

Abstract
Liver X receptor (LXR) signaling pathways regulate lipid metabolism and inflammation, which has generated widespread interest in developing synthetic LXR agonists as potential therapeutics for the management of atherosclerosis. In this study, we demonstrate that nanoparticles (NPs) containing the synthetic LXR agonist GW3965 (NP-LXR) exert anti-inflammatory effects and inhibit the development of atherosclerosis without causing hepatic steatosis. These NPs were engineered through self-assembly of a biodegradable diblock poly(lactide-co-glycolide)-b-poly(ethylene glycol) (PLGA-b-PEG) copolymer. NP-LXR was significantly more effective than free GW3965 at inducing LXR target gene expression and suppressing inflammatory factors in macrophages in vitro and in vivo. Addtionally, the NPs elicited negligible lipogenic gene stimulation in the liver. Using the Ldlr−/− mouse model of atherosclerosis, we saw abundant co-localization of fluorescently labeled NPs within plaque macrophages following systemic administration. Notably, six intravenous injections of NP-LXR over two weeks markedly reduced the CD68-positive cell (macrophage) content of plaques (by 50%) without increasing total cholesterol or triglycerides in the liver and plasma. Together, these findings identify GW3965-encapsulated PLGA-b-PEG NPs as a promising nanotherapeutic approach to combat atherosclerosis, providing the benefits of LXR agonists without their adverse effects on hepatic and plasma lipid metabolism.
Keywords: Nanoparticles, drug delivery, Liver X receptor (LXR), atherosclerosis
1. Introduction
Advances in vascular biology have revealed that inflammation plays a pivotal role in the pathophysiology of atherosclerosis, the major cause of cardiovascular diseases and the leading cause of mortality in the developed world.[1,2] Atherosclerotic plaques develop through a maladaptive, macrophage-driven, chronic inflammatory response to retained subendothelial apolipoprotein B-containing lipoproteins.[3] Defective resolution of this inflammation increases permeation of lipoproteins and further induction of endothelial adhesion molecules, followed by the recruitment of additional monocytes into the plaque that differentiate into macrophages and eventually transform into lipid-laden foam cells. Through apoptosis and decreased clearance by healthy macrophages (i.e., efferocytosis), these foam cells undergo secondary necrosis, leading to the formation and expansion of a necrotic core and the progression of atherosclerotic lesions into dangerous plaques that are vulnerable to rupture, which can in turn trigger myocardial infarction and stroke.[4]
Our improved understanding of the pathogenesis of atherosclerosis has suggested new opportunities for its treatment and prevention, among which nanoparticles (NPs) present an attractive approach.[5] The application of nanotechnology to medicine has already demonstrated clinical impact in terms of delivery strategies for a range of bioactive molecules, including therapeutic agents, nucleic acids, and imaging contrast agents.[6] Since the approval of liposomal doxorubicin (Doxil®) in 1995, nearly 250 nanotherapeutics have entered clinical practice or are in some stage of preclinical and clinical development. For example, BIND-014 is a targeted polymeric nanotherapeutic currently in Phase II clinical trials and has shown early promising results.[7] BIND-14 accumulates in tumors while avoiding uptake by healthy cells and can deliver a substantially higher dose of the drug to tumors compared with the unencapsulated drug.[7] There has been extensive exploration of NP-based drug delivery to enhance therapeutic efficacy, deliver drugs across a range of biological barriers, and circumvent certain drawbacks associated with the drug molecules themselves, such as low water solubility and poor pharmacokinetics.[8]
Here we report the development of a NP platform to deliver a liver X receptor (LXR) agonist to macrophages in atherosclerotic plaques. The LXRs (LXRα and LXRβ) are members of the nuclear hormone receptor family of transcription factors. LXR activation in macrophages exerts a number of atheroprotective effects by promoting cholesterol efflux through upregulation of ATP-binding cassette (ABC)A1 and G1, suppressing inflammation and improving its resolution through trans-repressing NF-kB pathways, and enhancing efferocytosis.[3,9,10] Recent studies have shown that administration of a LXR agonist in a mouse model of atherosclerosis results in attenuation and regression of atherosclerosis in vivo.[9,11,12] A major barrier to their clinical use, however, has been hypertriglyceridemia and hepatic steatosis caused by LXR-mediated induction of SREBP-1c.[13–15] To mitigate these adverse effects, we examined the utility of PLGA-b-PEG-based NPs encapsulating a synthetic LXR agonist GW3965 to enable plaque macrophage activation, inhibit atherosclerosis progression, and resolve plaque inflammation, while avoiding an increase in hepatic triglyceride production. NPs self-assembled from the biodegradable PLGA-b-PEG block copolymers represent a promising class of delivery vehicles due to several unique properties: PLGA-b-PEG copolymers i) are biocompatible, biodegradable, and used in many products already approved by the Food and Drug Administration (FDA), ii) are capable of encapsulating small- and macro-molecular payloads with a wide range of physiochemical properties, and iii) can be designed for controlled release through a combination of polymer degradation and drug diffusion.[16]
It is generally accepted that the long circulation time of NPs conferred by PEG can facilitate homing to the site of disease through the enhanced permeability and retention (EPR) produced by the “leaky” endothelium often found near atherosclerotic vessels.[5,17,18] For the present application, we designed PLGA-b-PEG NPs with a hydrophobic core containing the GW3965 LXR agonist molecules and a hydrophilic PEG corona (NP-LXR). In parallel, we also prepared another LXR agonist-encapsulated NP by self-assembling phosphatidylserine (PS) lipid with PLGA-b-PEG, which we designated a hybrid polymer/lipid NP (PSNP-LXR). PS is generally considered a marker for apoptosis (a key event in the progression of atherosclerosis) and functionally, the appearance of PS on the surface of apoptotic cells identifies them as targets for engulfment by phagocytic cells, such as macrophages.[19] Using a rat model of acute myocardial infarction (MI), Cohen and co-workers exploited PS-presenting liposomes to mimic the anti-inflammatory effects of apoptotic cells. That treatment has demonstrated the ability of PS-liposomes to prompt the immune system’s macrophages to shift to an anti-inflammatory mode, promoting angiogenesis and eliciting infarct repair.[20] These findings suggest that NPs incorporating PS may promote resolution of inflammation and induce the production of anti-inflammatory cytokines in plaque macrophages.
For both types of NPs, we compared their abilities to regulate the expression of key inflammation and lipid metabolism LXR-related genes with that of unencapsulated GW3965, both in vitro and in vivo. In particular, using a Ldlr−/− mice model, both GW3965 and its encapsulated NP form demonstrated multiple signs of atheroprotection in terms of macrophage content and inflammation; however, unlike the free drug, NPs did not adversely affect plasma or hepatic lipid profiles.
2. Results and Discussion
2.1. Synthesis and Characterization of NPs
The design and preparation of GW3965-encapsulated NPs are shown in Figure 1A. NPs self-assembled from PLGA-b-PEG were designated NP-LXR, while hybrid NPs self-assembled from PLGA-b-PEG and PS were designated PSNP-LXR. Transmission electron microscopy (TEM) images of NPs stained with uranyl acetate show that the particles were spherical and monodisperse (Figure 1B). Dynamic light scattering (DLS) determined the size of NP-LXR to be 156.6 ± 10.3 nm (mean ± SD) with a polydispersity index (PDI) ≤0.2. The incorporation of PS lipid slightly decreased their size (by approximately 18 nm), but the PDI remained similar. Calculated based on the drug content by RP-HPLC, the final entrapment and loading efficacy using a 20 wt% GW3965/polymer input were determined to be 58.8 ± 1.3% of the drug input weight and 9.8 ± 0.2% by the NP mass for NP-LXR, and 65.4 ± 5.8% and 10.9 ± 1% for hybrid PSNP-LXR, respectively (Figure 1C). A possible explanation for the differences in size and loading is that lipid molecules (e.g., PS) cover the surface of the PLGA hydrophobic core, resulting in more compact structures with smaller size and greater drug loading capacity. [21]
Figure 1.

(A) Chemical structure of GW3965-encapsulated NPs. The particle consists of a surface-associated fluorescent molecule, an outer PEG surface, and a biodegradable polymer matrix loaded with hydrophobic GW3965. (B) Size distribution of NP-LXR determined by DLS and representative TEM image. (C) Characterization summary of NP-LXR and PSNP-LXR. (D) In vitro release profile of GW3965 from NPs (n=4).
To measure drug release kinetics, NP samples were dialyzed against 2 L of frequently renewed phosphate-buffered saline (PBS) at pH 7.4 and 37°C to mimic physiological conditions. Quadruplicate aliquots of each NP sample (n=4) were withdrawn at indicated time points for reversed-phase high-performance liquid chromatography (RP-HPLC) analysis. In this NP platform, the GW3965 compound is homogeneously dispersed by encapsulation throughout the hydrophobic PLGA core, and possible mechanisms of release include diffusion and polymer degradation.[22] As shown in Figure 1D, 20.7% of the total encapsulated GW3965 was rapidly released from NP-LXR over the first 6 h followed by a sustained, slower release after 8 h. This controlled release of GW3965 from the NP-LXR extended over two weeks, attaining a maximum value of 84.4% thereafter. The release profile of PSNP-LXR suggested that the incorporation of lipid slightly interfered with the self-assembly process to marginally decrease rates of drug release. The initial release of GW3965 compound from PSNP-LXR over the first 8 hours was found to be 22.7% of the loaded drug content, then reaching a maximum value of 87.0% over 14 days. The above results demonstrate that the synthesized NPs are capable of dense loading and sustained release of GW3965.
The feasibility of NPs as a drug delivery platform was then tested through in vitro uptake studies in a RAW264.7 murine macrophage cell model. Fluorescently labeled PLGA-b-PEG, composed of end-to-end linkages between PLGA-b-PEG and Alexa Fluor® 680 (AF680), was used to prepare AF680-NP-LXR and AF680-PSNP-LXR. By tracking the fluorescent tags on NPs, the ability of AF680-NPs to enter cells was measured by flow cytometry. Untreated cells and non-labeled NPs were used as negative controls. The results indicated the successful internalization of the NPs in RAW264.7 cells after 2 h of incubation (Figure 2A). When RAW264.7 cells were treated with AF680-PSNP-LXR at an equivalent dose of GW3965, the fraction of fluorescence-positive signals tended to be higher compared with AF680-NP-LXR. The minor increase in cellular uptake of PSNP-LXR over NP-LXR can likely be attributed to its smaller size and specific recognition through the PS receptor on macrophages.
Figure 2.
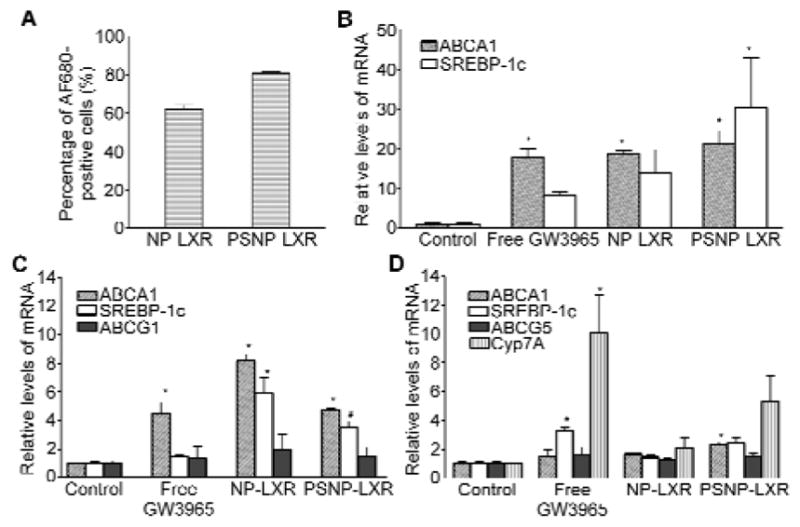
Regulation of LXR target gene expression by GW3965-encapsulated NPs in vitro and in vivo. (A) Flow cytometry analysis of AF680-labeled NPs demonstrating NP internalization into RAW264.7 cells. Data are represented as the mean ± standard deviation (n=3). (B) Murine thioglycolate-elicited peritoneal macrophages from C57BL/6J mice were treated for 18h with the indicated regimens at an equivalent GW3965 concentration of 1 μM. RNA was isolated and gene expression was analyzed by qRT-PCR. (C) Regulation of LXR target gene expression by the free GW3965 or the indicated NPs in aortic arch and (D) the liver in WD-fed Ldlr−/−mice (n=5). RNA was isolated and gene expression was analyzed by qRT-PCR. Data are presented as mRNA level relative to vehicle control. Each bar represents the mean ± standard deviation. *P<0.05 and #P<0.01, as compared with the control group.
2.2. Regulation of LXR Target Gene Expression by GW3965-Encapsulated NPs
Next we compared the effects of GW3965 in its conventional dosage form and in GW3965-encapsulated NPs on regulating the expression of LXR target genes in macrophages. LXRs have shown beneficial effects in mouse models of atherosclerosis by regulating inflammation and lipid metabolism.[23-25] In macrophages, LXRs control transcription of several genes involved in the cholesterol efflux pathway, including ABCA1 and ABCG1.[26] Certain lipogenic genes, such as SREBP-1c (sterol regulatory element binding protein), have also been identified as LXR targets.[27] As a result, LXR ligands have been implicated in triggering induction of the lipogenic pathway in mice, with activation of SREBP-1c in the liver, which leads to the adverse effects of steatosis and hypertriglyceridemia.[14,28]
We selected ABCA1 and SREBP-1c as target genes to examine the ability of GW3965-encapsulated NPs to activate LXR target genes in thioglycolate-elicited murine peritoneal macrophages. NPs encapsulating an equivalent dose of 1μM GW3965 or the non-NP agonist-only counterpart were incubated with the cells, and the expression of both genes was measured using quantitative real-time PCR (qRT-PCR) 18 h post-treatment. As shown in Figure 2B, non-PS-containing GW3965-encapsulated NPs were at least as effective as the free GW3965 in upregulating both ABCA1 and SREBP-1c expression in peritoneal macrophages. Furthermore, we noted that the incorporation of PS lipid within NPs encapsulating GW3965 further augmented cellular mRNA levels of both LXR target genes. In vitro cytotoxicity of the different NPs was also evaluated in peritoneal macrophages by lactate dehydrogenase (LDH) assays (Figure S1). Over 80% of cells survived when incubated with NPs encapsulating a dose of GW3965 equivalent to that used for the aforementioned qRT-PCR assay. Taken together, these results indicate that NP-LXR and PSNP-LXR might serve as efficient carriers for the delivery of LXR agonists in vivo.
The effect of GW3965-encapsulated NPs on target gene expression in the liver and aortic arch of Ldlr−/− mice was also investigated. The Ldlr−/− mouse model has been widely used to identify genes that modify atherosclerosis susceptibility and in the development of antiatherogenic therapies.[29] Ldlr−/− mice were fed an atherogenic western diet (WD) for 16 weeks to develop atherosclerotic plaques. Mice were randomly divided into four groups, and each group was treated with retro-orbital injections of PBS or of the following regimens with an equivalent dose of 6 mg/kg entrapped GW3965: (i) GW3965 dissolved in dimethyl sulfoxide (DMSO) (free GW3965), (ii) NP-LXR, or (iii) PSNP-LXR. After five consecutive daily injections, qRT-PCR assays of plaque-containing aortic arches revealed that the strongest induction of LXR target genes SREBP-1c, ABCA1, and ABCG1 was in response to NP-LXR, although treatment with PSNP-LXR also tended to induce the three genes more strongly than free GW3965 (Figure 2C).
In the liver, free GW3965 significantly induced the expression of SREBP-1c and Cyp7A (Figure 2D). Note that the effects in the liver of SREBP-1c and ABCA1 reflect not only target gene activation in parenchymal cells (~80% of the cellular contents), but also effects on the macrophage-like cells of the liver (e.g., Kupffer cells).[30,31] CYP7A and ABCG5, on the other hand, would reflect mainly effects in parenchymal cells.[30] Both NP-LXR and PSNP-LXR protected against the induction of SREBP-1c and CYP7A, with NP-LXR being more effective in this regard. These observations indicate that less GW3965 was taken up by either hepatocytes or Kupffer cells in the liver when NPs were used as delivery vehicles. The differences in efficacy between the two types of NPs may be related to the report by Huong et al., who showed enhanced uptake and accelerated clearance of PS-containing liposomes by macrophages in the hepatic sinusoids in rats and guinea pigs.[32]
Taken together, the results in Figure 2 show that both GW3965-loaded NPs and free GW3965 stimulate LXR-target gene expression in the atherosclerotic aortic arch, with the NP-LXR being the most potent, but that the NPs are less effective than free drug in inducing them in the liver.
2.3. Localization of NPs in Atherosclerotic Plaques and Liver
Since GW3965-encapsulated NPs affected LXR-target gene expression in vitro and in vivo in the aorta and liver, we sought to investigate the accumulation of i.v.-injected NPs in those tissues. Ldlr−/− mice were maintained on western diet (WD) for 16 weeks to induce atherosclerotic plaques. Mice were then administered either AF488-labeled NP-LXR or AF488-labeled PSNP-LXR via retro-orbital injection. Aortic roots and livers were harvested 4 h post-NP injection and sectioned for confocal microscopy. As illustrated in Figure 3A, NP-LXR showed substantial internalization within CD68-positive cells (i.e., macrophages) in the aortic root. Both types of NPs were also colocalized within CD68-positive cells in the liver (Figure 3A & B). As quantitatively determined by ImagePro Plus 3.0 software, there was greater co-localization of NP-LXR within CD68-positive cells in the aortic root (20.3% ± 0.17 for NP-LXR vs 7.6% ± 0.7 for PSNP-LXR) and lower co-localization in liver sections (0.85% ± 0.24 for NP-LXR vs 4.2% ± 1.08 for PSNP-LXR). This observation supports the superior ability of the NP-LXR relative to PSNP-LXR to activate LXR expression in plaques and to spare the liver (Figure 2D).
Figure 3.
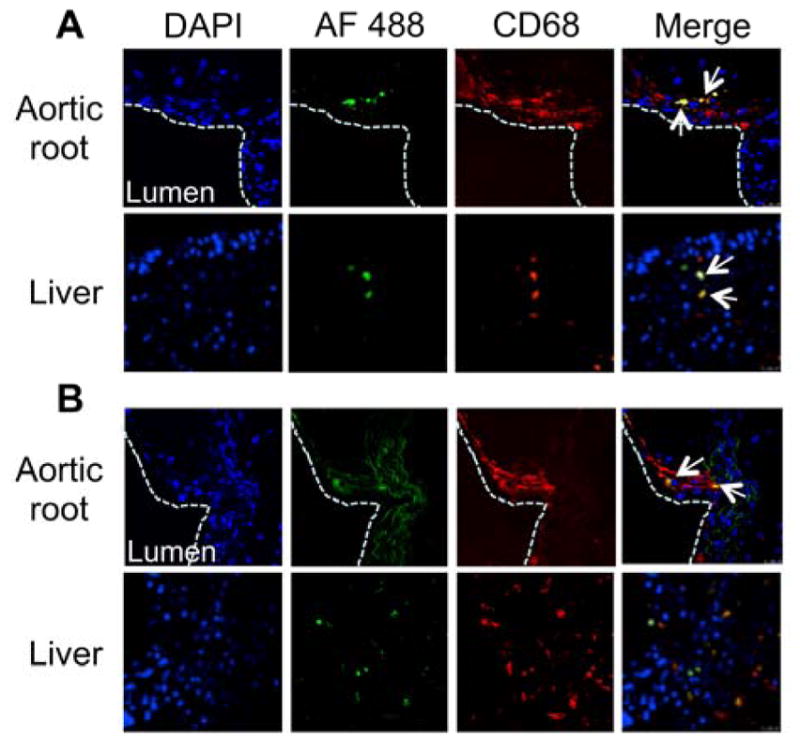
Co-localization of AF488-labeled NP-LXR (A) and PSNP-LXR (B) within CD68+ macrophages in the aortic root and liver. Ldlr−/− mice (n=5) with WD-induced atherosclerotic plaques were treated with the fluorescently labeled NPs at an equivalent dose of 10 mg/kg GW3965. The aortic root and liver were then harvested and sectioned for confocal imaging (magnification 40x). Each sample was imaged for DAPI-stained nuclei (blue), AF488 fluorescent signal (green) from NPs, and anti-CD68-stained cells (red). Images are representative of n=5. The white dotted line outlines the vascular lumen interior. The arrows point at cells expressing CD68 marker that were also positive for AF488 from NPs. Quantitative analysis showed higher localization of AF488-labeled NP-LXR in the aortic root and less accumulation in the liver.
2.4. In Vitro and In Vivo Anti-Inflammatory Effects of GW3965-Encapsulated NPs
Since LXR is known to have anti-inflammatory effects that may contribute to its anti-atherogenic action,[33] in the next set of experiments we evaluated whether the GW3965-encapsulated NPs were able to alter lipopolysaccharide (LPS)-induced inflammatory responses in mouse peritoneal macrophages. Isolated murine peritoneal macrophages were pretreated for 18 h with a dose of 1 μM GW3965 in DMSO or an equivalent dose of GW3965-encapsulated NPs, followed by stimulation with LPS to induce a pro-inflammatory response. ELISA assays were performed on the culture supernatant to measure the production of pro-inflammatory and atherogenic factors MCP-1 and TNFα. Treatment with the GW3965 in all delivery forms substantially suppressed secretion of MCP-1 and TNFα (Figure 4A). Furthermore, when GW3965 was delivered via NPs, the production of both pro-inflammatory factors declined slightly in comparison to the free drug. We also analyzed expression levels of the genes encoding MCP-1 and TNFα. As at the protein level, all delivery forms of GW3965 reduced the LPS-induction of the mRNAs (Figure 4B).
Figure 4.

Anti-inflammatory effects of GW3965-encapsulated NPs in vitro and in vivo. Peritoneal macrophages were incubated with PBS vehicle, free GW3965, or the indicated NPs at an equivalent GW3965 concentration of 1 μM. At 18 hours post incubation, the cells were treated for an additional 6 hours with 100ng/ml LPS. The upper medium was collected and tested for MCP-1 and TNFα production from cells by ELISA (A), and MCP-1 and TNFα gene expression was determined by qRT-PCR (B). (C) WT male mice (n=5) were i.p. administered vehicle, free GW3965 (10mg/kg), or an equivalent dose of GW3965-encapsulated NPs, followed by i.p. injections of Zymosan (100 μg). Four hours afterwards, peritoneum exudates were tested for MCP-1 and TNFα production by ELISA. Data are presented as the mean ± standard deviation. *P<0.01, as compared with the control group.
The anti-inflammatory capacity of GW3965-encapsulated NPs was also evaluated in a zymosan-induced peritonitis model in wild-type C57BL/6 mice. Mice were randomly divided into four groups (n=5) and intraperitoneally (i.p.) injected with PBS or the following regimens including a dose of 10 mg/kg GW3965: (i) GW3965 in DMSO (free GW3965), (ii) NP-LXR, or (iii) PSNP-LXR. One hour post injection, the mice were administered 100 μg zymosan A via i.p. injection to induce a local and acute inflammatory response. The peritoneal exudates were harvested 4 h later, and the production of MCP-1 and TNFα was determined. As shown in Figure 4C, the ELISA assay revealed that treatment with GW3965, either in solution or NP form, significantly inhibited the secretion of both pro-inflammatory factors. Both GW3965-encapsulated NPs tended to exhibit slightly more anti-inflammatory ability than the free GW3965, as evidenced by the small differences in TNFα production. This finding is consistent with the results of a recently published parallel study.[34] NP-LXR and PSNP-LXR showed similar efficacy in regulating the inflammatory response, indicating that the incorporation of PS lipid provided only limited additional effects in modulating inflammatory processes in vivo.
Overall, these proof-of-concept studies suggest that GW3965-encapsulated NPs would bring about similar changes in macrophage inflammation in atherosclerotic plaques.
2.5. Effect of NPs on atherosclerosis
The influence of GW3965-encapsulated NPs on the development of atherosclerosis was analyzed in Ldlr−/− mice that were fed a WD for 16 weeks. Mice were then randomly divided into four groups and administered PBS, free GW3965, NP-LXR, or PSNP-LXR (each at a dose of 10 mg/kg GW3965) via retro-orbital injection three times weekly for 2 weeks. This time point was chosen based on our previous report using the LXR agonist T0901317.[9]
First we investigated the effects of GW3965 in either solution or NP form on lipid levels in the plasma and liver (Table S1). Treatment with free GW3965 produced significant higher plasma and hepatic triglyceride levels compared with controls. This finding is consistent with previous descriptions of hypertriglyceridemia in mice associated with LXR agonist treatment.[14,15] In contrast, the group treated with NP-LXR showed lower hepatic total cholesterol and triglyceride levels in comparison with free GW3965. PSNP-LXR treatment also reduced hepatic triglyceride levels but raised hepatic total cholesterol levels compared to the free GW3965, presenting a less favorable lipid profile in the liver than NP-LXR. Plasma total cholesterol and triglyceride levels were significantly reduced in both NP groups in comparison with the free GW3965 group.
The effects of the different treatment regimens on target gene expression in the livers of Ldlr−/− mice were also assessed by qRT-PCR assay. As shown in Figure S2, administration of free GW3965 markedly induced the expression of hepatic ABCA1, SREBP-1c, and CYP7A. Notably, similar to the results in Figure 2D, there was a protective effect against the induction of SREBP-1c and CYP7A in the groups receiving GW3965-loaded NPs, as well as comparable effects on the expression of ABCA1 by NP-LXR and free drug.
CD68+ staining of aortic root sections revealed a significant reduction in the macrophage content of atherosclerotic plaques in all treatment groups (Figure 5A). Quantitative analysis of the CD68-positive areas (Figure 5B) showed that Ldlr−/− mice receiving NP-LXR showed a statistically significant (P<0.01) 50% reduction in the CD68+ area. PSNP-LXR NP and free GW3965 treatments were less effective: ~40% (P<0.01) and 30% (P<0.01) lower, respectively, compared with the control mice.
Figure 5.

GW3965-encapsulated NPs inhibit the progression of atherosclerotic lesions in Ldlr−/− mice. Ldlr-/− mice (n=5) with WD-induced atherosclerotic plaques were treated i.v. for 2 weeks with vehicle, free GW3965 (10 mg/kg), or an equivalent dose of GW3965-encapsulated NPs. (A) Representative CD68+ staining of aortic root lesions (magnification 4x). (B) Quantitative analysis of CD68+ lesion area by ImagePro Plus software. The aortic root sections were analyzed for the expression of LXR target genes ABCA1 and SREBP-1c (C), and inflammatory genes MCP-1 and TNFα (D). Data are represented as the mean ± standard deviation (n=5). *P<0.05 and **P<0.01, as compared with the control group.
In addition, we examined gene expression levels in the plaques. mRNA was extracted from the laser-captured CD68+ cells of aortic roots and quantified using qRT-PCR. As shown in Figure 5C, the strongest induction of ABCA1 and SREBP-1c was noted in response to NP-LXR. On the other hand, in agreement with the suggestion based on the peritonitis study (Figure 4), expression of MCP-1 was much more effectively suppressed by either GW3965-loaded NP than by the free GW3965 (Figure 5D). In comparison with the PBS control group, TNFα levels were slightly suppressed by NP-LXR treatment, while no change was observed in the PSNP-LXR-treated group.
The results in Table S1 and in Figure 5 suggest that in plaques the NP-LXR particles exert more favorable gene-targeting effects, including anti-inflammatory effects in the macrophages, while protecting hepatic and plasma lipid levels.
3. Conclusion
In summary, we have developed polymeric NPs capable of delivering GW3965, a standard LXR agonist, to atherosclerotic plaque macrophages. After systemic administration, these NPs co-localized within CD68+ plaque macrophages of Ldlr−/− mice. These NPs were also shown to improve resolution of inflammation in LPS-activated murine peritoneal macrophages and a zymosan-induced peritonitis model. Additionally, they are more potent than free GW3965 in inducing LXR gene expression in vitro and in vivo. Strikingly, short-term treatment of plaque-bearing Ldlr-/- mice with NP-LXR resulted in up to a 50% reduction in atherosclerotic lesion area without increasing total cholesterol or triglycerides in the liver and plasma. We believe the PLGA-b-PEG-based NPs to be biocompatible, biodegradable, and safe as drug carriers for clinical use, since both PLGA and PEG polymers have been approved by the FDA for medical applications. Thus, we can envision a therapeutic strategy in which NP-LXR could offer the benefits of LXR activation on atherosclerosis while decreasing the likelihood of adverse effects on the liver, the major impediment to their clinical application.
4. Experimental Section
Materials
N-hydroxysuccinimide (NHS), 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC), Zymosan A, N,N-diisopropylethylamine (DIEA), LDH kit, Brewer thioglycolate medium, and all other solvents and chemicals were purchased from Sigma-Aldrich. Alexa Fluor® (AF) 488 C5 maleimide and Alexa Fluor® (AF) 680 C2 maleimide were purchased from Invitrogen. Carboxy-terminated PLGA [50:50 poly(DL-lactide-coglycolide) (0.55–0.75 dL/g)] was purchased from Lactel Absorbable Polymers. The heterobifunctional PEG polymers NH2-PEG-COOH and NH2-PEG-SH (molecular weight of 3,400 Da) were purchased from JenKem Technology USA. PS lipid was purchased from Avanti Polar Lipids, Inc. 1H NMR spectra were recorded on a Bruker AVANCE-400 NMR spectrometer. The NP sizes and ζ-potentials were obtained by quasi-electric laser light scattering using a ZetaPALS dynamic light-scattering detector (15mW laser, incident beam ¼ 676 nm; Brookhaven Instruments). Transmission electron microscopy (TEM) was performed on a JEOL 2011 at 200 kV. The Agilent 1100 HPLC (Santa Clara, CA) was equipped with a UV detector and a reverse-phase column (Eclipse, 4.6 × 150 mm, 5 μm) with a gradient mobile phase of water and acetonitrile at a constant flow rate 1 mL/min.
Synthesis of PLGA-b-PEG (1)
Carboxylate-functionalized copolymer PLGA–b–PEG was synthesized by the conjugation of COOH–PEG–NH2 to PLGA–COOH. PLGA– COOH (250 mg, 0.005 mmol) in dichloromethane (DCM, 2.5 mL) was converted to PLGA–NHS in the presence of excess EDC (4.8 mg, 0.025 mmol) and NHS (3 mg, 0.035 mmol). PLGA–NHS was precipitated with an ice-cold mixture of diethyl ether and methanol. The resultant precipitate was centrifuged, and the pellet was dissolved in DCM followed by repeated precipitation/wash (two times). After drying under vacuum, PLGA–NHS (200 mg, 0.004 mmol) was dissolved in DCM (1.5 mL) followed by the addition of NH2–PEG–COOH (20.4 mg, 0.006 mmol) and DIEA (7.5 mg, 0.06 mmol). The reaction was stirred for 12 h at RT. The co-polymer was precipitated with cold diethylether/methanol. The pellet was redissolved in DCM and precipitate, followed by three washes with the same solvent to remove unreacted PEG. The resulting PLGA–b-PEG block copolymer was dried under vacuum and used for NP preparation without further treatment. 1H NMR (400 Hz, CDCl3 -d6, δ): 5.2 (m, 2H; CH), 4.8 (m, 2H; CH2), 3.7 (s, 4H; CH2), 1.6 (d, 3H; CH3).
Synthesis of NH2-PEG-AF680 and NH2-PEG-AF488 (2)
NH2-PEG-AF680 or NH2-PEG-AF488 was synthesized by reacting NH2-PEG-SH with respective maleimide-functionalized fluorophores at a molar ratio of 1.2:1. The reactions were stirred in the dark at room temperature overnight. Fluorescently labeled PEG was obtained by dialysis against water (MWCO 3500) and lyophilization.
Synthesis of PLGA-b-PEG-AF680 and PLGA-b-PEG-AF488 (3)
Fluorescently labeled PLGA–b–PEG (AF680 and AF488) were synthesized by the conjugation of PLGA–COOH to NH2-PEG-AF680 or NH2-PEG-AF488, respectively, as described above.
Preparation and Characterization of NPs
The NPs encapsulating GW3965 were formulated via the single emulsion solvent evaporation technique. In brief, PLGA-b-PEG was dissolved in dichloromethane (DCM), and GW3965 was dissolved in dimethyl sulfoxide (DMSO). The mixture of GW3965 and PLGA-b-PEG solutions was added into aqueous solution containing 1% PVA, followed by probe sonication to form the emulsion. Next, the emulsified mixture was poured into 15 mL water with or without PS, followed by stirring for 2 hours to allow the DCM solvent to evaporate and the particles to harden. The remaining organic solvent and free molecules were removed by washing the particle solution three times using an Amicon Ultra-4 centrifugal filter (Millipore, Billerica, MA) with a molecular weight cutoff of 100 kDa. The NPs were diluted 20-fold in either water or PBS, and their size and surface charge were determined using dynamic light scattering. Samples for TEM were stained with 1% uranyl acetate and observed using a JEOL 2011 at 200 kV. The GW3965 content in the NPs was analyzed by RT-HPLC (Agilent, CA). Drug loading is defined as the mass fraction of drug in the nanoparticles, whereas entrapment efficiency (EE) is the fraction of initial drug encapsulated by the nanoparticles.
Release Kinetics Study
To determine the GW3965 release profile, a suspension of NPs in PBS was aliquotted (100 μL) into several semipermeable minidialysis tubes (molecular mass cutoff 100 kDa; Pierce) and dialyzed against 2L of frequently renewed PBS (pH 7.4) at 37 °C with gentle stirring. At predetermined time points, quadruplicate aliquots of each NP sample (n=4) were withdrawn, and the GW3965 content was analyzed by RP-HPLC. The amounts of released GW3965 were calculated to generate a cumulative release curve.
Cell Culture
RAW264.7 cells were maintained in Dulbecco’s minimal essential medium (DMEM) supplemented with 10% FBS, streptomycin (100 μg/ml), penicillin (100 U/ml), and 4mM L-glutamine (ATCC, Manassas, VA) at 37°C in a humidified 5% CO2-containing atmosphere. Peritoneal macrophages were obtained from C57Bl/6J mice after i.p. injection of Brewer thioglycolate medium as described previously.[35] Cells (1 × 106) were plated on 6-well plates and cultured as above. For ligand treatments, cells were cultured for 18 h with the respective regimens with an equivalent GW3965 concentration of 1μM. For the anti-inflammatory experiments, cells were cultured as above followed by stimulation with LPS (100ng/ml) for an additional 6h to induce a pro-inflammatory response.
Cellular Internalization of LXR Agonist
RAW264.7 cells were seeded into 24-well plates at a density of 1 × 105 cells per well 24 hours prior to the experiment. NPs encapsulating a GW3965 concentration of 1 μM were then incubated with cells for the desired time points. Afterwards, the cells were washed with PBS, followed by trypsinization and centrifugation to remove excess trypsin-EDTA. Cells were then resuspended in PBS, and cellular uptake of NPs was assessed via flow cytometry (BD LSR II, BD Bioscience, San Jose, CA). Data were analyzed using BD FACSDiva software.
LDH Assay
Peritoneal macrophages were treated with NPs encapsulating a dose of GW3965 equivalent to that used for the qRT-PCR assay. To examine the cytotoxicity of the GW3965-encapsulated NPs, the activity of released LDH (a marker of membrane integrity) in the cell medium was determined using a commercial LDH Kit (Sigma-Aldrich) according to the manufacturer's instructions. Cell viability was expressed as a percentage of the control to which no treatment had been given.
Animals
All mice were on the C57BL/6 background and were obtained from Jackson Laboratories, Bar Harbor, Maine, USA. The animals were allowed free access to sterile food pellets and water. All in vivo studies were performed in accordance with National Institutes of Health Animal Care guidelines. The regimens described in Results/Discussion and the figure legends were administered to Ldlr−/− mice (n=50) via retro-orbital injection to investigate their accumulation in tissues, as well as their effects on plasma and hepatic lipid levels, LXR-related gene expression in the aortic tissues and liver, and atherosclerosis.
Murine Peritonitis
Male C57Bl/6J mice (6–8 weeks, n=20) were administered i.p. vehicle, free GW3965 (10mg/kg), or an equivalent dose of GW3965-encapsulated NPs. One hour afterwards the mice were injected i.p. with Zymosan A (100μg/mouse) to induce peritonitis.[36] Peritoneal exudates were harvested 4 h post-zymosan administration.
qRT-PCR
Total RNA abundance was determined by qRT-PCR (TaqMan) using 100pg total RNA. The primer and probe sequences are described in Table S2. Peritoneal macrophage and in vivo data were normalized to 28s and Cyclophilin, respectively, and presented as the fold difference over controls. The in vitro data were reported as mean standard deviation for duplicate samples, and the experiment was repeated twice. For the laser-captured cell data, the results were from independent samples, each representing a pool of CD68+ cell RNA from 6 slides/animal.
Lipid Analysis
Plasma and hepatic total cholesterol and triglycerides were measured using commercially available enzymatic kits (Wako Chemicals). Lipids were extracted from frozen livers by overnight incubation with isopropanol. Hepatic lipids were normalized to the hepatic protein content, which was measured with the DC Protein Assay Kit (Bio-Rad, Hercules, USA) according to the manufacturer’s instructions.
Laser Capture Microdissection (LCM)
To isolate CD68+ cells from plaques, laser capture microdissection was performed with PixCell II (Arcturus Bioscience, Mountain View, CA) as previously reported.[9] Briefly, 6-μm frozen sections were dehydrated in ethanol and xylene and then air dried. At 100-μm intervals, sections were immunostained for CD68 and used as templates for the next five serial sections. RNA was isolated using a PicoPure RNA Isolation Kit (Arcturus) and quantitatively analyzed using a Ribogreen RNA Quantitation kit (Invitrogen). Quality of extracted RNA was verified using an Agilent 2100 Bioanalyzer. Expression of the specific genes was analyzed by qRT-PCR.
Immunohistochemistry
Prior to harvesting, aortic roots and livers were perfused with 10% sucrose. The harvested tissues were embedded in optimum cutting temperature medium (OCT) compound and sectioned using a cryostat. The slides were stained for CD68 (AbD Serotec) as previously described.[37] Intimal lesions and stained areas were quantified by computer-aided morphometric analysis of digitized images (ImagePro Plus 7.0 software, Silver Spring, USA).
Statistical Analysis
Data are expressed as mean ± SEM and were analyzed by one-way ANOVA as appropriate for multiple comparisons. A P value less than 0.05 was considered significant.
Supplementary Material
Figure S1: LDH assay of murine thioglycolate-elicited peritoneal macrophages that were incubated with vehicle, placebo NPs, free GW3965, or the indicated NPs at an equivalent concentration of 1 μM GW3965. Data are represented as the mean ± standard deviation (n=2).
Figure S2: Effects of GW3965 (10 mg/kg) in either solution or NP form on hepatic mRNA expression in Ldlr−/− mice (n=5) with WD-induced atherosclerotic plaques. Data are represented as the mean ± standard deviation. *P<0.05, as compared with the control group.
Table S1. Effects of GW3965 (10 mg/kg) in either solution or NP form on total cholesterol (TC) and triglycerides (TG) in the plasma and liver of Ldlr−/− mice fed a high-fat diet. Data are expressed as mean ± standard deviation (n=5). *P<0.05, as compared with the control group.
Table S2. Sequences of forward primers, reverse primers, and TaqMan probes.
Acknowledgments
This work was supported by a Program of Excellence in Nanotechnology Award, Contract HHSN268201000045C, from the National Heart, Lung, and Blood Institute, National Institutes of Health (NIH). This work was also supported by NIH Grant CA151884, HL084312, and by a David Koch–Prostate Cancer Foundation Award in Nanotherapeutics. Dr. X.X. acknowledges postdoctoral support from an NIH National Research Service Award (NRSA) (1F32CA168163-02).
Footnotes
Supporting Information is available from the Wiley Online Library or from the authors.
Conflict of interest statement
In compliance with the Brigham and Women’s Hospital and Harvard Medical School institutional guidelines, O.C.F. discloses his financial interest in BIND Therapeutics, Selecta Biosciences, and Blend Therapeutics, three biotechnology companies developing nanoparticle technologies for medical applications. BIND, Selecta, and Blend did not support the aforementioned research, nor do these companies currenly have any rights to any technology or intellectual property developed as part of this research. The rest of the authors declare no conflicts of interest.
Contributor Information
Dr. Xue-Qing Zhang, Laboratory of Nanomedicine and Biomaterials, Department of Anesthesiology, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02115, USA
Dr. Orli Even-Or, Laboratory of Nanomedicine and Biomaterials, Department of Anesthesiology, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02115, USA, Leon H. Charney Division of Cardiology and the Marc and Ruti Bell Program in Vascular Biology, Department of Medicine, New York University School of Medicine, New York, NY 10016, USA
Dr. Xiaoyang Xu, Laboratory of Nanomedicine and Biomaterials, Department of Anesthesiology, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02115, USA, The David H. Koch Institute for Integrative Cancer Research. Massachusetts Institute of Technology. Cambridge, MA 02139, USA
Mariska van Rosmalen, Leon H. Charney Division of Cardiology and the Marc and Ruti Bell Program in Vascular Biology, Department of Medicine, New York University School of Medicine, New York, NY 10016, USA.
Lucas Lim, Laboratory of Nanomedicine and Biomaterials, Department of Anesthesiology, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02115, USA.
Prof. Omid C. Farokhzad, Email: ofarokhzad@zeus.bwh.harvard.edu, Laboratory of Nanomedicine and Biomaterials, Department of Anesthesiology, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02115, USA, King Abdulaziz University, Jeddah 21589, Saudi Arabia
Prof. Edward A. Fisher, Email: Edward.Fisher@nyumc.org, Leon H. Charney Division of Cardiology and the Marc and Ruti Bell Program in Vascular Biology, Department of Medicine, New York University School of Medicine, New York, NY 10016, USA
References
- 1.Hansson GK. N Engl J Med. 2005;352:1685. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 2.Libby P, Ridker PM, Hansson GK. Nature. 2011;473:317. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 3.Tabas I. Nat Rev Immunol. 2010;10:36. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Libby P. Nature. 2002;420:868. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 5.Lobatto ME, Fuster V, Fayad ZA, Mulder WJ. Nat Rev Drug Discov. 2011;10:835. doi: 10.1038/nrd3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang XQ, Xu X, Bertrand N, Pridgen E, Swami A, Farokhzad OC. Adv Drug Deliv Rev. 2012;64:1363. doi: 10.1016/j.addr.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hrkach J, Von Hoff D, Mukkaram AM, Andrianova E, Auer J, Campbell T, De Witt D, Figa M, Figueiredo M, Horhota A, Low S, McDonnell K, Peeke E, Retnarajan B, Sabnis A, Schnipper E, Song JJ, Song YH, Summa J, Tompsett D, Troiano G, Van Geen Hoven T, Wright J, LoRusso P, Kantoff PW, Bander NH, Sweeney C, Farokhzad OC, Langer R, Zale S. Sci Transl Med. 2012;4:128. doi: 10.1126/scitranslmed.3003651. [DOI] [PubMed] [Google Scholar]
- 8.Farokhzad OC, Langer R. ACS Nano. 2009;3:16. doi: 10.1021/nn900002m. [DOI] [PubMed] [Google Scholar]
- 9.Feig JE, Pineda-Torra I, Sanson M, Bradley MN, Vengrenyuk Y, Bogunovic D, Gautier EL, Rubinstein D, Hong C, Liu J, Wu C, van Rooijen N, Bhardwaj N, Garabedian M, Tontonoz P, Fisher EA. J Clin Invest. 2010;120:4415. doi: 10.1172/JCI38911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zelcer N, Tontonoz P. J Clin Invest. 2006;116:607. doi: 10.1172/JCI27883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joseph SB, McKilligin E, Pei L, Watson MA, Collins AR, Laffitte BA, Chen M, Noh G, Goodman J, Hagger GN, Tran J, Tippin TK, Wang X, Lusis AJ, Hsueh WA, Law RE, Collins JL, Willson TM, Tontonoz P. Proc Natl Acad Sci U S A. 2002;99:7604. doi: 10.1073/pnas.112059299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levin N, Bischoff ED, Daige CL, Thomas D, Vu CT, Heyman RA, Tangirala RK, Schulman IG. Arterioscler Thromb Vasc Biol. 2005;25:135. doi: 10.1161/01.ATV.0000150044.84012.68. [DOI] [PubMed] [Google Scholar]
- 13.Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL, Mangelsdorf DJ. Genes Dev. 2000;14:2819. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, Lustig KD, Shan B. Genes Dev. 2000;14:2831. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grefhorst A, Elzinga BM, Voshol PJ, Plosch T, Kok T, Bloks VW, van der Sluijs FH, Havekes LM, Romijn JA, Verkade HJ, Kuipers F. J Biol Chem. 2002;277:34182. doi: 10.1074/jbc.M204887200. [DOI] [PubMed] [Google Scholar]
- 16.Farokhzad OC. Expert Opin Drug Deliv. 2008;5:927. doi: 10.1517/17425247.5.9.927. [DOI] [PubMed] [Google Scholar]
- 17.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nat Nanotechnol. 2007;2:751. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 18.Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Adv Drug Deliv Rev. 2014;66:2. doi: 10.1016/j.addr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. J Immunol. 1992;148:2207. [PubMed] [Google Scholar]
- 20.Harel-Adar T, Ben Mordechai T, Amsalem Y, Feinberg MS, Leor J, Cohen S. Proc Natl Acad Sci U S A. 2010;108:1827. doi: 10.1073/pnas.1015623108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang L, Chan JM, Gu FX, Rhee JW, Wang AZ, Radovic-Moreno AF, Alexis F, Langer R, Farokhzad OC. ACS Nano. 2008;2:1696. doi: 10.1021/nn800275r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z, Feng SS. Biomacromolecules. 2006;7:1139. doi: 10.1021/bm050953v. [DOI] [PubMed] [Google Scholar]
- 23.Calkin AC, Tontonoz P. Arterioscler Thromb Vasc Biol. 2010;30:1513. doi: 10.1161/ATVBAHA.109.191197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tangirala RK, Bischoff ED, Joseph SB, Wagner BL, Walczak R, Laffitte BA, Daige CL, Thomas D, Heyman RA, Mangelsdorf DJ, Wang X, Lusis AJ, Tontonoz P, Schulman IG. Proc Natl Acad Sci U S A. 2002;99:11896. doi: 10.1073/pnas.182199799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Repa JJ, Mangelsdorf DJ. Nat Med. 2002;8:1243. doi: 10.1038/nm1102-1243. [DOI] [PubMed] [Google Scholar]
- 26.Joseph SB, Tontonoz P. Curr Opin Pharmacol. 2003;3:192. doi: 10.1016/s1471-4892(03)00009-2. [DOI] [PubMed] [Google Scholar]
- 27.Yoshikawa T, Shimano H, Amemiya-Kudo M, Yahagi N, Hasty AH, Matsuzaka T, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Osuga J, Harada K, Gotoda T, Kimura S, Ishibashi S, Yamada N. Mol Cell Biol. 2001;21:2991. doi: 10.1128/MCB.21.9.2991-3000.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joseph SB, Laffitte BA, Patel PH, Watson MA, Matsukuma KE, Walczak R, Collins JL, Osborne TF, Tontonoz P. J Biol Chem. 2002;277:11019. doi: 10.1074/jbc.M111041200. [DOI] [PubMed] [Google Scholar]
- 29.Smith JD, Breslow JL. J Intern Med. 1997;242:99. doi: 10.1046/j.1365-2796.1997.00197.x. [DOI] [PubMed] [Google Scholar]
- 30.Hoekstra M, Kruijt JK, Van Eck M, Van Berkel TJ. J Biol Chem. 2003;278:25448. doi: 10.1074/jbc.M301189200. [DOI] [PubMed] [Google Scholar]
- 31.Ma YJ, Xu LY, Rodriguez-Agudo D, Li, Heuman DM, Hylemon PB, Pandak WM, Ren SL. Am J Physiol-Endocrinol Metab. 2008;295:E1369. doi: 10.1152/ajpendo.90555.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huong TM, Ishida T, Harashima H, Kiwada H. Int J Pharm. 2001;215:197. doi: 10.1016/s0378-5173(00)00691-8. [DOI] [PubMed] [Google Scholar]
- 33.Im SS, Osborne TF. Circ Res. 2011;108:996. doi: 10.1161/CIRCRESAHA.110.226878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gadde S, Even-Or O, Kamaly N, Hasija A, Gagnon PG, Adusumilli KH, Erakovic A, Pal AK, Zhang XQ, Kolishetti N, Shi J, Fisher EA, Farokhzad OC. Adv Healthcare Mater. 2014 doi: 10.1002/adhm.201300688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Piao ZH, Kim MS, Jeong M, Yun S, Lee SH, Sun HN, Song HY, Suh HW, Jung H, Yoon SR, Kim TD, Lee YH, Choi I. Cell Immunol. 2012;280:1. doi: 10.1016/j.cellimm.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 36.Cash JL, White GE, Greaves DR. Methods Enzymol. 2009;461:379. doi: 10.1016/S0076-6879(09)05417-2. [DOI] [PubMed] [Google Scholar]
- 37.Trogan E, Feig JE, Dogan S, Rothblat GH, Angeli V, Tacke F, Randolph GJ, Fisher EA. Proc Natl Acad Sci U S A. 2006;103:3781. doi: 10.1073/pnas.0511043103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: LDH assay of murine thioglycolate-elicited peritoneal macrophages that were incubated with vehicle, placebo NPs, free GW3965, or the indicated NPs at an equivalent concentration of 1 μM GW3965. Data are represented as the mean ± standard deviation (n=2).
Figure S2: Effects of GW3965 (10 mg/kg) in either solution or NP form on hepatic mRNA expression in Ldlr−/− mice (n=5) with WD-induced atherosclerotic plaques. Data are represented as the mean ± standard deviation. *P<0.05, as compared with the control group.
Table S1. Effects of GW3965 (10 mg/kg) in either solution or NP form on total cholesterol (TC) and triglycerides (TG) in the plasma and liver of Ldlr−/− mice fed a high-fat diet. Data are expressed as mean ± standard deviation (n=5). *P<0.05, as compared with the control group.
Table S2. Sequences of forward primers, reverse primers, and TaqMan probes.