

Abstract
We synthesized a library of 48 analogs of the Mycobacterium tuberculosis cell growth inhibitor SQ109 in which the ethylene diamine linker was replaced by oxa-, thia- or heterocyclic species, and in some cases, the adamantyl group was replaced by a 1,2-carborane or the N-geranyl group by another hydrophobic species. Compounds were tested against Mycobacterium tuberculosis (H37Rv and/or Erdman), Mycobacterium smegmatis, Bacillus subtilis, Escherichia coli, Saccharomyces cerevisiae, Trypanosoma brucei and two human cell lines (human embryonic kidney, HEK293T, and the hepatocellular carcinoma, HepG2). Most potent activity was found against T. brucei, the causative agent of human African trypanosomiasis, and involved targeting of the mitochondrial membrane potential with 15 SQ109 analogs being more active than was SQ109 in cell growth inhibition, having IC50 values as low as 12 nM (5.5 ng/mL) and a selectivity index of ~300.
Keywords: Tuberculosis, sleeping-sickness, uncouplers, menaquinone, trypanosomes
The occurrence of drug resistance is a growing problem1,2. One serious threat is with tuberculosis since there are many millions of individuals infected with Mycobacterium tuberculosis, the causative agent of tuberculosis, resulting in ~1.5 million deaths per year3. Chemotherapy is lengthy and there is increasing resistance to antibiotics. New drugs and drug leads are thus needed. One of the oldest drugs for tuberculosis treatment is ethambutol (1), an ethylenediamine derivative, and in recent work some 74,000 analogs4,5 of ethambutol including the ethylenediamine SQ1094 (2) and the piperidine SQ6095 have shown promise. One mechanism of action of SQ109 has been proposed to be inhibition of the membrane protein MmpL36, a trehalose monomycolate transporter7. There have been no reports of spontaneous resistance to SQ109 but resistance to somewhat similar species involving MmpL3 has been reported8,9, and these mmpL3 mutants have modest cross-resistance to SQ1096. SQ109 also has activity against other bacteria (e.g. Helicobacter pylori10), fungi (e.g. Candida abicans11) as well as the malaria parasite Plasmodium falciparum, all of which lack the mmpL3 gene, so in these organisms there must be other targets/mechanisms of action. SQ109 analogs might thus be of interest as anti-infective leads against a range of organisms. Here, we elected to synthesize four types of SQ109-inspired species that might have activity against bacteria, fungi or protozoa.
We synthesized the SQ109 analogs (3-50) shown in Figures 1–3: a) 13 alkanolamine analogs (3-15, Figure 1); b) 3 thia analogs (16-18, Figure 1); c) 23 heterocycle-containing analogs (19-41, Figure 2) and d) 9 carborane-containing analogs (42-50, Figure 3). Full synthesis and characterization details are given in the Supporting Information.
Figure 1.

Alkanolamine and mercaptoethylamine analogs of SQ109 and their activities against Mycobacterium tuberculosis and Trypanosoma brucei. Mt = M. tuberculosis H37Rv; MtE = M. tuberculosis Erdman; Tb = Trypanosoma brucei. Values shown are in μg/mL and are MIC for the mycobacteria, and IC50 for T. brucei.
Figure 3.
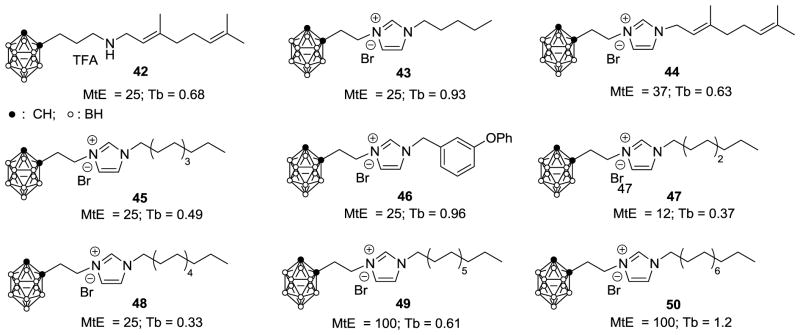
Carborane-containing analogs of SQ109 and their activities against Mycobacterium tuberculosis and Trypanosoma brucei. Mt = M. tuberculosis H37Rv; MtE = M. tuberculosis Erdman; Tb = Trypanosoma brucei; Values shown here are in μg/mL and are MIC for the mycobacteria and IC50 for T. brucei.
Figure 2.

Heterocyclic analogs of SQ109 and their activities against Mycobacterium tuberculosis and Trypanosoma brucei. Mt = M. tuberculosis H37Rv; MtE = M. tuberculosis Erdman; Tb = Trypanosoma brucei; Values shown here are in μg/mL and are MIC for the mycobacteria and IC50 for T. brucei.
In previous work, we found that compound 51 (Figure 1), the alkanolamine analog of SQ109, was more active (0.035 μg/mL) against M. tuberculosis) than was SQ109 (0.15 μg/mL)12. We therefore first synthesized and tested 13 alkanolamine-analogs of SQ109 (3-15, Figure 1) against M. tuberculosis, M. smegmatis, B. subtilis, S. cerevisiae, E. coli, T. brucei, HEK293T and HepG2 cells. MIC (Mycobacterium tuberculosis H37Rv, Mycobacterium tuberculosis Erdman), IC50 (M. smegmatis, B. subtilis, E. coli, S. cerevisiae, T. brucei) and CC50 (HEK293T and HepG2) values are given in Table 1 with the M. tuberculosis and T. brucei results shown, for convenience, below the structures in Figure 1.
Table 1.
Growth inhibition of various cells by SQ109 and its analogs.
| Tba | Mtb | MtEc | Msd | Bse | Ecf | Scg | HEK 293Th | HepG2i | SI(HEK 293T/Tb)j | SI(HepG 2/Tb)k | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | ND | 2.0 | ND | 1.0 | ND | ND | ND | ND | ND | ND | ND |
| 2 | 0.078±0.001 | 0.15 | 0.50 | 3.1 | 7.6 | 2.8 | 1.1 | 1.9 | 1.2 | 24 | 15 |
| 3 | 1.5±0.1 | 3.1 | 4.0 | ND | >120 | 9.0 | 25 | 9.2 | 4.8 | 6.0 | 3.1 |
| 4 | 1.1±0.1 | 25 | 16 | ND | >120 | 9.6 | 2.1 | 8.8 | 4.5 | 7.7 | 3.9 |
| 5 | 0.75±0.1 | 0.39 | 1.0 | ND | 0.8 | 0.6 | 0.7 | 11 | 9.4 | 15 | 13 |
| 6 | 0.50±0.1 | 3.1 | 16 | ND | 3.0 | 10 | 2.8 | 1.7 | 1.6 | 3.4 | 3.2 |
| 7 | 0.59±0.05 | 0.78 | 12 | ND | 2.1 | 12 | 3.2 | 4.6 | 2.9 | 7.8 | 4.9 |
| 8 | 0.33±0.06 | ND | 0.78 | 5.8 | 2.2 | 6.2 | 4.1 | 6.1 | 5.4 | 19 | 16 |
| 9 | 0.49±0.09 | ND | 0.78 | 5.7 | 3.3 | 8.0 | 4.1 | 9.5 | 5.6 | 20 | 12 |
| 10 | 0.23±0.03 | ND | 25 | 5.1 | 2.8 | 8.9 | 5.1 | 5.2 | 4.7 | 23 | 21 |
| 11 | 0.55±0.1 | ND | 38 | 5.2 | 3.9 | 9.2 | 6.1 | 5.5 | 3.6 | 10 | 6.6 |
| 12 | 7.7±1.5 | 50 | 100 | ND | >60 | >60 | >60 | 18 | 12.0 | 2.3 | 1.6 |
| 13 | 8.1±0.1 | 50 | ND | ND | >60 | >60 | >60 | 16 | 7.3 | 2.0 | 0.9 |
| 14 | 4.6±0.4 | 50 | ND | ND | >60 | >60 | >60 | 16 | 12 | 3.5 | 2.6 |
| 15 | 3.3±0.5 | 25 | ND | ND | >60 | >60 | >60 | 21 | 20 | 6.4 | 6.1 |
| 16 | 0.31±0.02 | 0.39 | 16 | 1.2 | 1.4 | 1.4 | 0.38 | 2.7 | 1.6 | 8.6 | 5.1 |
| 17 | 0.69±0.12 | 3.1 | 8.0 | 1.1 | 0.5 | 0.7 | 0.1 | 4.9 | 4.1 | 7.1 | 6.0 |
| 18 | 0.89±0.01 | 6.2 | 2.0 | 4.8 | 2.3 | 2.3 | 2.2 | 4.7 | 3.4 | 5.3 | 3.8 |
| 19 | 3.4±0.8 | 12 | 25 | 4.6 | 28 | >74 | >74 | 17 | 19 | 5.0 | 5.6 |
| 20 | 2.8±0.1 | 6.2 | ND | 5.6 | >90 | >90 | >90 | 12 | 12 | 4.2 | 4.2 |
| 21 | 0.39±0.01 | 1.6 | 6.2 | 1.2 | 0.38 | 2.0 | 1.5 | 1.7 | 1.5 | 4.4 | 3.8 |
| 22 | 0.047±0.007 | 3.1 | 12 | 1.5 | 1.2 | 36 | 9.1 | 3.2 | 4.3 | 68 | 91 |
| 23 | 0.023±0.005 | 3.1 | 1.6 | 0.7 | 2.1 | 33 | 17 | 4.1 | 5.4 | 180 | 240 |
| 24 | 0.21±0.02 | 0.78 | 0.50 | 3.2 | 3.1 | 13 | 8.4 | 4.9 | 6.2 | 24 | 30 |
| 25 | 0.058±0.011 | ND | 3.1 | 1.2 | 2.1 | 12 | 5.7 | 2.6 | 3.1 | 45 | 53 |
| 26 | 0.31±0.05 | ND | 6.2 | 1.5 | 2.0 | 12 | 4.8 | 5.1 | 5.5 | 17 | 18 |
| 27 | 0.0055±0.0001 | ND | 3.1 | 0.9 | 0.88 | 7.2 | 4.4 | 1.6 | 2.0 | 290 | 370 |
| 28 | 0.0076±0.0004 | ND | 6.2 | 0.9 | 0.41 | 5.4 | 2.2 | 1.3 | 1.5 | 170 | 200 |
| 29 | 0.12±0.01 | ND | 6.2 | 1.6 | 0.78 | 5.6 | 3.8 | 3.7 | 3.7 | 30 | 30 |
| 30 | 0.019±0.001 | ND | 4.6 | 1.0 | 0.48 | 3.4 | 2.7 | 1.6 | 1.9 | 86 | 100 |
| 31 | 0.017±0.001 | ND | 6.2 | 1.0 | 0.37 | 2.0 | 1.6 | 1.6 | 1.5 | 92 | 86 |
| 32 | 0.12±0.02 | ND | 9.3 | 3.5 | 0.56 | 3.6 | 0.51 | 4.4 | 4.2 | 38 | 36 |
| 33 | 0.016±0.001 | ND | 4.6 | 0.8 | 0.59 | 6.7 | 1.9 | 1.8 | 2.1 | 110 | 130 |
| 34 | 0.15±0.02 | ND | 3.1 | 1.8 | 0.69 | 4.0 | 2.1 | 3.2 | 3.5 | 21 | 23 |
| 35 | 0.022±0.001 | ND | 12 | 2.3 | 3.3 | 35 | 12 | 3.2 | 5.4 | 97 | 160 |
| 36 | 0.021±0.004 | ND | 6.2 | 0.8 | 0.22 | 1.0 | 1.4 | 1.5 | 1.5 | 70 | 70 |
| 37 | 0.026±0.002 | ND | 12 | 0.5 | 0.24 | 1.0 | 1.8 | 1.9 | 1.6 | 73 | 61 |
| 38 | 3.0±0.5 | ND | 17 | 16 | 1.2 | 85 | 46 | 19 | ND | 6.3 | ND |
| 39 | 1.4±0.1 | ND | 3.1 | 1.1 | 0.36 | 4.6 | 4.5 | 5.2 | 6.3 | 3.8 | 4.7 |
| 40 | 0.93±0.10 | ND | 6.2 | 1.9 | 1.1 | 6.4 | 3.5 | 3.6 | 3.4 | 3.9 | 3.6 |
| 41 | 0.40±0.06 | ND | 25 | 7.3 | 3.8 | 19 | 16 | 6.3 | 4.9 | 16 | 12 |
| 42 | 0.68±0.07 | ND | 25 | 1.3 | 0.75 | 2.1 | 3.4 | 3.5 | 3.0 | 5.1 | 4.4 |
| 43 | 0.93±0.17 | ND | 25 | 3.8 | 1.6 | 11 | 29 | 7.2 | 5.1 | 7.7 | 5.5 |
| 44 | 0.63±0.09 | ND | 37 | 1.9 | 0.31 | 5.0 | 7.3 | 7.9 | 7.8 | 13 | 12 |
| 45 | 0.49±0.08 | ND | 25 | 0.8 | 0.2 | 1.8 | 3.0 | 4.1 | 3.7 | 8.4 | 7.6 |
| 46 | 0.96±0.09 | ND | 25 | ND | ND | ND | ND | 14 | 14 | 15 | 15 |
| 47 | 0.37±0.09 | ND | 12 | ND | ND | ND | ND | 2.5 | 3.1 | 6.8 | 8.4 |
| 48 | 0.33±0.05 | ND | 25 | ND | ND | ND | ND | 1.9 | 1.4 | 5.8 | 4.3 |
| 49 | 0.61±0.16 | ND | 100 | ND | ND | ND | ND | 4.7 | 4.2 | 7.6 | 6.8 |
| 50 | 1.2±0.2 | ND | 100 | ND | ND | ND | ND | 7.5 | 7.9 | 6.4 | 6.7 |
| 51 | 0.26±0.07 | 0.035 | ND | 1.6 | 16 | 2.8 | 1.8 | 1.3 | 1.0 | 4.9 | 3.8 |
Tb = Trypanosoma brucei, IC50;
Mt = M. tuberculosis H37Rv, MIC;
MtE = M. tuberculosis Erdman, MIC;
Ms = M. smegmatis, IC50;
Bs = B. subtilis, IC50;
Ec = E. coli, IC50;
Sc = S. cerevisiae, IC50;
human embryonic kidney, HEK293T, CC50;
human hepatocellular carcinoma, HepG2, CC50;
SI = CC50(HEK293T)/IC50(Tb);
SI = CC50(HepG2)/IC50(Tb).
All units for MIC, IC50 and CC50 are μg/mL. The T. brucei results show mean and standard deviations of two independent experiments (R2 for pIC50=0.99); the fitting errors for Ms, Bs, Ec and Sc obtained from dose-response curves (8 half-log dilutions) were 9%, 9%, 11% and 14%, respectively. M. tuberculosis inhibition MICs were estimated visually from 2x serial dilutions while human cell growth inhibition was determined from fitting dose-response curves to a rectangular hyperbolic function.
There were several compounds with promising activity against M. tuberculosis. The most active compound was 5, an analog of SQ109 (2) in which the ethylenediamine nitrogen attached to the adamantane group was replaced by an oxygen, and the geranyl (C10) side-chain by a farnesyl (C15) group. The MIC was 0.39 μg/mL for M. tuberculosis H37Rv and 1.0 μg/mL for M. tuberculosis Erdman (MtE), Figure 1 and Table 1, to be compared with 0.1–0.5 μg/mL for SQ109 (2), in both strains and 0.035 μg/mL for 51 in M. tuberculosis H37Rv12. The reduced side-chain species 6 was ~10–20x less active than was the farnesyl analog. The isopentenyl ethanolamine analog (3) was also less active than was 5, and reduction (4) reduced activity further. Incorporation of a 1-Me or 1 i-Pr group (8, 9) decreased activity when compared with 51. The presence of a 1-OH group (7) also resulted in decreased activity (0.78 μg/mL) over that found with SQ109. The O-methylated analogs (10, 11) showed worse activity against MtE compared with the 1-OH species. Replacement of the isoprenoid side-chains with aromatic groups (12-15) blocked all activity and in other work12 we found the diether analog of 2 was also inactive12. These results indicate that optimum activity is found with a single nitrogen and that the order of activity of these alkanolamines is geranyl≫farnesyl≫isopentenyl, and that the reduced side-chain containing species are all less active than the unsaturated species. In the other assays (B. subtilis, E. coli and S. cerevisiae) the most potent cell growth inhibitor (Table 1) was 5, the N-farnesyl ethanolamine.
With the trypanosomatid parasite T. brucei, we found that SQ109 itself had quite potent activity against bloodstream form (BSF) parasites with an IC50 of 0.078 μg/mL and a selectivity index (SI), defined as SI = CC50 (HEK293T)/IC50 (T. brucei) or CC50 (HepG2)/IC50 (T. brucei) in the 15–24 range, Table 1. The most active SQ109 analogs were 10 (IC50 = 0.23 μg/mL), 8 (IC50 = 0.33 μg/mL) and 6 (IC50 = 0.50 μg/mL) with selectivity indices of 23, 21 (10), 19, 16 (8) and ~3–4 (6), so these analogs are less promising than is SQ109 against T. brucei. We also tested the SQ109 analog reported previously (51) to have potent activity against M. tuberculosis, but again it was slightly less active and had a worse SI as compared to SQ109 (Table 1).
We next investigated the 3 thia-analogs of SQ109 (16-18) in which the N attached to adamantane in SQ109 (O in the more active ethanolamine analog) was replaced by an S or SO2 group (providing different H-bonding possibilities), and in two cases the geranyl group was reduced to the per-hydro species. Cell growth inhibition results are shown in Table 1.
As can be seen in Figure 1 and Table 1, the thio-ether 16 had potent activity against M. tuberculosis H37Rv with an MIC of 0.39 μg/mL. 16 is the closest analog to SQ109 in the compounds studied here and also had activity against M. smegmatis (1.2 μg/mL), S. cerevisiae (0.38 μg/mL) and E. coli (1.4 μg/mL). Interestingly, in these organisms, the reduced species was even more active (Table 1). The sulfone had weak activity in all assays. The results in M. tuberculosis are consistent with the results found for the alkanolamines 5, 6 in that best activity is observed with the unsaturated side-chain containing species. With T. brucei, the most active thia-analog was 16 (IC50 = 0.31 μg/mL; SI 5–9), followed by 17 (IC50 = 0.69 μg/mL; SI 6–7) and 18 (IC50 = 0.89 μg/mL, SI = 4–5).
The results described above are of interest in that we show, for the first time, that SQ109 has activity against the parasitic protozoan T. brucei, but unlike the situation found with the alkanolamine analogs of SQ109 reported previously12, none of the new analogs showed improved activity (over that seen with 51) against M. tuberculosis, although 5, 16 and 17 were all more active than was SQ109 against the Gram negative bacterium, E. coli (5, IC50 = 0.60 μg/mL; 16, IC50 = 1.4 μg/mL; 17, IC50 = 0.70 μg/mL, versus IC50 = 2.8 μg/mL for SQ109; Table 1), although the computed selectivity indices (using HEK293T and HepG2) are poor (~5).
In previous work12 we also found that another SQ109 analog, a choline-derivative containing a quaternary ammonium instead of a protonable N, had the most potent activity against a different parasitic protozoan, the malaria parasite P. falciparum, in addition to being a very potent inhibitor of respiration, in M. smegmatis12. We thus reasoned that other cationic analogs of SQ109 might have better anti-bacterial and/or anti-protozoal activity, so we made and tested two further sets of analogs. We first synthesized a series of 23 SQ109 analogs with primarily protonatable (or fixed charge) heterocycle linker groups replacing the ethylenediamine fragment. The heterocycles investigated were neutral (the 1,2,3-triazoles 19, 20); protonatable (guanidines and amidines, 21, 40, 41; and an imidazole, 38), or they contained a fixed positive charge (imidazoliums and pyridiniums, 23-39). The two neutral triazoles had low activity against M. tuberculosis (19, MIC = 12, 25 μg/mL; 20, MIC = 6.2 μg/mL) and M. smegmatis (IC50 ~5 μg/mL) and essentially no activity against the other bacteria or the fungus.
Of the other heterocyclic compounds investigated, most had some activity against M. tuberculosis Erdman (and M. tuberculosis H37Rv), Figure 2 and Table 1. However, there were only 3 compounds (21, 23, 24) in which at least one of the M. tuberculosis MIC values was <2 μg/mL. Both 21 and 23 contain as a common structural feature the O-CH2-CH2-N group found in the potent alkanolamines and in both cases, the nitrogen is expected to have either a formal +1 charge (23) or a large positive charge density (21, due to the strong basicity of the ligand and charge delocalization), so both resemble the protonated ethanolamines. In 24, the aliphatic “linker” group is absent, but we now see that this potent inhibitor resembles SQ109 in another way in that it contains the N-C-C-N group found in the ethylenediamine fragment which, in SQ109 is expected to carry a +1 charge (at pH~7), again delocalized most likely over both nitrogens. As can be seen in Table 1, many of the other heterocyclic analogs have activity against the other bacteria as well as the fungus S. cerevisiae, but they also inhibited the growth of the two human cell lines (Table 1), resulting in poor selectivity indices.
The results obtained against T. brucei were, however, much more encouraging, Table 1. Specifically, we found that there were 15 analogs of SQ109 that had better IC50 and SI values than did SQ109 (IC50 = 0.078 μg/mL; SI ~15–24, Table 1). A typical set of dose-response curves for the top five T. brucei cell growth inhibitors, together with their corresponding effects on HEK293T and HepG2 cell growth, are shown in Figure S1, and selectivity index versus T. brucei cell growth inhibition results (for both human cell lines) are shown in Figure S2. The best T. brucei IC50 value was 5.5 ng/mL, with corresponding SI values of 290 and 370 (Table 1). Clearly, these results are encouraging and as noted above, are reminiscent of the activity of the choline analog of SQ109 against P. falciparum in the intra-erythrocytic assay where an IC50 = 80 nM (35 ng/mL) was found (corresponding to a SI~400)12, plus, activity against the two human cell lines is similar to that seen with SQ109 (which is already in advanced clinical trials for tuberculosis).
Next, we sought to see whether improved activity might be found by replacing the adamantyl group by a 1-o-carboranyl group, which is similar to the adamantyl group in terms of size, shape and hydrophobicity13. We produced the 9 carboranes (42-50) shown in Figure 3. None had potent activity against M. tuberculosis Erdman, Table 1. However, in almost all cases there was activity against M. smegmatis, B. subtilis, S. cerevisiae and more surprisingly against E. coli, with the ~2 μg/mL IC50 values found for 42, 45 against E. coli being of interest since we found worse activity against this Gram negative with the other analogs. Reasons for the enhanced activity against E. coli are unknown. Three compounds (45, 47, 48) also had IC50 < 0.5 μg/mL against T. brucei, although none approached the activity (and hence, SI values) seen with the adamantane-containing analogs.
The most potent compound against M. tuberculosis Erdman is thus 24 with an MIC of 0.50 μg/mL, and 24 also has a 0.78 μg/mL MIC against M. tuberculosis H37Rv (Table 1). What is of interest about 24 is that it closely resembles the structure of SQ109 in that there are adamantyl and geranyl groups and a N-C-C-N linker but here, the linker is an imidazolium, not an ethylenediamine group. The heterocycles (19-41) as a class have most potent activity against the trypanosomatid parasite T. brucei and are also most active against the 2 human cell lines. However, when selectivity index values are calculated it can be seen that 27, 28 have the best IC50 values of ~5–7 ng/mL and SI~300. All of the compounds with the best SI (22-37) also have fixed charge centers, raising the question as to their possible mechanism of action.
In earlier work we found that SQ109 acted as an uncoupler in E. coli as well as in M. smegmatis and we proposed that this uncoupling activity was important for its activity against M. tuberculosis12. Similar results have now been reported for a broader range of compounds which are now proposed to act as uncouplers, in M. tuberculosis14, targeting ΔpH, Δψ, or both. We therefore tested SQ109 and the most interesting potential lead, 27, in T. brucei, to see if similar effects were seen with either or both BSF and procyclic forms (PCF). We first tested whether SQ109 had effects on the proton motive force (more specifically, the inner mitochondrial membrane potential, Δψ) using the safranine method15,16 with BSF parasites. Figure 4A shows that addition of 10 μM (3.3 μg/mL) SQ109 or 10 μM (4.5 μg/mL) 27 decreased Δψ, which was further reduced by addition of 8 μM (2 μg/mL) FCCP (carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone), a potent protonophore uncoupler. Similar results were obtained with PCF, Figure 4B. T. brucei mitochondria were able to phosphorylate ADP, as demonstrated by the small decrease in Δψ after its addition, Figure 4C. This activity was inhibited by the ATP synthase inhibitor oligomycin. In addition, the mitochondria were able to transport Ca2+, as shown by the decrease in the Δψ after addition of CaCl2, and the Δψ returned to basal levels after addition of the Ca2+-chelator EGTA. Further addition of SQ109 or 27 followed by FCCP again collapsed the Δψ, Figure 4C. Both SQ109 and 27 collapsed Δψ in a dose-dependent manner (Figure S3) and SQ109 alone or solvent (0.2 % DMSO) had no effect. These results show that mitochondria in permeabilized T. brucei are able to develop a Δψ, phosphorylate ATP and transport Ca2+ and that SQ109 and 27 collapse Δψ. These effects on the proton motive force are rapid and are very similar to those observed for SQ109 in bacterial systems17,18 and are likely to make a significant contribution to SQ109 and 27 inhibiting cell growth.
Figure 4.

Effects of SQ109 or 27 on Δψ in digitonin-permeabilized T. brucei. (A) BSF trypanosomes (2×108 cells) were added to reaction buffer (125 mM sucrose, 65 mM KCl, 10 mM Hepes-KOH buffer, pH 7.2, 1 mM MgCl2, 2.5 mM potassium phosphate; 2 mL) containing 20 μM EGTA, 1 mM ATP, 500 μM orthovanadate and 5 μM safranine, and the reaction started with 40 μM digitonin. (B, C) T. brucei PCF (5×107 cells) were added to the reaction buffer (2.4 mL) containing 2 mM succinate and 5 μM safranine, and the reaction initiated with or without (yellow trace in B) 50 μM digitonin. SQ109 (3.3 μg/mL), and 27 (4.5 μg/mL) (equimolar amounts), FCCP (8 μM), ADP (10 μM), oligomycin (Oligo, 2 μg/ml), CaCl2 (12 μM), EGTA (200 μM) were added where indicated. No changes were detected in the absence of digitonin indicating lack of secondary effects of the drugs
In addition to their effects on Δψ, it seemed possible that some compounds might act by inhibiting quinone biosynthesis, in some systems, just as other SQ109 analogs did with the prenyl transferase MenA (1,4-dihydroxy-2-naphthoate octaprenyltransferase). We tested a representative set of compounds from the alkanolamine (5), imidazolium (22, 27), imidazole (39) and carborane groups (48) against an expresssed E. coli MenA using the method reported previously12. Compounds 22 and 48 had no activity (IC50>40 μM, 20 μg/mL), the IC50 for 27 was 19 μM (8.5 μg/mL), for 5, 9.0 μM (3.6 μg/mL), while that for 39 was 1.5 μM (0.54 μg/mL), suggesting that MenA inhibition with 39 could be of importance in MtE cell growth inhibition (MIC = 3.1 μg/mL). However, 39 has a poor SI.
Overall, the results reported above are of interest since we synthesized a broad range of analogs of the M. tuberculosis growth inhibitor, the ethylene diamine SQ109, and tested their activity against bacteria, a fungus, as well as a protozoan parasite. Protonatable or cationic species had the most activity and the most potent leads against M. tuberculosis (MIC~0.4–0.5 μg/mL) contained ethanolamine, mercaptoethylamine or imidazolium linkers. The carboranes were less active against M. tuberculosis but surprisingly, had activity (IC50~2 μg/mL) against the Gram negative, E. coli. However, we did not obtain compounds that were more active against M. tuberculosis than was the ethanolamine analog of SQ109 reported earlier. However, we did discover that the parent compound SQ109 had activity against the trypanosomatid parasite, T. brucei, the causative agent of human African trypanosomiasis, and that two SQ109 analogs had IC50 values in the ~5–7 ng/mL range against this organism with SI values of ~300.
METHODS
Chemical Syntheses: General Methods
All chemicals were reagent grade. 1H NMR and 13C NMR spectra were obtained on Varian (Palo Alto, CA) Unity spectrometers at 400 and 500 MHz for 1H and at 100 and 125 MHz for 13C. Elemental analyses were carried out in the University of Illinois Microanalysis Laboratory. HPLC/MS analyses were performed by using an Agilent LC/MSD Trap XCT Plus system (Agilent Technologies, Santa Clara, CA) with an 1100 series HPLC system including a degasser, an autosampler, a binary pump, and a multiple wavelength detector. All final compounds were ≥90% pure as determined by quantitative spin count NMR (qNMR) and structures were characterized by 1H NMR and HRMS. The synthesis and characterization of all new compounds (3-50) are shown in the Supporting Information.
T. brucei 427 (bloodstream forms) growth inhibition assay
T. brucei strain 427 bloodstream forms were cultivated at 37 °C with 5% CO2 in HMI-9 medium supplemented with 10% fetal bovine serum (FBS). T. brucei parasites (5 × 104/mL) were seeded in 384 well plates with or without a serial compound dilution. After 72 h of incubation, the parasites were exposed to 120 μM of resazurin sodium salt (Sigma, St. Louis, MO, USA) and were incubated for another 5 h. Then, the parasites were fixed with 4% paraformaldehyde (PFA) and the assay plates were read by using a Victor 3™ fluorimeter (PerkinElmer, Waltham, MA, USA) at an excitation wavelength of 530 nm and emission of 590 nm. Pentamidine was used as a reference drug and DMSO 0.5% was used as a drug-negative control. Two independent sets of experiments were carried out and the mean and standard deviations are shown in Table 1; the R2 for the pIC50 correlation was 0.99.
Supplementary Material
Acknowledgments
This work was supported by NIH grants GM065307, AI049151 and AI104120; a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (NO. 2007-00559), Gyeonggi-do and KISTI.
Footnotes
Author Contributions
K.L. Y.W. and E.O. designed research; K.L. Y.W. and A.G. synthesized compounds; Y.W. G.Y. S.-Y.B. G.R. C.S. D.C. M.C. and J.-H.N. performed cell growth inhibition experiments; G.H. and R.D. performed mitochondrial membrane potential experiment; Y.W. and E.O. analyzed data. Y.W. and E.O. wrote the paper.
The authors declare no competing financial interest.
Full details of all assays; representative dose-response curves; selectivity index results; mitochondrial membrane bioenergetics; full synthesis and characterization of SQ-109 and its analogs as well as qNMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Centers for Disease Control and Prevention. [accessed Dec 15, 2014];Threat Report 2013: Antibiotic/Antimicrobial Resistance. http://www.cdc.gov/drugresistance/threat-report-2013/
- 2.World Health Organization. [accessed Dec 15, 2014];Antimicrobial Resistance: Global Report on Surveillance. 2014 http://www.who.int/drugresistance/documents/surveillancereport/en/
- 3.World Health Organization. [accessed Dec 15, 2014];Global Tuberculosis Report. 2014 http://www.who.int/tb/publications/global_report/en/
- 4.Protopopova M, Hanrahan C, Nikonenko B, Samala R, Chen P, Gearhart J, Einck L, Nacy CA. Identification of a new antitubercular drug candidate, SQ109, from a combinatorial library of 1,2-ethylenediamines. J Antimicrob Chemother. 2005;56(5):968–974. doi: 10.1093/jac/dki319. [DOI] [PubMed] [Google Scholar]
- 5.Bogatcheva E, Hanrahan C, Nikonenko B, de los Santos G, Reddy V, Chen P, Barbosa F, Einck L, Nacy C, Protopopova M. Identification of SQ609 as a lead compound from a library of dipiperidines. Bioorg Med Chem Lett. 2011;21(18):5353–5357. doi: 10.1016/j.bmcl.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tahlan K, Wilson R, Kastrinsky DB, Arora K, Nair V, Fischer E, Barnes SW, Walker JR, Alland D, Barry CE, Boshoff HI. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core ofMycobacterium tuberculosis. Antimicrob Agents Chemother. 2012;56(4):1797–1809. doi: 10.1128/AAC.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Varela C, Rittmann D, Singh A, Krumbach K, Bhatt K, Eggeling L, Besra GS, Bhatt A. MmpL genes are associated with mycolic acid metabolism in Mycobacteria and Corynebacteria. Chem Biol. 2012;19(4):498–506. doi: 10.1016/j.chembiol.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Owens CP, Chim N, Goulding CW. Insights on how the Mycobacterium tuberculosis heme uptake pathway can be used as a drug target. Future Med Chem. 2013;5(12):1391–1403. doi: 10.4155/fmc.13.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Owens CP, Chim N, Graves AB, Harmston CA, Iniguez A, Contreras‡ A, Liptak MD, Goulding CW. The Mycobacterium tuberculosis secreted protein Rv0203 transfers heme to membrane proteins MmpL3 and MmpL11. J Biol Chem. 2013;288(30):21714–21728. doi: 10.1074/jbc.M113.453076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Makobongo MO, Einck L, Peek RM, Merrell DS. In vitro characterization of the anti-bacterial activity of SQ109 against Helicobacter pylori. PLoS One. 2013;8(7):e68917. doi: 10.1371/journal.pone.0068917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barbosa, et al. In vitro antifungal susceptibility testing of drug candidate SQ109 Candida albicans. Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC); San Francisco, CA. 2006. [Google Scholar]
- 12.Li K, Schurig-Briccio LA, Feng X, Upadhyay A, Pujari V, Lechartier B, Fontes FL, Yang H, Rao G, Zhu W, Gulati A, No JH, Cintra G, Bogue S, Liu YL, Molohon K, Orlean P, Mitchell DA, Freitas-Junior L, Ren F, Sun H, Jiang T, Li Y, Guo RT, Cole ST, Gennis RB, Crick DC, Oldfield E. Multitarget drug discovery for tuberculosis and other infectious diseases. J Med Chem. 2014;57(7):3126–3139. doi: 10.1021/jm500131s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brynda J, Mader P, Šícha V, Fábry M, Poncová K, Bakardiev M, Grüner B, Cígler P, Řezáčová P. Carborane-based carbonic anhydrase inhibitors. Angew Chem Int Ed. 2013;52(51):13760–13763. doi: 10.1002/anie.201307583. [DOI] [PubMed] [Google Scholar]
- 14.Li W, Upadhyay A, Fontes FL, North EJ, Wang Y, Crans DC, Grzegorzewicz AE, Jones V, Franzblau SG, Lee RE, Crick DC, Jackson M. Novel insights into the mechanism of inhibition of MmpL3, a target of multiple pharmacophores in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2014;58(11):6413–6423. doi: 10.1128/AAC.03229-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vercesi AE, Bernardes CF, Hoffmann ME, Gadelha FR, Docampo R. Digitonin permeabilization does not affect mitochondrial function and allows the determination of the mitochondrial membrane potential of Trypanosoma cruzi in situ. J Biol Chem. 1991;266(22):14431–14434. [PubMed] [Google Scholar]
- 16.Huang G, Vercesi AE, Docampo R. Essential regulation of cell bioenergetics in Trypanosoma brucei by the mitochondrial calcium uniporter. Nat Commun. 2013;4:2865. doi: 10.1038/ncomms3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haagsma AC, Podasca I, Koul A, Andries K, Guillemont J, Lill H, Bald D. Probing the interaction of the diarylquinoline TMC207 with its target mycobacterial ATP synthase. PLoS One. 2011;6(8):e23575. doi: 10.1371/journal.pone.0023575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boshoff HI, Myers TG, Copp BR, McNeil MR, Wilson MA, Barry CE. 3rd, The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: novel insights into drug mechanisms of action. J Biol Chem. 2004;279(38):40174–40184. doi: 10.1074/jbc.M406796200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.