

Abstract
A series of novel diarylacrylonitrile and trans-stilbene analogues of resveratrol has been synthesized and evaluated for their anticancer activities against a panel of 60 human cancer cell lines. The diarylacrylonitrile analogues 3b and 4a exhibited the most potent anticancer activity of all the analogues synthesized in this study, with GI50 values of < 10 nM against almost all the cell lines in the human cancer cell panel. Compounds 3b and 4a were also screened against the acute myeloid leukemia (AML) cell line, MV4-11, and were found to have potent cytotoxic properties that are likely mediated through inhibition of tubulin polymerization. Results from molecular docking studies indicate a common binding site for 4a and 3b on the 3,3-tubulin heterodimer, with a slightly more favorable binding for 3b compared to 4a; this is consistent with the results from the microtubule assays, which demonstrate that 4a is more potent than 3b in inhibiting tubulin polymerization in MV4-11 cells. Taken together, these data suggest that diarylacrylonitriles 3b and 4a may have potential as antitubulin therapeutics for treatment of both solid and hematological tumors.
Introduction
Chemicals that perturb microtubule and tubulin dynamics are widely used in cancer chemotherapy.1 Taxane, vinca and colchicine domains on tubulin are the major binding sites for anti-cancer drugs such as paclitaxel, docetaxel, cabazitazel, vincristine, vinblastine and colchicine.2 The mode of action of these drugs is via inhibition of tubulin polymerization or by preventing depolymerization of tubulin, resulting in mitotic arrest. Lately, anti-mitotic agents that bind to the colchicine domain on tubulin have received much attention, and drugs such as combretastatin A-4 (CA-4) (Fig. 1; structure A) and its analogues have been extensively studied for their anti-cancer activity.3–6 CA-4, a cis-stilbene analogue, has been isolated from the South African native willow tree, Combretum caffrum, and is a potent anti-proliferative agent.7 CA-4 inhibits the polymerization of tubulin by interacting with the colchicine binding site on tubulin, resulting in mitotic arrest. CA-4 is also a promising anti-angiogenic molecule, and its water soluble phosphate salt is currently in phase III clinical trials for treatment of anaplastic thyroid cancer.8
Fig. 1.

Structures of combretastatin A-4 and resveratrol related anticancer agents.
The structurally related trans-stilbene analogue, resveratrol is a phytolexin first isolated from white hellebore (Veratrum grandiflorum O. Loes) and from about 70 other plant species.9 Resveratrol has been reported as a potential chemotherapeutic agent, due to its striking inhibitory effects on cellular events associated with cancer initiation, promotion, and progression.10
Chen and co-workers have synthesized and evaluated a series of methoxylated resveratrol derivatives for their anti-cancer properties against three different human cancer cell lines, i.e. K562, HT29, and HePG2. They reported that (E)-3,5,4′-trimethoxystilbene (Fig. 1; structure B) and (E)-3,4,5,4′-tetramethoxystilbene (Fig. 1; structure C) were the most effective anti-cancer agents among the analogues investigated, and showed improved cytotoxicity compared to resveratrol itself.11
Intriguing results have been reported with the O-methylated resveratrol analogue (E)-3,4,5,4′-tetramethoxystilbene (Fig. 1; structure C). This compound was found to potently inhibit the proliferation of a variety of cancer cells, including HeLa cervical cancer cells, MCF-7 and MDA-MB-435/LCC6 breast cancer cells, HepG2 hepatoma cells, LnCaP prostate cancer cells and HT-29 colon cancer cells.12 Recently, our laboratory has reported on some novel O-methylated resveratrol substrates for human hepatic, intestinal, and renal UDP-glucuronosyl transferases. These compounds were found to have increased bioavailability via altered conjugation, and were considered as alternative scaffolds for the development of new bioactive resveratrol analogues.13
More recently, we have reported on a series of (Z)-benzothiophene acrylonitrile derivatives of resveratrol (Fig. 1; structure D); these compounds are potent anti-cancer agents which are not substrates for cellular P-glycoprotein.14c In this respect, Ohsumi et al. have reported that (E)-substituted diarylacrylonitrile analogues structurally related to the combretastatins (Fig. 1; structure E) are also effective anti-cancer agents against murine solid tumors.15
In our continuing quest to improve the potencies of natural anti-cancer molecules through chemical modification, we have now synthesized a small library of trans-stilbenes and related (Z)-diarylacrylonitrile analogues structurally related to resveratrol, and have evaluated them against a panel of 60 human tumor cell lines and against MV-411 acute myeloid leukemia (AML) cells for their anti-cancer activity. In these molecules, the trans-stilbene structural moiety present in resveratrol have been modified by introducing a cyano group at one of the sp2 carbons of the stilbene double bond, and a variety of different aromatic substituents have been introduced into the phenyl rings to improve anticancer activity. We also describe the tubulin binding properties of the most potent of these novel trans-stilbene analogues in comparison to the tubulin binding properties of the corresponding cis-stilbene analogue.
Chemistry
A series of (Z)-substituted diarylacrylonitrile analogues (3a–3h) were synthesized by reacting a variety of substituted benzyl carbaldehydes (2a–2e) with an appropriately substituted phenylacetonitrile (1a–1d) in 5% sodium methoxide/methanol. The reaction mixture was stirred at room temperature for 2–3 h to allow the reaction to go to completion, during which time the desired product crashed out of the solution. The resulting precipitate was filtered, washed with water and dried to yield the desired compound in yields ranging from 70–95% (Scheme 1).
Scheme 1.
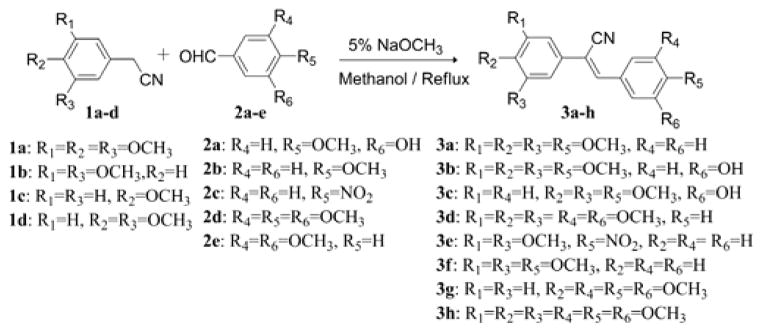
Synthesis of (Z)-substituted diarylacrylonitrile analogues (3a–3h).
(E)-Substituted diarylacrylonitrile analogues 4a and 4b were obtained by refluxing a methanolic solution of the (Z)-isomers 3b and 3c in under ultraviolet light at 254 nm for 24 h. The time course of the reaction was monitored by GC-MS. Once the reaction was complete, the solution was cooled to room temperature and the resulting precipitate was filtered off to yield the (E)-substituted diarylacrylonitrile analogues 4a and 4b (Scheme 2).
Scheme 2.
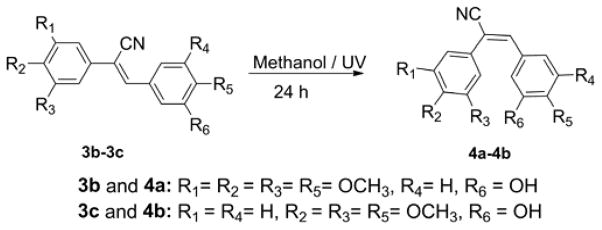
Synthesis of (E)-substituted diarylacrylonitrile analogues (4a and 4b).
In related studies, Ruan et al. have reported the antitumor activity of resveratrol derivatives possessing a chalcone moiety (Fig. 1; structure F); these analogues exhibited potent anti-proliferative and antitubulin activities, and compound F (Fig. 1) inhibited the growth of cancer cell lines HepG2, B16-F10, and A549 with IC50 values of 0.2, 0.1, and 1.4 μg/mL, respectively.16 In view of this finding, and the potent anticancer activity of the aryl-substituted acrylonitrile analogues of structure E (Fig. 1), a series of hybrid resveratrol derivatives possessing a phenylacrylonitrile moiety attached to the C2-position of the (E)-3,5,4′-trimethoxystilbene scaffold were investigated. The synthetic strategy for preparing such compounds is given in Scheme 3. The synthetic procedure for synthesizing the key intermediate 5 (Scheme 3) involves O-methylation of the hydroxyl groups of resveratrol with MeI/K2CO3 in acetone, to form (E)-1,3-dimethoxy-5-(4-methoxystyryl)-benzene, followed by C2-formylation with a slight molar excess of POCl3 in DMF at 0 °C for 30 min, to yield trans-2-formyl-3,43,5-trimethoxystilbene (5).17,18
Scheme 3.

Synthesis of (Z)-3-(2,4-dimethoxy-6-(4-methoxystyryl)-2-phenylacrylonitrile analogues (7a–7g). (a) MeI, K2CO3, acetone; (b) POCl3, DMF, 0 °C, 69% yield; (c) NaOMe, EtOH, 6h reflux.
With intermediate 5 in hand, a novel series of (Z)-3-(2,4-dimethoxy-6-(4-methoxystyryl)phenyl)-2-phenylacrylonitrile analogues (7a–7g) could be synthesized by reacting intermediate 5 with appropriately substituted phenylacetonitriles (6a–6d, 1a–1c) in 5% sodium methoxide/methanol. The reaction mixture was stirred at room temperature for 2–3 h to allow the reaction to go to completion, during which time the desired product was precipitated. The resulting solid was filtered off, washed with water and dried to yield the desired compound in yields ranging from 55–80% (Scheme 3). Confirmation of the structure and purity of these analogues was obtained from 1H, 13C-NMR and mass spectroscopic analysis. According to previous literature, base-catalyzed condensation of aryl/heteroaryl aldehydes with aryl/heteroaryl acetonitriles leads exclusively to the formation of the (Z)-isomer.14a–14c We confirmed the (Z) configuration of analogues 7a–7g by carrying out carbon-proton coupling experiments to determine the magnitude of the coupling constant (JCH) of the CN carbon doublet that results from coupling with the adjacent olefinic proton.14b
Biological evaluation
A In vitro growth inhibition studies
Primary in vitro screening of all the synthesized compounds was carried out against a panel of 60 human tumor cell lines utilizing the sulforhodamine B (SRB) assay procedure described by Rubinstein et al.19,20 Compounds 3a–3h were initially screened at 10−5 M concentration to determine growth inhibition properties. Only compounds that showed more than 60% growth inhibition at 10−5 M in at least eight cell lines from the panel of 60 cell lines were selected for a complete dose-response study with five different concentrations, i.e. 10−4 M, 10−5 M, 10−6 M, 10−7 M and 10−8 M.
In the (Z)-diarylacrylonitrile series of compounds (3a–3h), only compounds 3a–3d were selected for full dose-response studies (Table 1). When the 4-methoxyphenyl group on ring B of compound 3a was replaced with a 3,5-dimethoxyphenyl moiety (3d), the average GI50 activity declined from 18 nM to 13 μM. Also, when the 3,4,5-trimethoxyphenyl group on ring A of compound 3a was replaced with a 3,5-dimethoxyphenyl moiety (3f) the average GI50 activity declined from 18 nM to 400 nM. Interestingly, introduction of a 3-hydroxy group into ring B of compound 3a to afford compound 3b improved the average GI50 value of 18 nM against all the cancer cell lines in the NCI panel to <10 nM. Comparing the growth inhibition activities of 3d and 3f with their more active counterparts 3a and 3b suggests that both the 3,4,5-trimethoxy group on ring A and the 4-methoxy-3-hydroxy group on ring B contribute significantly to the anticancer activity of the (Z)-diarylacrylonitrile analogues.
Table 1.
Growth inhibition (GI50)a data for compounds 3a, 3d, 3f, 3c, 4b, 3b, 4a, 7b, 7e and CA4 against a panel of 60 human cancer cell lines
| Panel/Cell line | 3a | 3d | 3f | 3c | 4b | 3b | 4a | 7b | 7e | CA4b |
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| GI50 (μM) | GI50 (μM) | GI50 (μM) | GI50 (μM) | GI50 (μM) | GI50 (μM) | GI50 (μM) | GI50 (μM) | GI50 (μM) | GI50 (μM) | |
| Leukemia | ||||||||||
| CCRF-CEM | 0.03 | 51.2 | 0.32 | 0.03 | 0.04 | <0.01 | <0.01 | 2.24 | 0.75 | 0.003 |
| HL-60(TB) | 0.02 | 68.6 | 0.28 | 0.03 | 0.03 | <0.01 | <0.01 | 0.58 | 1.66 | 0.002 |
| K-562 | <0.01 | 3.14 | 0.31 | 0.02 | 0.03 | <0.01 | <0.01 | 0.43 | 0.43 | 0.002 |
| MOLT-4 | 0.03 | 28.2 | 0.43 | 0.05 | 0.07 | <0.01 | <0.01 | 3.30 | 4.38 | 0.003 |
| RPMI-8226 | 0.03 | 6.64 | 0.33 | 0.04 | 0.05 | <0.01 | <0.01 | 2.85 | 1.16 | 0.003 |
| SR | <0.01 | 4.14 | 0.29 | 0.02 | 0.03 | <0.01 | <0.01 | 0.28 | 0.91 | 0.003 |
| Non-Small Cell Lung Cancer | ||||||||||
| A549 | 0.01 | 6.35 | 0.38 | 0.21 | 0.25 | <0.01 | <0.01 | 3.56 | 0.91 | 0.016 |
| HOP-62 | 0.03 | 7.63 | 0.51 | 0.03 | 0.03 | <0.01 | NA | 2.79 | 0.97 | 0.003 |
| HOP-92 | 0.02 | 3.57 | NA | 1.69 | 0.86 | <0.01 | <0.01 | NA | NA | 0.003 |
| NCI-H23 | 0.05 | 43.0 | 0.53 | 0.07 | 0.08 | <0.01 | <0.01 | NA | NA | 0.003 |
| NCI-H522 | <0.01 | 20.5 | 0.09 | <0.01 | 0.01 | <0.01 | <0.01 | 0.25 | 0.24 | 0.002 |
| Colon Cancer | ||||||||||
| COLO 205 | 0.01 | 5.16 | 0.27 | 1.24 | 1.26 | 2.99 | 0.30 | 2.28 | 1.24 | 0.100 |
| HCC-2998 | 0.04 | 97.0 | 0.34 | 1.81 | 0.17 | 0.02 | <0.01 | 5.47 | 2.49 | 0.063 |
| HCT-116 | <0.01 | 6.05 | 0.33 | 0.04 | 0.03 | <0.01 | <0.01 | 1.29 | 0.49 | 0.003 |
| HCT-15 | <0.01 | 4.47 | 0.33 | 0.04 | 0.03 | <0.01 | <0.01 | 0.76 | 0.65 | 0.004 |
| HT29 | <0.01 | 4.24 | 0.28 | 0.57 | 0.62 | 3.18 | 0.32 | 0.48 | 0.69 | 0.100 |
| KM12 | <0.01 | 4.97 | 0.38 | 0.02 | 0.04 | <0.01 | <0.01 | 0.64 | 0.78 | 0.005 |
| SW-620 | 0.01 | 5.17 | 0.42 | 0.04 | 0.04 | <0.01 | <0.01 | 0.71 | 0.50 | 0.003 |
| CNS Cancer | ||||||||||
| SF-268 | 0.04 | 25.3 | 0.84 | 0.06 | 0.08 | <0.01 | <0.01 | 4.18 | 4.71 | 0.006 |
| SF-295 | <0.01 | 5.33 | 0.22 | 0.10 | 0.58 | 0.05 | 0.01 | 2.51 | 0.36 | 0.004 |
| SF-539 | <0.01 | 6.60 | 0.22 | 0.02 | 0.02 | <0.01 | <0.01 | 1.55 | 0.27 | 0.003 |
| SNB-19 | 0.06 | 5.46 | 0.98 | 0.05 | 0.04 | <0.01 | <0.01 | 8.77 | >100 | 0.004 |
| SNB-75 | <0.01 | 2.67 | 0.29 | 0.02 | 0.02 | <0.01 | <0.01 | 2.28 | 0.47 | 0.008 |
| U251 | 0.02 | 7.82 | 0.343 | 0.04 | 0.05 | 0.01 | <0.01 | 2.77 | 0.64 | 0.004 |
| Melanoma | ||||||||||
| LOX IMVI | 0.01 | 5.31 | 0.58 | 0.06 | 0.07 | <0.01 | <0.01 | NA | NA | 0.003 |
| M14 | 0.01 | 4.45 | 0.28 | 0.03 | 0.02 | <0.01 | <0.01 | 1.14 | 1.38 | 0.003 |
| MDA-MB-435 | <0.01 | 1.63 | 0.17 | <0.01 | <0.01 | <0.01 | <0.01 | 0.28 | 0.24 | NA |
| SK-MEL-2 | 0.08 | 5.44 | 0.88 | NA | NA | <0.01 | <0.01 | 2.13 | NA | 0.004 |
| SK-MEL-28 | NA | 14.0 | 0.49 | 1.11 | 1.27 | 1.01 | <0.01 | 4.22 | 0.75 | 0.008 |
| SK-MEL-5 | 0.01 | 4.70 | 0.27 | 0.04 | 0.05 | <0.01 | <0.01 | 2.44 | 2.28 | 0.004 |
| UACC-62 | <0.01 | 2.42 | 0.40 | 0.02 | NA | <0.01 | <0.01 | NA | 0.70 | 0.006 |
| Ovarian Cancer | ||||||||||
| IGROV1 | 0.02 | 14.6 | 0.53 | 0.06 | 0.07 | <0.01 | <0.01 | 5.28 | 2.35 | 0.015 |
| OVCAR-3 | <0.01 | 6.69 | 0.30 | 0.02 | 0.02 | <0.01 | <0.01 | 1.38 | 0.63 | 0.002 |
| OVCAR-4 | 0.01 | 7.48 | 0.73 | 1.56 | 1.07 | <0.01 | <0.01 | 3.59 | 0.50 | 0.014 |
| NCI/ADR-RES | <0.01 | 3.25 | 0.27 | 0.02 | 0.02 | <0.01 | <0.01 | 0.71 | 0.65 | NA |
| SK-OV-3 | <0.01 | 14.4 | 0.37 | 0.05 | 0.05 | 0.01 | <0.01 | 4.73 | 1.35 | 0.059 |
| Renal Cancer | ||||||||||
| 786-0 | 0.02 | 13.0 | 0.42 | 0.36 | 0.24 | <0.01 | 0.02 | 3.25 | 1.72 | 0.100 |
| A498 | <0.01 | 1.29 | 0.25 | 0.02 | 0.02 | <0.01 | <0.01 | 1.37 | 0.38 | 0.006 |
| ACHN | 0.01 | 9.78 | 0.53 | 0.08 | 0.09 | <0.01 | <0.01 | 4.25 | 0.83 | 0.006 |
| CAKI-1 | NA | 6.19 | 0.54 | NA | 0.42 | 0.03 | <0.01 | 2.20 | 0.50 | 0.026 |
| UO-31 | <0.01 | 1.92 | 0.49 | 0.06 | 0.09 | <0.01 | <0.01 | 3.44 | 0.81 | 0.019 |
| Breast Cancer | ||||||||||
| MCF7 | <0.01 | 4.35 | 0.31 | 0.03 | 0.03 | <0.01 | <0.01 | 0.72 | 0.39 | NA |
| MDA-MB-231/ATCC | 0.03 | 4.83 | 0.43 | 0.04 | 0.05 | <0.01 | <0.01 | 2.83 | 2.78 | NA |
| HS 578T | <0.01 | 16.7 | 0.31 | 0.03 | 0.03 | <0.01 | <0.01 | 1.34 | 0.68 | NA |
| MDA-MB-468 | 0.02 | 3.44 | 0.29 | 0.04 | 0.04 | <0.01 | <0.01 | 1.70 | 1.72 | NA |
NA: Not analyzed,
GI50: 50% growth inhibition, concentration of drug resulting in a 50% reduction in net protein increase compared with control cells.
NCI data for CA4 NSC 613729.3
Interestingly, Ohsumi et al. has previously reported that the (E)-diarylacrylonitrile analogue 4a potently inhibits the proliferation of colon 26 cancer cells with an IC50 of 23 nM.15 Thus, (E)-isomers 4a and 4b (Scheme 3) were synthesized to compare their growth inhibitory values with those of their Z-counterparts, 3b and 3c. The average GI50 values of the E/Z pair of isomers 4b and 3c were similar (177 nM and 223 nM, respectively; Table 1), while the other pair of E/Z isomers 3b and 4a were both found to be the most potent compounds in this series from the five dose study data, with GI50 values of < 10 nM against almost all the NCI human cancer cell lines examined. Importantly, compounds 3b and 4a were significantly more effective against the growth of several cancer cell lines when compared to CA4 (Table 1). These include non-small cell lung cancer A549/ATCC, colon cancer HCC-2998, ovarian cancer (IGROV1, IGROV4, SK-OV-3), renal cancer (786-0, UO-31) cell lines (Table 1).
In the (Z)-3-(2,4-dimethoxy-6-(4-methoxystyryl)phenyl)-2-phenylacrylonitrile analogue series (7a–7g), two compounds, 7b and 7e, showed good anticancer activity in the single dose cancer cell screen, and were subsequently evaluated in the five dose testing paradigm. Both compounds were found to be effective against four particular cancer cell lines: viz. SR, NCI H522, SF-539 and MDA-MB-435 with GI50 values less than 300 nM (Table 1). However, the average GI50 value of compounds 7b and 7e against all 60 cell lines was only 2.79 and 1.19 μM, respectively. Compound 7e (GI50 = 0.91 μM) was found to be somewhat more potent than the previously reported reseveratrol-chalcone molecule F (Figure 1) (GI50 = 1.40 μM) against non-small lung cancer cell line A549.16
Thus, compounds 3b and 4a are the most potent compounds of all the resveratrol analogues synthesized in this study, with GI50 values of < 10 nM against almost all the cell lines in the human cancer cell panel.
B In vitro toxicity study on AML cells and tubulin activity
Compounds 3c, 4b, and 3b and 4a were found to be very effective cytotoxic agents against the leukemia cell sub-panel in the 60 cancer cell screen (Table 1). Notably, compounds 3b and 4a exhibited GI50 values of < 10 nM across all six leukemia cell lines. We have also tested the cytotoxicity of these four lead compounds against MV-411 AML cells (Fig. 2), and have conducted tubulin binding assays on these compounds in the same cell line (Fig. 3).
Fig. 2.

Lead compounds 3b, 4a, 3c and 4b exhibit potent anti-leukemia activity against MV-411 cells. MV-411 cells were treated with the indicated compounds for 24 and 48 h. Cell viability was determined by Annexin V staining. Percent viability was calculated as the percent of Annexin v-/7-AAD- cells relative to control. N = 5; error bars represent the SD.
Fig. 3.

Microtubule depolymerization assays with lead compounds 3b, 3c, 4a and 4b. P = pellet, S = supernatant.
MV4-11 cells were treated with increasing concentrations of the above four lead compounds for 24 and 48 hours. Figure 2 shows the dose-response curves for each of the four compounds at both time points. We found that after 48 hours of drug treatment 4a was the most potent anti-leukemic compound causing 50 percent cell death at a concentration of 2.5 nM (Fig. 2a). Compound 3b exhibited an LD50 value of 38.6 nM (Fig. 2b), and compounds 3c and 4b afforded LD50 values of 353 nM and 409 nM, respectively (Figs. 2c and 2d). These data suggest that compounds 4a and 3b hold promise as potential treatments for AML.
We also investigated whether the above four lead compounds could interfere with microtubule polymerization utilizing an immunoblot assay.21,22 MV4-11 cells were treated with three concentrations (25, 50 and 100 nM) of 3b, 3c, 4a and 4b for 2 hours. Cell-based tubulin depolymerization assays were then performed. The polymerized 3-tubulin in the pellets (P) and unpolymerized 3-tubulin in the supernatants (S) were detected by Western blotting using antibody against 3-tubulin. The data demonstrate that lead compounds 4a and 3b bind to tubulin directly to inhibit polymerization. Consistent with the superior anti-leukemic activity observed for 4a over 3b in MV4-11 cells, 4a demonstrated a more potent inhibition of MT polymerization when compared to 3b (Fig. 3).
C In silico molecular docking studies
Since the (E) and (Z) isomers 4a and 3b were found to be the most potent compounds (GI50 <10 nM) against almost all the cancer cell lines tested, these molecules were chosen for molecular docking studies utilizing the available crystal structure of tubulin, in order to identify their binding sites on this protein. The crystal structure of the tubulin-colchicine complex was chosen as the target for docking analysis (PDB ID 1SA0). Colchicine was removed from the coordinate file and the coordinates of only chains A and B, corresponding to a 3,3-tubulin heterodimer were used for the docking studies. Atomic coordinates for all compounds were generated using MarvinSketch (ChemAxon), and both the ligand and target protein coordinates files were prepared for docking using the Dock Prep module in the UCSF-Chimera package23. Compound 3d, which was found to be significantly less potent than either 4a or 3b in our biological assays (GI50 > 1 μM), was also evaluated. Docking was performed using SwissDock (http://www.swissdock.ch/), based on the docking algorithm EADock DSS.24,25
Previously, we have successfully used this method for docking small-molecule inhibitors to human Y-family polymerases.26,27 Several trial docking runs were carried out to test the validity of our protocol. We consistently obtained a binding mode for colchicine in our trial docking runs that is essentially identical to what was reported in the actual crystal structure (data not shown). Docking with 4a, 3b and 3d was performed using the most exhaustive and unbiased option in SwissDock, in order to sample the maximum number of binding modes. The best hits based on the SwissDock FullFitness scoring function (FF) from three repeated docking runs were considered further.
The crystal structure of the tubulin-colchicine complex shows a single colchicine ligand bound to the 3-subunit at the interface with the 3-subunit of each tubulin 3,3-heterodimer. Our docking results indicated that both 3b and 4a bind to the colchicine site in the tubulin heterodimer (Figure 4). In the crystal structure, colchicine does not have polar contacts with any residues of either the 3 or the 3 subunit of tubulin. Colchicine binds in the 4–5 Å wide cavity of its binding site and interacts with the protein through van der Waals’ interactions with the mostly non-polar side chains and backbone atoms of tubulin.
Fig. 4.
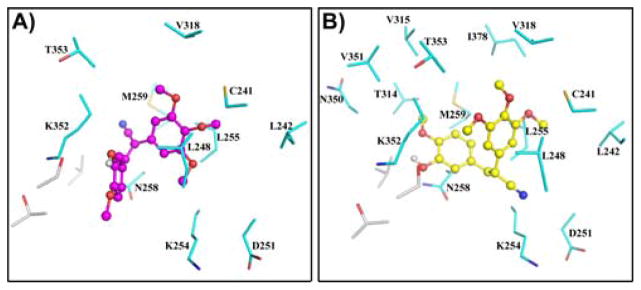
Compounds 4a and 3b bound in the colchicine binding site on tubulin. The most favored docked poses of 3b (magenta) and 4a (yellow) are shown in panels A and B, respectively, as ball-and-stick models in the binding site on the 3,3-tubulin heterodimer. 3-Tubulin residues are shown as grey sticks, while the 3-tubulin residues are cyan. All structural representations shown in this, and subsequent figures, were generated using PyMol (DeLano Scientific, San Carlos, CA).
A comparison of the residues lining the binding cavity of the three compounds, colchicine, 4a and 3b revealed that all three compounds share most of the van der Waals’ contacts to the protein (see Table in Supplementary Data) with only some minor differences. Compound 3b has fewer non-polar contacts to tubulin, when compared to both colchicine and 4a. Compound 3d (the least potent compound) has even fewer contacts with tubulin residues than 3b. This is reflected in the FF and 3G scores of the three compounds (Table 2), with the FF score for 4a being consistently higher than that for 3b, while that for 3d was the lowest. Additionally, compound 4a appears to be buried deeper in the binding cavity compared to both 3b and 3d (Figure 5), and this is supported by the greater calculated surface area of the protein within contact distance of 4a (2165 Å) compared to that for 3b (1924 Å).
Table 2.
SwissDock statistics for the docking runs with compounds 3b, 4a and 3d (scores shown are averages from 3 docking runs).
| Comp | FF score (kcal/mol) | ΔG (kcal/mol) |
|---|---|---|
| 3b | -4225.4 | -8.0 |
| 4a | -4231.2 | -8.3 |
| 3d | -4218.2 | -7.6 |
Fig. 5.

Relative binding of compounds 3b, 4a and 3d in the colchicine binding site on tubulin. The colchicine-binding pocket on the 3-subunit of tubulin is shown as a solid molecular surface, which accounts for most of the van der Waals’ interactions with compounds 3b (magenta), 4a (yellow) and 3d (cyan). The atoms of 3-tubulin have been removed for better clarity. The A-ring moiety of the (Z)-isomer 3b can be seen clearly projecting out of the binding cavity. In the case of the (E)-isomer 4a, both A and B rings are placed snugly into the cavity, forming a more extensive interacting surface with the protein as compared to 3b. Compound 3d, which is the least potent of the three compounds, is the least buried in the binding pocket, compared to both 3b and 4a.
Conclusion
A series of aromatic substituted trans-stilbenes and diarylacrylonitrile analogues have been synthesized and evaluated for their anticancer activities against a panel of 60 human cancer cell lines. The studies demonstrate that resveratrol analogues that have been modified by introducing a cyano group onto the double bond of the stilbene scaffold and incorporating methoxy substituents into the phenyl rings generally improves the growth inhibition properties of these analogues against human cancer cell lines when compared to aromatic substituted resveratrol analogues. Compounds 3b and 4a were the most potent compounds of all the diarylacryonitrile analogues synthesized in this study, with GI50 values of < 10 nM against almost all the cell lines in the human cancer cell panel. The most active compounds from the human cancer cell screens were also screened against the acute myeloid leukemia cell line, MV4-11, and were found to have potent anti-cancer properties that are likely mediated through interference with tubulin polymerization. Results from molecular docking studies indicate a common binding site for 3b and 4a on the 3,3-tubulin heterodimer, with a slightly more favorable binding for 4a compared to 3b, which is consistent with the results from microtubule depolymerization assays, which demonstrate that 4a is more potent than 3b in inhibiting tubulin polymerization in MV4-11 cells. Taken together, these data suggest that diarylacrylonitriles 3b and 4a may have potential as therapeutics for treatment of both solid and hematological tumors.
Supplementary Material
Acknowledgments
The authors thank the NIH/National Cancer Institute (CA140409 to PAC and CA183895 to RLE), and the Arkansas Research Alliance for financial support; MLG is funded by the US National Institutes of Health (NIH) through the NIH Director’s New Innovator Award Program, 1 DP2 OD007399-01. We are also grateful to the NCI Developmental Therapeutic Program (DTP) for anticancer screening data.
References
- 1.Thorpe PE. Clinical cancer research: an official journal of the American Association for Cancer Research. 2004;10:415–427. doi: 10.1158/1078-0432.ccr-0642-03. [DOI] [PubMed] [Google Scholar]
- 2.Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M. Nature. 2004;428:198–202. doi: 10.1038/nature02393. [DOI] [PubMed] [Google Scholar]
- 3.Demchuk DV, Samet AV, Chernysheva NB, Ushkarov VI, Stashina GA, Konyushkin LD, Raihstat MM, Firgang SI, Philchenkov AA, Zavelevich MP, Kuiava LM, Chekhun VF, Blokhin DY, Kiselyov AS, Semenova MN, Semenov VV. Bioorganic & Medicinal Chemistry. 2014;22:738–755. doi: 10.1016/j.bmc.2013.12.015. [DOI] [PubMed] [Google Scholar]
- 4.Banimustafa M, Kheirollahi A, Safavi M, Kabudanian Ardestani S, Aryapour H, Foroumadi A, Emami S. European Journal of Medicinal Chemistry. 2013;70:692–702. doi: 10.1016/j.ejmech.2013.10.046. [DOI] [PubMed] [Google Scholar]
- 5.Madadi NR, Penthala NR, Song L, Hendrickson HP, Crooks PA. Tetrahedron Letters. 2014;55:4207–4211. doi: 10.1016/j.tetlet.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carr M, Greene LM, Knox AJS, Lloyd DG, Zisterer DM, Meegan MJ. European Journal of Medicinal Chemistry. 2010;45:5752–5766. doi: 10.1016/j.ejmech.2010.09.033. [DOI] [PubMed] [Google Scholar]
- 7.Pettit GR, Singh SB, Boyd MR, Hamel E, Pettit RK, Schmidt JM, Hogan F. Journal of medicinal chemistry. 1995;38:1666–1672. doi: 10.1021/jm00010a011. [DOI] [PubMed] [Google Scholar]
- 8.Siemann DW, Chaplin DJ, Walicke PA. Expert Opin Investig Drugs. 2009;18:189–197. doi: 10.1517/13543780802691068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baur JA, Sinclair DA. Nature reviews Drug discovery. 2006;5:493–506. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- 10.Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, Fong HH, Farnsworth NR, Kinghorn AD, Mehta RG, Moon RC, Pezzuto JM. Science. 1997;275:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- 11.Chen Y, Hu F, Gao Y, Jia S, Ji N, Hua E. Res Chem Intermed. 2013:1–14. doi: 10.1007/s11164-013-1382-y. [DOI] [Google Scholar]
- 12.Chen L-k, Qiang P-f, Xu Q-p, Zhao Y-h, Dai F, Zhang L. Acta Pharmacol Sin. 2013;34:1174–1182. doi: 10.1038/aps.2013.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greer AK, Madadi NR, Bratton SM, Eddy SD, Mazerska Z, Hendrickson H, Crooks PA, Radominska-Pandya A. Chemical research in toxicology. 2014;27:536–545. doi: 10.1021/tx400408x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Jalily PH, Hadfield JA, Hirst N, Rossington SB. Bioorganic & medicinal chemistry letters. 2012;22:6731–6734. doi: 10.1016/j.bmcl.2012.08.089. [DOI] [PubMed] [Google Scholar]; (b) Penthala NR, Sonar VN, Horn J, Leggas M, Yadlapalli JS, Crooks PA. MedChem Comm. 2013;4:1073–1078. doi: 10.1039/C3MD00130J. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Penthala NR, Janganati V, Bommagani S, Crooks PA. MedChemComm. 2014;5:886–890. [Google Scholar]
- 15.Ohsumi K, Nakagawa R, Fukuda Y, Hatanaka T, Morinaga Y, Nihei Y, Ohishi K, Suga Y, Akiyama Y, Tsuji T. Journal of medicinal chemistry. 1998;41:3022–3032. doi: 10.1021/jm980101w. [DOI] [PubMed] [Google Scholar]
- 16.Ruan BF, Lu X, Tang JF, Wei Y, Wang XL, Zhang YB, Wang LS, Zhu HL. Bio organic & Medicinal Chemistry. 2011;19:2688–2695. doi: 10.1016/j.bmc.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 17.Madadi NR, Reddy TRY, Penthala NR, Parkin S, Crooks PA. Acta Crystallographica Section E: Structure Reports Online. 2010;66:o1792–o1792. doi: 10.1107/S1600536810018520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Madadi NR, Parkin S, Crooks PA. Acta crystallo graphica Section E, Structure reports online. 2012;68:o730. doi: 10.1107/S1600536812005843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rubinstein LV, Shoemaker RH, Paull KD, Simon RM, Tosini S, Skehan P, Scudiero DA, Monks A, Boyd MR. Journal of the National Cancer Institute. 1990;82:1113–1118. doi: 10.1093/jnci/82.13.1113. [DOI] [PubMed] [Google Scholar]
- 20.Madadi NR, Penthala NR, Janganati V, Crooks PA. Bioorganic & medicinal chemistry letters. 2014;24:601–603. doi: 10.1016/j.bmcl.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gundersen GG, Kalnoski MH, Bulinski JC. Cell. 1984;38:779–789. doi: 10.1016/0092-8674(84)90273-3. [DOI] [PubMed] [Google Scholar]
- 22.Baas PW, Black MM. The Journal of cell biology. 1990;111:495–509. doi: 10.1083/jcb.111.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. Journal of computational chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 24.Grosdidier A, Zoete V, Michielin O. Nucleic acids research. 2011;39:W270–277. doi: 10.1093/nar/gkr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grosdidier A, Zoete V, Michielin O. Journal of computational chemistry. 2011 doi: 10.1002/jcc.21797. [DOI] [PubMed] [Google Scholar]
- 26.Coggins GE, Maddukuri L, Penthala NR, Hartman JH, Eddy S, Ketkar A, Crooks PA, Eoff RL. ACS chemical biology. 2013;8:1722–1729. doi: 10.1021/cb400305r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ketkar A, Zafar MK, Maddukuri L, Yamanaka K, Banerjee S, Egli M, Choi JY, Lloyd RS, Eoff RL. Chemical research in toxicology. 2013;26:221–232. doi: 10.1021/tx300392m. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.