucleus, suggesting a need to suppress P-gp protein to avoid rapid extrusion of doxorubicin from the cell before the drug can penetrate the nucleus. When PEI coated silica particles loaded with doxorubicin and complexed to siP-gp (PEI-DOX-siPgp) were cultured with KB-V1 cells, a significant increase of doxorubicin in the nucleus was observed. The IC50 of KB-V1 cells was 2.5 times lower when treated with PEIDOX-siPgp compared to cells treated with free doxorubicin, even though it was not as low as the IC50 of nonresistant cells. Experiments targeting simultaneously Bcl-2 and P-gp while administering doxorubicin on KB-V1 cells did not seem to improve cytotoxicity of doxorubicin and failed in restoring sensitivity of resistant cancer cells to normal levels. This indicates that the dual targeting (Bcl-2 and P-gp) strategy to overcome cancer resistance is cell type specific. Mesoporous silica nanoparticles for the inhalatory administration route to treat lung cancer cells were synthesized. This route is favored because it avoids systemic toxicity and first-pass metabolic degradation [21]. Doxorubicin and cisplatin were loaded into the nanoparticles pores. Two different reduced siRNAs were conjugated to pyridylthiol-silica nanoparticles via disulfide bonds. Nanoparticles were decorated with LHRH peptide to target A549 human adenocarcinoma cells. LHRH was conjugated to HS-PEG-COOH to obtain LHRH-PEG-SH, which was then covalently linked to silica nanoparticles. The hydrodynamic diameter of the fully functionalized nanoparticle measured by dynamic light scattering was ≈200nm. Cell viability of cells cultured with silica nanoparticles for 24 h, decreased down to 95% at a concentration of 1 mg/mL. This low cytotoxicity, compared with other formulations used for delivery of siRNA, is probably due to the lack of positively charged polymer on the surface. Confocal experiments showed release of doxorubicin in the perinuclear region. According to RT-PCR results, the effectiveness of siBcl-2-silica and siMRP1-silica nanoparticles in silencing genes in the condition above mentioned was of 56% for Bcl-2 and 58% for MRP1. Cell viability of cells treated with a mixture of targeted nanoparticles carrying siBcl-2, siMRP1, doxorubicin, and cisplatin for 24h at a drug concentration of 1 μg/mL decreased down to 25% compared to a decrease down to 85% when treated with a mixture of free cisplatin and Doxorubicin. IC50 dose of a mixture of free drugs was 30 times greater when compared to cells treated with the aforementioned mixture.
To be able to track the fate of the co-delivery system once in contact with cells, Li et al. [90] opted to use quantum dots. Quantum dots have been widely used in biomedicine as imaging and delivery systems [32,91—93]. CdSe/ZnSe QD were coated with β-cyclodextrin, which was previously funtionalized with l-arginine or l-histidine to confer positive charge and biocompatibility. Doxorubicin was encapsulated within the β-cyclodextrin rings. siRNA against MRP1 was complexed onto the nanoparticle surface. A release study of doxorubicin from the QD-DOX showed that more doxorubicin was released at endosomal pH (5.0) compared to physiological pH (7.4). The authors attributed this to the fact that doxorubicin is protonated at low pH leading to increased solubility. siRNA-DOX-QD were internalized by doxorubicin resistant HeLa cells (HeLa/Dox) within 1 h of incubation. The nanoparticles were confined to vesicles and attached to the membrane, probably due to its positive charge. Within 3 h, nanoparticles were released to the cytosol after rupture of the vesicles and rapidly dispersed throughout the cytoplasm. By using confocal, they observed that doxorubicin was able to reach the nucleus when cells were incubated with siMRP1-DOX-QD, indicating that these particles were also able to silence the gene encoding for the P-gp protein. Cell viability of doxorubicin resistant HeLa cells was assessed by treatment with various formulations maintaining the concentration of doxorubicin fixed at 1 μg/mL during 72 h. Cytotoxicity of siMDR1-DOX-QD presented a 3 fold increase compared to DOX-QD, and a 5 fold increase when compared to free doxorubicin.
Conclusions, limitations, and future directions
The use of nanotechnology to develop nanodelivery systems has allowed researchers to overcome limitations of antineoplastic drugs by increasing the solubility of the drug and decreasing the toxicity to healthy tissues. Due to the tunable size of the nanocarriers, they can be formulated to penetrate tumors from the blood stream by the Enhance Permeation and Retention (EPR) effect while evading renal clearance from the body. This decreases the non-specific distribution by increasing penetration to the tumor, increasing efficiency of the treatment. Even though the initial limitations of antineoplastic drugs were improved by the use of nanocarriers, new limitations have arisen. Cells have developed strategies to avoid cell death by increasing their resistance to chemotherapy through the activation of efflux pumps to clear drugs from inside the cell and by increasing anti-apoptotic pathways. By encapsulating drugs into nanoparticles that bypass the efflux pumps [8], drug efflux is reduced, hence increasing the intracellular concentration of the drug. The activation of anti-apoptotic pathways is a defense mechanism that rescues cells from cell death. siRNA has the ability to disrupt cellular pathways by knocking down genes, opening the door to new treatments of diseases caused by aberrant gene expression. Rapid systemic clearance of siRNA by the renal system, lack of selectivity toward the desired tissue, and poor cell uptake have been reported, decreasing the effectiveness of gene therapy. The use of nanocarriers prevents both renal clearance and RNase degradation by protecting siRNA chains, increasing their half life in blood. However, to be able to sensitize cancer cells, while retaining all the aforementioned advantages of using nanocarriers, both the siRNA and drug must be delivered in the same device. While some types of cancer cells were completely sensitized to antineoplastic drugs by siRNA therapy, others maintained their resistance to the treatment by compensating for the loss of one type of resistance mechanism by increasing other multidrug resistance mechanisms. Double sensitization, targeting two multidrug resistance mechanisms simultaneously while administering the drug, is a promising strategy to overcome multidrug resistance, but also prevents healthy cells from protecting themselves from antineoplastic agents, increasing the toxicity of the drug. Moreover, it appears that the effectiveness of decreasing cell viability by double sensitization is cell specific. Even though these strategies have been effective for sensitizing cancer cells, they target machinery that is common to both cancer and healthy cells. Specific targeting strategies to treat only cancer cells would be needed in order to reduce side effects of drugs in vivo when double-sensitizing cells. By functionalizing nanocarriers with different types of polymers, antibodies, ligands or small molecules, selectivity and cellular uptake can be significantly increased. It has been well established that cancer cells have the ability to avoid cell death by activating different anti-apoptotic pathways. These pathways seem to be cell type specific and assault type specific. Due to the heterogeneity of tumors and the diversity of cancer cells, a “one type fits all” method of treatment seems unlikely to eradicate prove viable. However, a multi-sensitization individualized therapy designed specifically for each type of cancer cell may be more likely to prove viable to overcome cancer cell resistance.
Acknowledgments
Supported by grants from the National Institutes of Health, Center for Oncophysics (CTO PSOC U54-CA-143837), and the National Science Foundation (no. 1033746).
Biographies

Mar Creixell is a postdoctoral research assistant in the Chemical and Biomedical Engineering Departments at The University of Texas at Austin, conducting research under Prof. Nicholas Peppas'direction. She obtained Degree in Pharmacy from the University of Barcelona in 2006 and a M.S. in Biotechnology and Pharmaceutical Industry from the PompeuFabra University in 2008. Subsequently, she began her Ph.D. work in Dr. Carlos Rinaldi's lab in the Chemical Engineering Department at the University of Puerto Rico at Mayagüez. She received a Ph.D. in Biomedicine in 2011 by the University of Barcelona.

Nicholas A. Peppas is the Fletcher S. Pratt Chair in engineering, a chaired professor of Chemical Engineering, Biomedical Engineering, and Pharmacy, and the Director of the Center for Biomaterials, Drug Delivery, Bio-nanotechnology and Molecular Recognition at The University of Texas at Austin. He is a member of the National Academy of Engineering, the Institute of Medicine of the National Academies, and the National Academy of Pharmacy of France. He received his diploma in engineering (DEng) from the National Technical University of Athens, Greece in 1971 and his ScD from MIT in 1973, both in chemical engineering. He holds honorary doctorates from the Universities of Ghent, Parma and Athens.
References
- 1.Mu L, Feng SS. J Control Release. 2003;86:33–48. doi: 10.1016/s0168-3659(02)00320-6. [DOI] [PubMed] [Google Scholar]
- 2.Primeau AJ, Rendon A, Hedley D, Lilge L, Tannock IF. Clin Cancer Res. 2005;11:8782–8788. doi: 10.1158/1078-0432.CCR-05-1664. [DOI] [PubMed] [Google Scholar]
- 3.Hambley TW. Coord Chem Rev. 1997;166:181–223. [Google Scholar]
- 4.Izquierdo MA, Shoemaker RH, Flens MJ, Scheffer GL, Wu L, Prather TR, Scheper RJ. Int J Cancer. 1996;65:230–237. doi: 10.1002/(SICI)1097-0215(19960117)65:2<230::AID-IJC17>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 5.Gottesman MM. Annu Rev Med. 2002;53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 6.Leslie EM, Deeley RG, Cole SPC. Toxicol Appl Pharmacol. 2005;204:216–237. doi: 10.1016/j.taap.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 7.Shapira A, Livney YD, Broxterman HJ, Assaraf YG. Drug Resist Updat. 2011;14:150–163. doi: 10.1016/j.drup.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 8.Iversen TG, Skotland T, Sandvig K. Nano Today. 2011;6:176–185. doi: 10.1021/nn2021953. [DOI] [PubMed] [Google Scholar]
- 9.Phillips MA, Gran ML, Peppas NA. Nano Today. 2010;5:143–159. doi: 10.1016/j.nantod.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liechty WB, Peppas NA. Eur J Pharm Biopharm. 2012;80:241–246. doi: 10.1016/j.ejpb.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adams JM, Cory S. Curr Opin Immunol. 2007;19:488–496. doi: 10.1016/j.coi.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burnett JC, Rossi JJ. Chem Biol. 2012;19:60–71. doi: 10.1016/j.chembiol.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen SH, Zhaori G. Eur J Clin Invest. 2011;41:221–232. doi: 10.1111/j.1365-2362.2010.02400.x. [DOI] [PubMed] [Google Scholar]
- 14.Aliabadi HM, Landry B, Sun C, Tang T, Uluda H. Biomaterials. 2011;33:2546–2569. doi: 10.1016/j.biomaterials.2011.11.079. [DOI] [PubMed] [Google Scholar]
- 15.Singha K, Namgung R, Kim WJ. Nucleic Acid Ther. 2011;21:133–147. doi: 10.1089/nat.2011.0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Devi GR. Cancer Gene Ther. 2006;13:819–829. doi: 10.1038/sj.cgt.7700931. [DOI] [PubMed] [Google Scholar]
- 17.Kato T, Natsume A, Toda H, Iwamizu H, Sugita T, Hachisu R, Watanabe R, Yuki K, Motomura K, Bankiewicz K, Wakabayashi T. Gene Ther. 2010;17:1363–1371. doi: 10.1038/gt.2010.88. [DOI] [PubMed] [Google Scholar]
- 18.Nakamura K, Abu Lila AS, Matsunaga M, Doi Y, Ishida T, Kiwada H. Mol Ther. 2011;19:2040–2047. doi: 10.1038/mt.2011.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh A, Boldin-Adamsky S, Thimmulappa RK, Rath SK, Ashush H, Coulter J, Blackford A, Goodman SN, Bunz F, Watson WH, Gabrielson E, Feinstein E, Biswal S. Cancer Res. 2008;68:7975–7984. doi: 10.1158/0008-5472.CAN-08-1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yadav S, van Vlerken LE, Little SR, Amiji MM. Cancer Chemother Pharmacol. 2009;63:711–722. doi: 10.1007/s00280-008-0790-y. [DOI] [PubMed] [Google Scholar]
- 21.Taratula O, Garbuzenko OB, Chen AM, Minko T. J Drug Target. 2011;19:900–914. doi: 10.3109/1061186X.2011.622404. [DOI] [PubMed] [Google Scholar]
- 22.Galmarini D, Galmarini CM, Galmarini FC. Crit Rev Oncol Hematol. 2012 [Google Scholar]
- 23.Kerbel RS, Klement G, Pritchard KI, Kamen B. Ann Oncol. 2002;13:12–15. doi: 10.1093/annonc/mdf093. [DOI] [PubMed] [Google Scholar]
- 24.Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, Nikolskaya T, Serebryiskaya T, Beroukhim R, Hu M, Halushka MK, Sukumar S, Parker LM, Anderson KS, Harris LN, Garber JE, Richardson AL, Schnitt SJ, Nikolsky Y, Gelman RS, Polyak K. Cancer Cell. 2007;11:259–273. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 25.Gillet JP, Gottesman MM. Methods Mol Biol. 2010;596:47–76. doi: 10.1007/978-1-60761-416-6_4. [DOI] [PubMed] [Google Scholar]
- 26.Jang SH, Wientjes MG, Lu D, Au JLS. Pharm Res. 2003;20:1337–1350. doi: 10.1023/a:1025785505977. [DOI] [PubMed] [Google Scholar]
- 27.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. J Control Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 28.Allen TM, Cullis PR. Science. 2004;303:1818–1822. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 29.Nat Biotechnol. 2000;18:IT18–IT20. [PubMed] [Google Scholar]
- 30.Lowe SW, Ruley HE, Jacks T, Housman DE. Cell. 1993;74:957–967. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- 31.Longo-Sorbello G, Bertino Haematologica. 2001;86:121–127. [PubMed] [Google Scholar]
- 32.Huang JG, Leshuk T, Gu FX. Nano Today. 2011;6:478–492. [Google Scholar]
- 33.Borst P, Evers R, Kool M, Wijnholds J. J Natl Cancer Inst. 2000;92:1295–1302. doi: 10.1093/jnci/92.16.1295. [DOI] [PubMed] [Google Scholar]
- 34.Zhao XQ, Xie JD, Chen XG, Sim HM, Zhang X, Liang YJ, Singh S, Talele TT, Sun Y, Ambudkar SV, Chen ZS, Fu LW. Mol Pharmacol. 2012 doi: 10.1124/mol.111.076299. http://dx.doi.org/10.1124/mol.111.076299. [DOI] [PMC free article] [PubMed]
- 35.Wang Y, Chen Q, Jin S, Deng W, Li S, Tong Q, Chen Y. Scand J Gastroenterol. 2012;0:1–7. doi: 10.3109/00365521.2012.683042. [DOI] [PubMed] [Google Scholar]
- 36.Ebert SP, Myette RL, Wetzel BG, Conseil l, Cole SPC, Sawada GA, Loo TW, Bartlett MC, Clarke DM, Detty MR. J Med Chem. 2012;55:4683–4699. doi: 10.1021/jm3004398. [DOI] [PubMed] [Google Scholar]
- 37.Setia N, Abbas O, Sousa Y, Garb JL, Mahalingam M. Mod Pathol. 2012:1530–0285. doi: 10.1038/modpathol.2012.71. ISSN. [DOI] [PubMed] [Google Scholar]
- 38.McNeil CM, Sergio CM, Anderson LR, Inman CK, Eggleton SA, Murphy NC, Millar EKA, Crea P, Kench JG, Alles MC, Gardiner-Garden M, Ormandy CJ, Butt AJ, Henshall SM, Musgrove EA, Sutherland RL. J Steroid Biochem Mol Biol. 2006;102:147–155. doi: 10.1016/j.jsbmb.2006.09.028. [DOI] [PubMed] [Google Scholar]
- 39.Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, Wang X. Genes Dev. 2003;17:1475–1486. doi: 10.1101/gad.1093903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun TM, Du JZ, Yao YD, Mao CQ, Dou S, Huang SY, Zhang PZ, Leong KW, Song EW, Wang J. ACS Nano. 2011;5:1483–1494. doi: 10.1021/nn103349h. [DOI] [PubMed] [Google Scholar]
- 41.Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ. Cell. 2009;137:835–848. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Strebhardt K, Ullrich A. Nat Rev Cancer. 2006;6:321–330. doi: 10.1038/nrc1841. [DOI] [PubMed] [Google Scholar]
- 43.Baldwin RL, Tran H, Karlan BY. Cancer Res. 2003;63:1413–1419. [PubMed] [Google Scholar]
- 44.Knapp DC, Mata JE, Reddy MT, Devi GR, Iversen PL. Anticancer Drugs. 2003;14:39–47. doi: 10.1097/00001813-200301000-00006. [DOI] [PubMed] [Google Scholar]
- 45.Huang Y, Anderle P, Bussey KJ, Barbacioru C, Shankavaram U, Dai Z, Reinhold WC, Papp A, Weinstein JN, Weinstein JN, Sadée W. Cancer Res. 2004;64:4294–4301. doi: 10.1158/0008-5472.CAN-03-3884. [DOI] [PubMed] [Google Scholar]
- 46.Wu J, Lu Y, Lee A, Pan X, Yang X, Zhao X, Lee RJ. J Pharm Pharm Sci. 2007;10:350–357. [PubMed] [Google Scholar]
- 47.Song XR, Cai Z, Zheng Y, He G, Cui FY, Gong DQ, Hou SX, Xiong SJ, Lei XJ, Wei YQ. Eur J Pharm Sci. 2009;37:300–305. doi: 10.1016/j.ejps.2009.02.018. [DOI] [PubMed] [Google Scholar]
- 48.Ganta S, Amiji M. Mol Pharm. 2009;6:928–939. doi: 10.1021/mp800240j. [DOI] [PubMed] [Google Scholar]
- 49.Kim C, Shah BP, Subramaniam P, Lee KB. Mol Pharm. 2011;8:1955–1961. doi: 10.1021/mp100460h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beh CW, Seow WY, Wang Y, Zhang Y, Ong ZY, Ee PLR, Yang YY. Biomacromolecules. 2009;10:41–48. doi: 10.1021/bm801109g. [DOI] [PubMed] [Google Scholar]
- 51.Zahedi P, De Souza R, Huynh L, Piquette-Miller M, Allen C. Mol Pharm. 2011;8:260–269. doi: 10.1021/mp100323z. [DOI] [PubMed] [Google Scholar]
- 52.Davis ME, Chen Z, Shin DM. Nat Rev Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 53.Fowers KD, Kopeček J. Macromol Biosci. 2012;12:502–514. doi: 10.1002/mabi.201100350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dickerson E, Blackburn W, Smith M, Kapa L, Lyon LA, McDonald J. BMC Cancer. 2010;10:10. doi: 10.1186/1471-2407-10-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caldorera-Moore M, Peppas NA. Adv Drug Deliv Rev. 2009;61:1391–1401. doi: 10.1016/j.addr.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Caldorera-Moore ME, Liechty WB, Peppas NA. Acc Chem Res. 2011;44:1061–1070. doi: 10.1021/ar2001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liechty WB, Kryscio DR, Slaughter BV, Peppas NA. Annu Rev Chem Biomol Eng. 2010;1:149–173. doi: 10.1146/annurev-chembioeng-073009-100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cao N, Cheng D, Zou S, Ai H, Gao J, Shuai X. Biomaterials. 2011;32:2222–2232. doi: 10.1016/j.biomaterials.2010.11.061. [DOI] [PubMed] [Google Scholar]
- 59.Cheng D, Cao N, Chen J, Yu X, Shuai X. Biomaterials. 2012;33:1170–1179. doi: 10.1016/j.biomaterials.2011.10.057. [DOI] [PubMed] [Google Scholar]
- 60.Huang HY, Kuo WT, Chou MJ, Huang YY. J Biomed Mater Res Part A. 2011;97A:330–338. doi: 10.1002/jbm.a.33055. [DOI] [PubMed] [Google Scholar]
- 61.Wang Y, Gao S, Ye WH, Yoon HS, Yang YY. Nat Mater. 2006;5:791–796. doi: 10.1038/nmat1737. [DOI] [PubMed] [Google Scholar]
- 62.Patil YB, Swaminathan SK, Sadhukha T, Ma L, Panyam J. Biomaterials. 2010;31:358–365. doi: 10.1016/j.biomaterials.2009.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xiong XB, Lavasanifar A. ACS Nano. 2011;5:5202–5213. doi: 10.1021/nn2013707. [DOI] [PubMed] [Google Scholar]
- 64.Zhu C, Jung S, Luo S, Meng F, Zhu X, Park TG, Zhong Z. Biomaterials. 2010;31:2408–2416. doi: 10.1016/j.biomaterials.2009.11.077. [DOI] [PubMed] [Google Scholar]
- 65.Kaneshiro TL, Lu ZR. Biomaterials. 2009;30:5660–5666. doi: 10.1016/j.biomaterials.2009.06.026. [DOI] [PubMed] [Google Scholar]
- 66.Arun R, Ashok KCK, Sravanthi VVNSS. Sci Pharm. 2008;76:567–598. [Google Scholar]
- 67.Creixell M, Bohórquez AC, Torres-Lugo M, Rinaldi C. ACS Nano. 2011;5:7124–7129. doi: 10.1021/nn201822b. [DOI] [PubMed] [Google Scholar]
- 68.Lai CJ, Bao R, Tao X, Wang J, Atoyan R, Qu H, Wang DG, Yin L, Samson M, Forrester J, Zifcak B, Xu GX, DellaRocca S, Zhai HX, Cai X, Munger WE, Keegan M, Pepicelli CV, Qian C. Cancer Res. 2010;70:3647–3656. doi: 10.1158/0008-5472.CAN-09-3360. [DOI] [PubMed] [Google Scholar]
- 69.Garcia-Fuentes M, Torres D, Alonso MJ. Int J Pharm. 2005;296:122–132. doi: 10.1016/j.ijpharm.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 70.Martins S, Costa-Lima S, Carneiro T, Cordeiro-da-Silva A, Souto EB, Ferreira DC. Int J Pharm. 2012;430:216–227. doi: 10.1016/j.ijpharm.2012.03.032. [DOI] [PubMed] [Google Scholar]
- 71.Pardeike J, Hommoss A, Müller RH. Int J Pharm. 2009;366:170–184. doi: 10.1016/j.ijpharm.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 72.Subedi RK, Kang KW, Choi HK. Eur J Pharm Sci. 2009;37:508–513. doi: 10.1016/j.ejps.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 73.Barenholz Y. J Control Release. 2012;160:117–134. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 74.Chang RS, Suh MS, Kim S, Shim G, Lee S, Han SS, Lee KE, Jeon H, Choi HG, Choi Y, Kim CW, Oh YK. Biomaterials. 2011;32:9785–9795. doi: 10.1016/j.biomaterials.2011.09.017. [DOI] [PubMed] [Google Scholar]
- 75.Chen Y, Bathula SR, Li J, Huang L. J Biol Chem. 2010;285:22639–22650. doi: 10.1074/jbc.M110.125906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen Y, Wu JJ, Huang L. Mol Ther. 2010;18:828–834. doi: 10.1038/mt.2009.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kang SH, Cho HJ, Shim G, Lee S, Kim SH, Choi HG, Kim CW, Oh YK. Pharm Res. 2011;28:3069–3078. doi: 10.1007/s11095-011-0569-4. [DOI] [PubMed] [Google Scholar]
- 78.Shim G, Han SE, Yu YH, Lee S, Lee HY, Kim K, Kwon IC, Park TG, Kim YB, Choi YS, Kim CW, Oh YK. J Control Release. 2011;155:60–66. doi: 10.1016/j.jconrel.2010.10.017. [DOI] [PubMed] [Google Scholar]
- 79.Yu YH, Kim E, Park DE, Shim G, Lee S, Kim YB, Kim CW, Oh YK. Eur J Pharm Biopharm. 2011;80:268–273. doi: 10.1016/j.ejpb.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 80.Koynova R, Wang L, MacDonald RC. Mol Pharm. 2008;5:739–744. doi: 10.1021/mp800011e. [DOI] [PubMed] [Google Scholar]
- 81.Saad M, Garbuzenko OB, Minko T. Nanomedicine. 2008;3:761–776. doi: 10.2217/17435889.3.6.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu G, Swierczewska M, Lee S, Chen X. Nano Today. 2010;5:524–539. doi: 10.1016/j.nantod.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Thanh NTK, Green LAW. Nano Today. 2010;5:213–230. [Google Scholar]
- 84.Yildirimer L, Thanh NTK, Loizidou M, Seifalian AM. Nano Today. 2011;6:585–607. doi: 10.1016/j.nantod.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rigby P. Nano Today. 2007;2:12. [Google Scholar]
- 86.Sealy C. Nano Today. 2006;1:19. doi: 10.1016/j.nantod.2021.101171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Soenen SJ, Rivera-Gil P, Montenegro JM, Parak WJ, De Smedt SC, Braeckmans K. Nano Today. 2011;6:446–465. [Google Scholar]
- 88.Chen AM, Zhang M, Wei D, Stueber D, Taratula O, Minko T, He H. Small. 2009;5:2673–2677. doi: 10.1002/smll.200900621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Meng H, Liong M, Xia T, Li Z, Ji Z, Zink JI, Nel AE. ACS Nano. 2010;4:4539–4550. doi: 10.1021/nn100690m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li JM, Wang YY, Zhao MX, Tan CP, Li YQ, Le XY, Ji LN, Mao ZW. Biomaterials. 2012;33:2780–2790. doi: 10.1016/j.biomaterials.2011.12.035. [DOI] [PubMed] [Google Scholar]
- 91.Zrazhevskiy P, Gao X. Nano Today. 2009;4:414–428. doi: 10.1016/j.nantod.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hong H, Zhang Y, Sun J, Cai W. Nano Today. 2009;4:399–413. doi: 10.1016/j.nantod.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Koo H, Huh MS, Ryu JH, Lee DE, Sun IC, Choi K, Kim K, Kwon IC. Nano Today. 2011;6:204–220. [Google Scholar]