

Abstract
β-Catenin is a critical transducer of the Wnt signal pathway and plays an important role in many developmental and cellular processes. Deregulation of β-catenin signaling has been observed in a broad range of human tumors. In this report, we investigated whether tyrosine kinase inhibitor STI-571 could inhibit the β-catenin signaling activity and hence suppress cell proliferation. Our results demonstrated that STI-571 effectively inhibited the constitutive activity of β-catenin signaling in human colon cancer cells as well as the Wnt1-induced activation of β-catenin signaling in HOS, HTB-94, and HEK 293 cells. Furthermore, STI-571 was shown to effectively suppress the proliferation of human colon cancer cells. Finally, we demonstrated that the Wnt1-mediated activation of a GAL4-β-catenin heterologous transcription system was effectively inhibited by STI-571. Thus, our findings suggest that tyrosine phosphorylation may play an important role in regulating β-catenin signaling activity, and inhibition of this signaling pathway by STI-571 may be further explored as an important target for alternative/adjuvant treatments for a broader range of human cancer.
Keywords: Cancer, β-Catenin, Gleevec, STI-571, Tyrosine kinase inhibitor, Wnt signal
1. Introduction
Recent studies suggest that deregulation of β-catenin signaling may play an important role in tumorigenesis [1-3]. Originally identified as a cytoplasmic protein that interacts with cell adhesion molecules, such as E-cadherin, β-catenin is an essential signal transducer of the Wnt/Wingless pathway [4,5]. Wnt ligands initiate their signaling by binding to the frizzled receptors, as well as the recently identified co-receptors LRP5 and LRP6, leading to phosphorylation of the disheveled protein. It then associates with Axin and the adenomatous polyposis coli (APC) tumor suppressor, preventing GSK3b from phosphorylating β-catenin. Unphosphorylated β-catenin is stabilized by escaping recognition by ubiquitin/proteosome complex, and eventually translocates to the nucleus where it engages transcription factors LEF/TCF-4 to activate the expression of downstream targets, such as c-Myc and cyclin D1 [3,6-11]. The involvement of β-catenin signaling in tumorigenesis was first established in colorectal cancer, where the β-catenin protein is stabilized by inactivating mutations of the APC tumor suppressor gene or oncogenic mutations of the β-catenin gene in the vast majority of colon cancers [1]. Recently β-catenin mutations have been detected in a variety of human tumors [12,13]. Oncogenic forms of β-catenin have been shown to induce tumor formation in transgenic animals [14,15], whereas mutations in β-catenin gene have been frequently detected in tumors induced by either carcinogens or activated oncogenes [16,17]. These collective genetic data strongly suggest that deregulation of β-catenin signaling may be involved in the development of a broad range of human malignancies.
Protein kinases play an important role in regulating cell proliferation [18,19]. Aberrant activation of some kinases has been associated with neoplastic transformation and/or tumorigenesis [19]. Targeting these kinases may represent an important therapeutic alternative for human cancer [20-22]. Recently, one such kinase inhibitor, STI-571, has gained widespread attention. STI-571 (a.k.a., Gleevec, Glivec, CGP57148, or imatinib mesylate), a small selective inhibitor of the Bcr-Abl, c-kit, and PDGF receptor tyrosine kinases, has been approved for the treatment of chronic myeloid leukemia (CML) and gastrointestinal stromal tumors (GISTs) [23-27]. Promising anti-tumor effects of STI-571 have also been reported in dermatofibrosarcoma protuberans, many of which harbor a COL1A1-PDGFB fusion protein [28,29]. As discussed above, many types of human cancer frequently exhibit a significant nuclear and/or cytoplasmic accumulation of β-catenin protein, a hallmark of deregulated β-catenin activity [3,30]. With the exception of colorectal cancer, genetic alterations of β-catenin are only detected in a small fraction of non-colon tumors, and the upstream regulators of the β-catenin signaling pathway are rarely mutated. Therefore, the molecular mechanisms underlying β-catenin deregulation remains undefined in most human tumors. Several recent studies suggest that β-catenin is a tyrosine-phosphorylated protein and some growth factors (such as HGF, IGF-1 and IGF-2) may play a role in regulating β-catenin signaling activity [31-40]. In this study, we investigated whether tyrosine kinase inhibitor STI-571 exhibits any inhibitory effect on β-catenin signaling.
2. Materials and methods
2.1. Cell lines and chemicals
Cell lines HEK 293, HOS (a human osteosarcoma cell line), and HTB-94 (a human chondrosarcoma cell line) were purchased from ATCC (Manassas, VA). Human colon cancer lines, HCT116 and SW480, were kindly provided by Bert Vogelstein of Johns Hopkins Medical Institutes, Baltimore, MD. HOS cells were maintained in complete MEM Eagle supplemented with 10% fetal bovine serum (FBS, Mediatech, Herndon, VA), 100 units of penicillin, and 100 μg of streptomycin, 1 × non-essential amino acids (Mediatech), 1 mM sodium pyruvate (Mediatech) at 37 °C in 5% CO2. HEK 293 and HTB-94 were maintained in complete DMEM containing 10% FBS and 100 units of penicillin, and 100 μg of streptomycin. HCT116 and SW480 were maintained in complete McCoy’s 5A supplemented with 10% FBS and 100 units of penicillin, and 100 μg of streptomycin. STI-571 was provided by Novartis Pharmaceuticals AG (Basel, Switzerland). Unless otherwise indicated, all chemicals were purchased from Sigma-Aldrich (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA).
2.2. Construction of the Wnt1 adenoviral vector and preparation of Wnt1-conditioned medium
The cDNA encoding mouse Wnt1 was tagged with a HA epitope at its C-terminus, and was subcloned into the pAdTrack-CMV vector, resulting in pAd-Track-Wnt1. Recombinant adenovirus (i.e. AdWnt1) expressing Wnt1 was generated as previously described [41]. To prepare Wnt1-conditioned medium (Wnt1-CM), the purified AdWnt1 was used to infect exponentially growing HCT116 cells in 75 cm2 cell culture flasks (MOI = 20). Adenovirus-containing medium was removed and washed off at 6 h after infection, and replaced with 15 ml of serum-free DMEM for each 75 cm2 cell culture flask. Wnt1-CM was collected at 24 h and after passing through 0.2-μm filters, was kept at 4 °C.
2.3. Establishment of 293-GEM stable lines as a functional reporter system of the β-catenin signaling activity
To construct a GAL4-β-Catenin (GBC) chimeric transcription factor, the DNA-binding domain (i.e. amino acids 1 – 147) of the yeast transcription factor GAL4 was fused in-frame with human β-catenin [42], resulting pCMV-GBC. A reporter vector, pGRE-GFP, which expressed green fluorescent protein (GFP) under the control of a GAL4-responsive promoter, was generously provided by Bert Vogelstein of Johns Hopkins Oncology Center [43]. In order to establish a 293-GEM stable line, exponentially growing HEK 293 cells were co-transfected with 1.0 μg of pGRE-GFP plasmid DNA and 0.2 μg of pCMV-GBC plasmid per 25 cm2 tissue culture flask by using LipofectAMINE according to the manufacturer’s instructions (Life Technologies, Gaithersburg, MD). At 24 h after transfection, cells were trypsinized and replated into 96-well cell culture plates at multiple dilutions. Stable clones were selected in the presence of G418 at a final concentration of 0.3 mg/ml for 2 weeks. Clones derived from single cells were grown up for further characterization [42].
2.4. Transfection and luciferase assay
Exponentially growing cells were seeded in 25 cm2 cell culture flasks, and transfected with 2 μg per flask of β-catenin/TCF4-responsive luciferase reporter, pTOP-Luc [44] or a control reporter, pGL3-Control (Promega, Madison, WI) using LipofectAmine (Life Technologies, Gaithersburg, MD). At 16 h after transfection, cells were replated to 48-well plates. At 12 h after replating, cells were treated with STI-571 only (for HCT116 and SW480), or STI-571 in the presence of Wnt1-CM (for HEK 293), or infection with AdWnt1 (for HOS and HTB-94). At 24–48 h after treatment, cells were lysed and cell lysates were collected for luciferase assays using Promega’s Luciferase Assay Kit. Each assay condition was performed in triplicate.
2.5. Cell proliferation assay
Exponentially growing HCT116 and SW480 cells were seeded at subconfluence in 48-well cell culture plates, and treated with STI-571 at a concentration range of 0–30 μM. At 24, 48, 72, and 96 h after treatment, cells were collected by trypsinization. Viable cells were counted in the presence of Trypan Blue (Mediatech). Each assay condition was carried out in triplicate. The average of viable cells was calculated from the cell counting from the triplicate in each assay condition.
3. Results
3.1. β-Catenin/TCF4-mediated transcription activity is inhibited by STI-571
To test the possible effect of STI-571 on β-catenin signaling activity, we first introduced a TCF4-responsive reporter into two human colorectal cancer lines, HCT116 and SW480. Both cell lines contain a high constitutive activity of β-catenin signaling because the HCT116 line harbors an oncogenic mutation of the β-catenin gene, whereas SW480 contains an inactivating mutation of the APC gene [1, 44]. It has been shown that TCF4-responsive reporter pTOP-Luc was highly active in these cell lines. As illustrated in Fig. 1, in the presence of STI-571 the TCF4 reporter activity was significantly inhibited. Specifically, the TCF4 reporter activity in HCT116 cells decreased to 55, 34, 24, and 14% of that of the control in the presence of 15, 20, 25, and 30 μM of STI-571, respectively (Fig. 1A). Interestingly, in SW480 cells, 15 μM STI-571 did not exert any detectable inhibition on TCF4 reporter; however, the effect of STI-571 at higher doses was more pronounced in SW480 cells when compared to HCT116 (Fig. 1B). For instance, when SW480 cells were treated with 25 μM of STI-571, TCF4 reporter activity decreased to 7%, as opposed to 24% in HCT116 cells treated with the same concentration of STI-571. This phenomenon was reproducibly observed in three independent batches of experiments. Conversely, using the same concentrations of STI-571, we did not observe any significant effect on pFOP-Luc, a mutant TCF4-responsive reporter in these cell lines (data not shown). Furthermore, the STI-571-mediated inhibition was seemingly specific, as the activity of pGL3-Control, a control reporter that was constitutively active under the control of SV40 promoter, was not affected under the same conditions. These results suggest that STI-571 can effectively inhibit the constitutive activity of β-catenin signaling in representative human colon cancer lines.
Fig. 1.
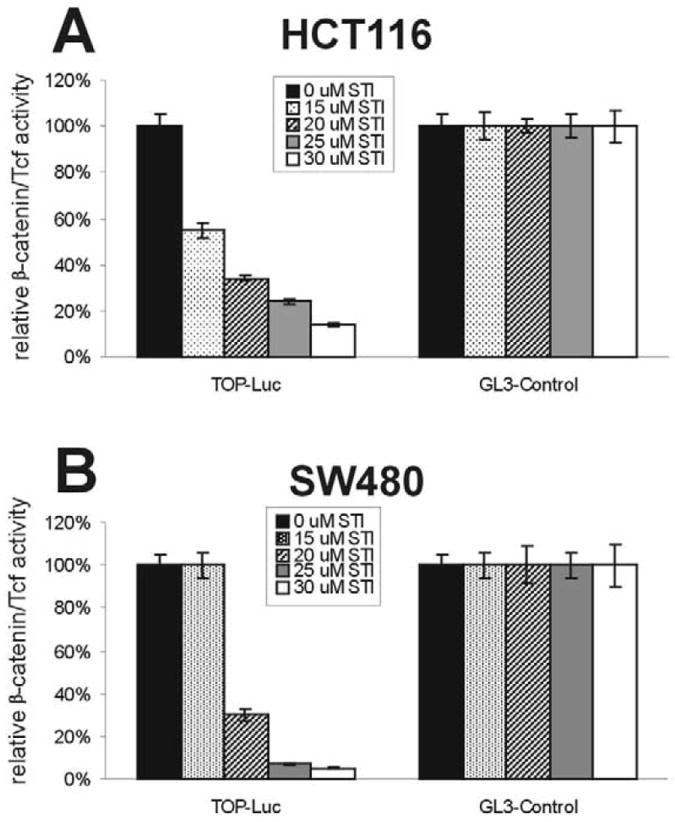
STI-571 inhibits β-catenin/TCF4-mediated transcription activity in human colon cancer cell lines. Subconfluent HCT116 (A) and SW480 (B) were transfected with the TCF4 reporter pTOP-Luc or the positive control reporter pGL3-Control. The transfected cells were replated in 48-well culture plates and treated with STI-571 at the indicated concentrations. At 24 h after treatment, cells were lysed for luciferase activity assays. Each assay condition was done in triplicate.
3.2. Wnt1-induced β-catenin signaling activity is inhibited by STI-571
We next studied whether STI-571 could inhibit Wnt1-induced β-catenin signaling activity in other human cells. Using a recombinant adenoviral vector expressing mouse Wnt1, we were previously able to reconstitute Wnt1-mediated activation of β-catenin signaling in the human osteosarcoma line, HOS, and the human chondrosarcoma line, HTB-94 [42]. In HOS cells, Wnt1-induced β-catenin activity decreased to 57 and 30% in the presence of 20 and 25 μM of STI-571, respectively (Fig. 2A). Interestingly, HTB-94 cells were seemingly more sensitive to STI-571 inhibition. The β-catenin/TCF4 activity was inhibited by approximately 60 and 90% when HTB-94 cells were treated with 5 and 10 μM of STI-571, respectively (Fig. 2B).
Fig. 2.
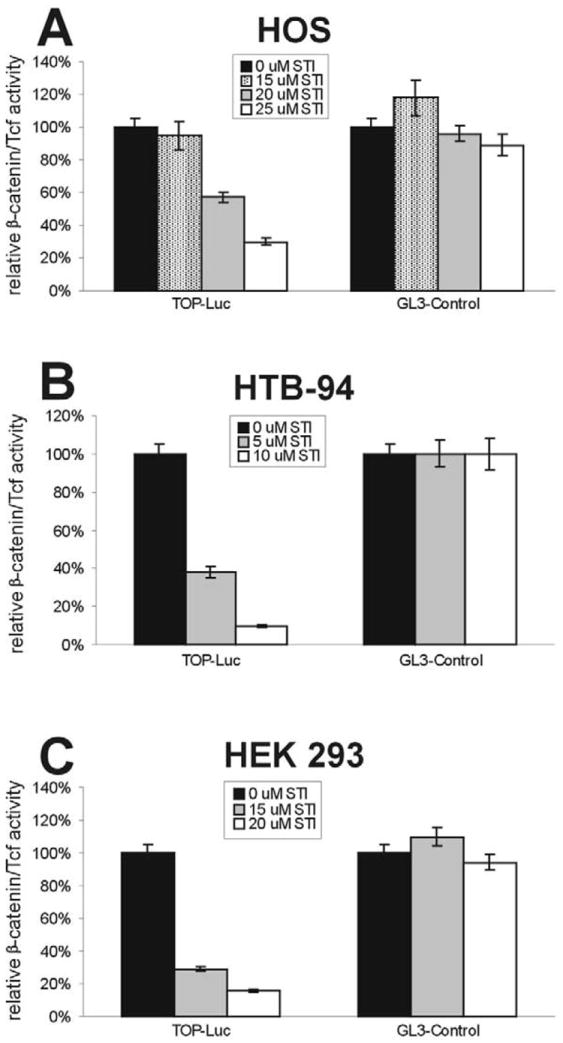
STI-571 inhibits Wnt1-induced activation of β-catenin signaling. Subconfluent HOS (A), HTB-94 (B), and HEK 293 (C) were transfected with the TCF4 reporter pTOP-Luc or the positive control reporter pGL3-Control. The transfected cells were replated in 48-well culture plates and treated with STI-571 (at the indicated concentrations) with the infection of AdWnt1 (A and B), or in the presence of Wnt1-conditioned medium (C). At 48 h after treatment, cells were lysed for luciferase activity assays. Each assay condition was done in triplicate.
Next, we tested whether Wnt1-induced β-catenin signaling could be inhibited by STI-571 in HEK 293 cells. The above experiments involved using human tumor cells in which the deregulation of β-catenin signaling is either well established (i.e. in HCT116 and SW480) or not known (i.e. in HOS and HTB-94). We sought to test the effect of STI-571 on β-catenin signaling in the cells without any possible alterations in the Wnt/β-catenin signaling pathway. Although HEK 293 cells are transformed human embryonic kidney cells, we have demonstrated that the β-catenin signaling pathway is intact in this cell line [44,45]. Using Wnt1-conditioned medium, we were able to reconstitute the activation of β-catenin signaling in HEK 293 cells. As shown in Fig. 2C, Wnt1-CM-induced β-catenin activity was inhibited by approximately 70 and 85% in the presence of 15 and 20 μM of STI-571, respectively. These results confirm that STI-571 can effectively inhibit the Wnt/β-catenin signaling pathway.
3.3. Proliferation of human cancer lines is inhibited by STI-571
We tested whether STI-571-mediated inhibition of β-catenin/TCF4 activity could affect the proliferation of human cancer cells. It has been well established that the activation of β-catenin signaling is a critical early step for colon cancer development. We were interested in testing whether the growth of colon cancer cells could be significantly inhibited by STI-571. Experimentally, subconfluent HCT116 and SW480 cells were treated with different concentrations (0–30 μM) of STI-571. Viable cells were counted at 24, 48, 72, and 96 h after treatment. As shown in Fig. 3, cell proliferation was, in general, inhibited by STI-571 in a time- and dose-dependent manner. For instance, at 30 μM of STI-571 the number of viable HCT116 cells decreased to approximately 36, 21, and 7% of that of the controls at 48, 72, and 96 h post treatment (Fig. 3A). Similar growth inhibition by STI-571 (especially at 30 μM) was observed in SW480 cells (Fig. 3B). Interestingly, we observed a similar growth inhibitory effect of STI-571 on HTB-94 and HOS cell lines (data not shown). These results suggest that inhibition of β-catenin/TCF4 activity may be, at least in part, responsible for the STI-571-mediated growth suppression of human tumor cells.
Fig. 3.
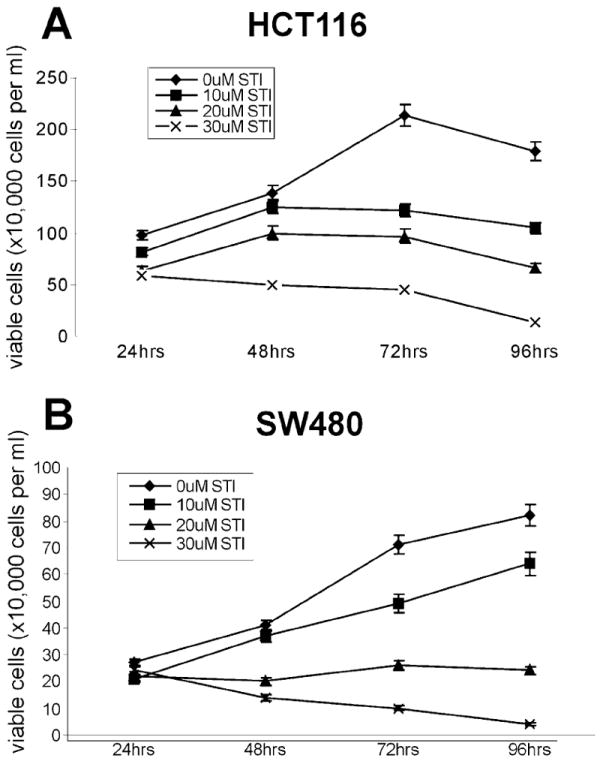
STI-571 inhibits proliferation of human colon cancer cells. Exponentially growing HCT116 (A) and SW480 (B) cells were treated with STI-571 at the indicated concentrations. At 24, 48, 72, and 96 h after treatment, cells were collected by trypsinization. Viable cells were counted after Trypan Blue staining. Each assay condition was done in triplicate.
3.4. Wnt1-mediated heterologous transactivation in 293-GEM cells is inhibited by STI-571
Finally, we sought to test whether STI-571 could effectively inhibit the Wnt1-mediated heterologous transactivation using our previously established functional assay system of β-catenin signaling [42]. This system consisted of a chimeric transcription factor (by fusing Gal4-DNA binding domain with full-length β-catenin) and a reporter (expressing GFP, driven by a Gal4-responsive promoter), both of which were stably introduced into HEK 293 cells (designated as 293-GEM cells) and were shown to be tightly regulated by Wnt signal [42]. As shown in Fig. 4A, GFP expression in 293-GEM cells were readily detectable at as early as 24 h after stimulation with the Wnt1-conditioned medium. However, the Wnt1-CM stimulated GFP expression was significantly inhibited in the presence of 10 and 20 μM of STI-571 (Fig. 4B). These results corroborate well with the findings using the TCF4 reporter (Fig. 2C), strongly suggesting that STI-571 can effectively inhibit the β-catenin signaling activity. With activation of β-catenin, signaling has been reported in a broad range of human tumors, our encouraging findings may merit further pre-clinical and clinical investigations on the use of STI-571 as a (at least adjuvant) chemotherapeutic agent for human cancer.
Fig. 4.
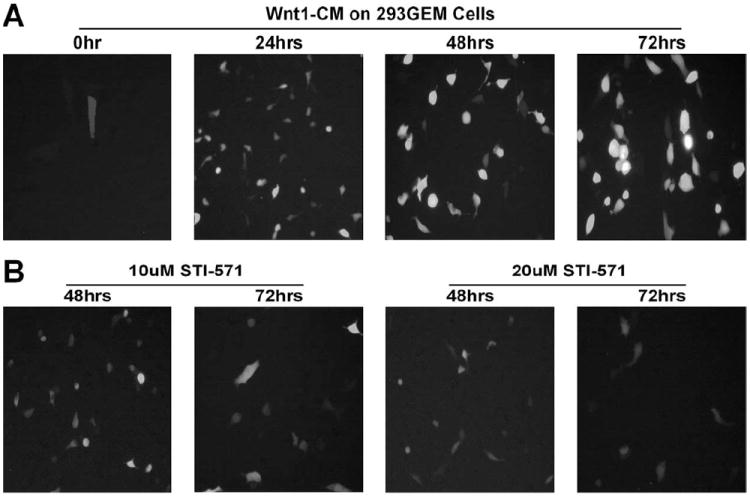
STI-571 inhibits Wnt1-induced β-catenin activation in 293-GEM cells. Exponentially growing 293-GEM cells were exposed to Wnt1-conditioned medium (Wnt1-CM) in the absence (A) or presence (B) of STI-571 (at 10 or 20 μM). Signal intensity of green fluorescent protein was recorded using fluorescent microscopy at the indicated time after treatment.
4. Discussion
In this study, we sought to test whether tyrosine kinase inhibitor STI-571 could inhibit β-catenin signaling activity, and hence suppress cell proliferation. Using a β-catenin/TCF4-responsive reporter, we demonstrated that STI-571 could effectively inhibit the constitutive activity of β-catenin signaling in human colon cancer cell lines (i.e. HCT116 and SW480), as well as the Wnt1-induced activation of β-catenin signaling in HOS, HTB-94, and HEK 293 cells. Furthermore, using a concentration range comparable with several published studies [46-49], we presented evidence that STI-571 could effectively suppress the proliferation of human colon cancer cell lines HCT116 and SW480. Finally, we demonstrated that STI-571 could effectively inhibit the Wnt1-mediated activation of a GAL4-β-catenin heterologous transcription system, namely the 293-GEM cells [42]. These findings suggest that tyrosine phosphorylation may play an important role in regulating β-catenin signaling activity.
Recent studies suggest that deregulation of β-catenin signaling may play an important role in human tumorigenesis [1,3]. With the exception of colon cancer, mechanisms underlying β-catenin deregulation are largely unknown in the vast majority of human tumors [12]. Several studies indicate that β-catenin can become a tyrosine-phosphorylated protein upon the activation of several protein tyrosine kinases, and β-catenin is an excellent substrate for protein tyrosine kinases, such as the Src kinase [50,51]. In fact, β-catenin is a major tyrosine-phosphorylated protein during mouse oocyte maturation and pre-implantation [52]. It has been reported that up-regulation of β-catenin tyrosine phosphorylation was more frequently observed in colon cancerous tissues than in the matching normal mucosa [53]. Several studies demonstrated that epidermal growth factor (EGF) is able to induce tyrosine phosphorylation of β-catenin, and in many cases, β-catenin is shown to associate with EGF receptor/c-erbB-2 gene product [54-62]. It has also been extensively documented that β-catenin protein becomes tyrosine-phosphorylated upon stimulation with hepatocyte growth factor (HGF)/scatter factor (SF) [31-33,63-64]. Moreover, β-catenin has been found in the complex containing HGF/SF receptor c-MET [63,65]. A recent study has demonstrated that oncogenic mutants of MET receptor tyrosine kinase can effectively activate the β-catenin signaling pathway. Other factors, including vascular endothelial growth factor (VEGF) [34-36], insulin-like growth factor 1 (IGF-1) [37,38], IGF-2 [39], and Gas6 (the ligand for the Axl RTK family) [40], have been shown to stimulate the tyrosine phosphorylation of β-catenin protein and to activate β-catenin signaling. Conversely, several forms of protein tyrosine phosphatases (PTPs), such as PTPμ and receptor PTPβ/ξ, have been shown to associate with β-catenin complex and to modulate tyrosine phosphorylation of the β-catenin protein [66-68]. The potential biological importance of the tyrosine-phosphorylated β-catenin remains to be defined. The current prevailing viewpoint is that tyrosine phosphorylation of β-catenin protein would result in its decreased association with E-cadherin, leading to the disruption of junctional assembly and an increased propensity for cancer cells to become invasive and metastasize [50,51,69]. However, growth factors that activate receptor protein tyrosine kinases have also been shown to stabilize cytoplasmic β-catenin protein and to activate downstream targets of the β-catenin signaling pathway [31-40], suggesting that tyrosine phosphorylation plays a role not only in regulating β-catenin’s involvement in cell adhesion but also β-catenin signaling activity. Interestingly, it has been recently reported that phosphorylation of Tyr-654 at the C-terminal portion of β-catenin decreases its binding to E-cadherin and also stimulates association of β-catenin with the basal transcription machinery [70,71]. Thus, it would be interesting to elucidate the nodal point in which β-catenin/TCF4 signaling activity is regulated by tyrosine phosphorylation.
The introduction of STI-571 as an agent targeting the causative molecular event in CML has been heralded as a major advance in the treatment of cancer [22-24,72]. Although originally developed as an inhibitor of constitutively active Bcr-abl kinase, STI-571 is also able to inhibit Abl kinase, c-kit tyrosine kinase, and the platelet-derived growth factor (PDGF) receptor kinases [22,24]. It is conceivable that other tyrosine kinases may also serve as possible cellular targets of STI-571, especially when higher concentrations of STI-571 are used. Given the fact that deregulation of β-catenin is a frequent event in many types of human cancer [3], targeted inhibition of β-catenin signaling may represent an important alternative for the treatment of human cancer. Our results suggest that STI-571 could be used as a potential inhibitory agent of β-catenin signaling activity. Our findings are consistent with an early study, in which the combination of sulindac (a non-steroidal anti-inflammatory drug with established chemopreventive activity in colon cancer) and EKI-569 (an irreversible inhibitor of EGF receptor kinase) exhibited a pronounced chemopreventive effect in APC/Min−/+ mice, a murine model of human familial adenomatous polyposis [73]. Furthermore, while we were preparing this report, Attoub et al. demonstrated that STI-571 was capable of inhibiting the proliferation of human colon cancer cell lines [49]. Thus, our findings corroborated well with these experimental results, and yet may provide a possible mechanistic explanation to the above observations. Taken together, our results suggest that tyrosine phosphorylation may play an important role in regulating β-catenin signaling activity, and inhibition of this signaling pathway by STI-571 may be further explored as an important alternative/adjuvant treatment of human cancer.
Acknowledgments
The authors are grateful for Novartis Pharmaceuticals AG, Basel, Switzerland for providing STI-571/Gleveec. The authors wish to thank Bert Vogelstein of Johns Hopkins for providing the GAL4-responsive GFP reporter, and HCT116 and SW480 cell lines. The reported work was supported in part by research grants from the Brinson Foundation and the Schweppe Foundation.
References
- 1.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 2.Behrens J. Control of β-catenin signaling in tumor development. Ann N Y Acad Sci. 2000;910:21–33. doi: 10.1111/j.1749-6632.2000.tb06698.x. [DOI] [PubMed] [Google Scholar]
- 3.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- 4.Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev. 1997;11:3286–3305. doi: 10.1101/gad.11.24.3286. [DOI] [PubMed] [Google Scholar]
- 5.Miller JR. The Wnts. Genome Biol. 2002;3 doi: 10.1186/gb-2001-3-1-reviews3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He T-C, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 7.Tetsu O, McCormick F. β-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 8.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, et al. The cyclin D1 gene is a target of the β-catenin/LEF-1 pathway. Proc Natl Acad Sci USA. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smalley MJ, Dale TC. Wnt signalling in mammalian development and cancer. Cancer Metastasis Rev. 1999;18:215–230. doi: 10.1023/a:1006369223282. [DOI] [PubMed] [Google Scholar]
- 10.van Noort M, Clevers H. TCF transcription factors, mediators of Wnt-signaling in development and cancer. Dev Biol. 2002;244:1–8. doi: 10.1006/dbio.2001.0566. [DOI] [PubMed] [Google Scholar]
- 11.Moon RT, Bowerman B, Boutros M, Perrimon N. The promise and perils of Wnt signaling through β-catenin. Science. 2002;296:1644–1646. doi: 10.1126/science.1071549. [DOI] [PubMed] [Google Scholar]
- 12.Polakis P. The oncogenic activation of β-catenin. Curr Opin Genet Dev. 1999;9:15–21. doi: 10.1016/s0959-437x(99)80003-3. [DOI] [PubMed] [Google Scholar]
- 13.Peifer M, Polakis P. Wnt signaling in oncogenesis and embryogenesis—a look outside the nucleus. Science. 2000;287:1606–1609. doi: 10.1126/science.287.5458.1606. [DOI] [PubMed] [Google Scholar]
- 14.Gat U, DasGupta R, Degenstein L, Fuchs E. De Novo hair follicle morphogenesis and hair tumors in mice expressing a truncated β-catenin in skin. Cell. 1998;95:605–614. doi: 10.1016/s0092-8674(00)81631-1. [DOI] [PubMed] [Google Scholar]
- 15.Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, et al. Intestinal polyposis in mice with a dominant stable mutation of the β-catenin gene. EMBO J. 1999;18:5931–5942. doi: 10.1093/emboj/18.21.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, et al. Somatic mutations of the β-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci USA. 1998;95:8847–8851. doi: 10.1073/pnas.95.15.8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsujiuchi T, Tsutsumi M, Sasaki Y, Murata N, Konishi Y. Mutations of adenomatous polyposis coli and β-catenin genes during progression of lung tumors induced by N-nitrosobis(2-hydroxypropyl) amine in rats. Cancer Res. 2000;60:6611–6616. [PubMed] [Google Scholar]
- 18.Pawson T, Hunter T. Signal transduction and growth control in normal and cancer cells. Curr Opin Genet Dev. 1994;4:1–4. doi: 10.1016/0959-437x(94)90084-1. [DOI] [PubMed] [Google Scholar]
- 19.Blume-Jensen P, Hunter T. Oncogenic kinase signaling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 20.Sawyers CL. Rational therapeutic intervention in cancer: kinases as drug targets. Curr Opin Genet Dev. 2002;12:111–115. doi: 10.1016/s0959-437x(01)00273-8. [DOI] [PubMed] [Google Scholar]
- 21.Cohen P. Protein kinases—the major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002;1:309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 22.Shawver LK, Slamon D, Ullrich A. Smart drugs: tyrosine kinase inhibitors in cancer therapy. Cancer Cell. 2002;1:117–123. doi: 10.1016/s1535-6108(02)00039-9. [DOI] [PubMed] [Google Scholar]
- 23.Druker BJ, Lydon NB. Lessons learned from the development of an abl tyrosine kinase inhibitor for chronic myelogenous leukemia. J Clin Invest. 2000;105:3–7. doi: 10.1172/JCI9083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sawyers CL. Disabling Abl-perspectives on Abl kinase regulation and cancer therapeutics. Cancer Cell. 2002;1:13–15. doi: 10.1016/s1535-6108(02)00022-3. [DOI] [PubMed] [Google Scholar]
- 25.Druker BJ. Perspectives on the development of a molecularly targeted agent. Cancer Cell. 2002;1:31–36. doi: 10.1016/s1535-6108(02)00025-9. [DOI] [PubMed] [Google Scholar]
- 26.Apperley JF, Gardembas M, Melo JV, Russell-Jones R, Bain BJ, Baxter EJ, et al. Response to imatinib mesylate in patients with chronic myeloproliferative diseases with rearrangements of the platelet-derived growth factor receptor beta. N Engl J Med. 2002;347:481–487. doi: 10.1056/NEJMoa020150. [DOI] [PubMed] [Google Scholar]
- 27.Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 28.Simon MP, Pedeutour F, Sirvent N, Grosgeorge J, Minoletti F, Coindre JM, et al. Deregulation of the platelet-derived growth factor B-chain gene via fusion with collagen gene COL1A1 in dermatofibrosarcoma protuberans and giant-cell fibroblastoma. Nat Genet. 1997;15:95–98. doi: 10.1038/ng0197-95. [DOI] [PubMed] [Google Scholar]
- 29.Rubin BP, Schuetze SM, Eary JF, Norwood TH, Mirza S, Conrad EU, et al. Molecular targeting of platelet-derived growth factor B by imatinib mesylate in a patient with metastatic dermatofibrosarcoma protuberans. J Clin Oncol. 2002;20:3586–3591. doi: 10.1200/JCO.2002.01.027. [DOI] [PubMed] [Google Scholar]
- 30.Morin PJ. β-Catenin signaling and cancer. Bioessays. 1999;21:1021–1030. doi: 10.1002/(SICI)1521-1878(199912)22:1<1021::AID-BIES6>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 31.Papkoff J, Aikawa M. WNT-1 and HGF regulate GSK3β activity and β-catenin signaling in mammary epithelial cells. Biochem Biophys Res Commun. 1998;247:851–858. doi: 10.1006/bbrc.1998.8888. [DOI] [PubMed] [Google Scholar]
- 32.Hiscox S, Jiang WG. Hepatocyte growth factor/scatter factor disrupts epithelial tumour cell- cell adhesion: involvement of β-catenin. Anticancer Res. 1999;19:509–517. [PubMed] [Google Scholar]
- 33.Danilkovitch-Miagkova A, Miagkov A, Skeel A, Nakaigawa N, Zbar B, Leonard EJ. Oncogenic mutants of RON and MET receptor tyrosine kinases cause activation of the β-catenin pathway. Mol Cell Biol. 2001;21:5857–5868. doi: 10.1128/MCB.21.17.5857-5868.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F, et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 1999;98:147–157. doi: 10.1016/s0092-8674(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 35.Ilan N, Mahooti S, Rimm DL, Madri JA. PECAM-1 (CD31) functions as a reservoir for and a modulator of tyrosine- phosphorylated β-catenin. J Cell Sci. 1999;112(18):3005–3014. doi: 10.1242/jcs.112.18.3005. [DOI] [PubMed] [Google Scholar]
- 36.Cohen AW, Carbajal JM, Schaeffer RC., Jr VEGF stimulates tyrosine phosphorylation of β-catenin and small-pore endothelial barrier dysfunction. Am J Physiol. 1999;277:H2038–H2049. doi: 10.1152/ajpheart.1999.277.5.H2038. [DOI] [PubMed] [Google Scholar]
- 37.Playford MP, Bicknell D, Bodmer WF, Macaulay VM. Insulin-like growth factor 1 regulates the location, stability, and transcriptional activity of β-catenin. Proc Natl Acad Sci USA. 2000;97:12103–12108. doi: 10.1073/pnas.210394297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Satyamoorthy K, Li G, Vaidya B, Patel D, Herlyn M. Insulin-like growth factor-1 induces survival and growth of biologically early melanoma cells through both the mitogen-activated protein kinase and β-catenin pathways. Cancer Res. 2001;61:7318–7324. [PubMed] [Google Scholar]
- 39.Morali OG, Delmas V, Moore R, Jeanney C, Thiery JP, Larue L. IGF-II induces rapid β-catenin relocation to the nucleus during epithelium to mesenchyme transition. Oncogene. 2001;20:4942–4950. doi: 10.1038/sj.onc.1204660. [DOI] [PubMed] [Google Scholar]
- 40.Goruppi S, Chiaruttini C, Ruaro ME, Varnum B, Schneider C. Gas6 induces growth, β-catenin stabilization, and T-cell factor transcriptional activation in contact-inhibited C57 mammary cells. Mol Cell Biol. 2001;21:902–915. doi: 10.1128/MCB.21.3.902-915.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He T-C, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou L, An N, Jiang W, Haydon RC, Cheng H, Zhou Q, et al. Fluorescence-based functional assay for Wnt/β-catenin signaling activity. BioTechniques. 2002;33:1126–1138. doi: 10.2144/02335dd07. [DOI] [PubMed] [Google Scholar]
- 43.Da Costa LT, Jen J, He T-C, Chan TA, Kinzler KW, Vogelstein B. Converting cancer genes into killer genes. Proc Natl Acad Sci USA. 1996;93:4192–4196. doi: 10.1073/pnas.93.9.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, et al. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 45.He T-C, Chan TA, Vogelstein B, Kinzler KW. PPARδ is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell. 1999;99:335–345. doi: 10.1016/s0092-8674(00)81664-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tuveson DA, Willis NA, Jacks T, Griffin JD, Singer S, Fletcher CD. STI571 inactivation of the gastrointestinal stromal tumor c-KIT oncoprotein: biological and clinical implications. Oncogene. 2001;20:5054–5058. doi: 10.1038/sj.onc.1204704. [DOI] [PubMed] [Google Scholar]
- 47.Joensuu H, Dimitrijevic S. Tyrosine kinase inhibitor imatinib (STI571) as an anticancer agent for solid tumours. Ann Med. 2001;33:451–455. doi: 10.3109/07853890109002093. [DOI] [PubMed] [Google Scholar]
- 48.Sjoblom T, Shimizu A, O’Brien KP, Pietras K, DalCin P, Buchdunger E. Growth inhibition of dermatofibrosarcoma protuberans tumors by the platelet-derived growth factor receptor antagonist STI571 through induction of apoptosis. Cancer Res. 2001;61:5778–5783. [PubMed] [Google Scholar]
- 49.Attoub S, Rivat C, Rodrigues S, Van Bocxlaer S, Bedin M, Bruyneel E, et al. The c-kit tyrosine kinase inhibitor STI571 for colorectal cancer therapy. Cancer Res. 2002;62:4879–4883. [PubMed] [Google Scholar]
- 50.Hinck L, Nathke IS, Papkoff J, Nelson WJ. β-Catenin: a common target for the regulation of cell adhesion by Wnt-1 and Src signaling pathways. Trends Biochem Sci. 1994;19:538–542. doi: 10.1016/0968-0004(94)90057-4. [DOI] [PubMed] [Google Scholar]
- 51.Daniel JM, Reynolds AB. Tyrosine phosphorylation and cadherin/catenin function. Bioessays. 1997;19:883–891. doi: 10.1002/bies.950191008. [DOI] [PubMed] [Google Scholar]
- 52.Ohsugi M, Butz S, Kemler R. β-Catenin is a major tyrosine-phosphorylated protein during mouse oocyte maturation and preimplantation development. Dev Dyn. 1999;216:168–176. doi: 10.1002/(SICI)1097-0177(199910)216:2<168::AID-DVDY7>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 53.Takayama T, Shiozaki H, Doki Y, Oka H, Inoue M, Yamamoto M, et al. Aberrant expression and phosphorylation of β-catenin in human colorectal cancer. Br J Cancer. 1998;77:605–613. doi: 10.1038/bjc.1998.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoschuetzky H, Aberle H, Kemler R. β-Catenin mediates the interaction of the cadherin-catenin complex with epidermal growth factor receptor. J Cell Biol. 1994;127:1375–1380. doi: 10.1083/jcb.127.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shibamoto S, Hayakawa M, Takeuchi K, Hori T, Oku N, Miyazawa K, et al. Tyrosine phosphorylation of β-catenin and plakoglobin enhanced by hepatocyte growth factor and epidermal growth factor in human carcinoma cells. Cell Adhes Commun. 1994;1:295–305. doi: 10.3109/15419069409097261. [DOI] [PubMed] [Google Scholar]
- 56.Ochiai A, Akimoto S, Kanai Y, Shibata T, Oyama T, Hirohashi S. c-erbB-2 gene product associates with catenins in human cancer cells. Biochem Biophys Res Commun. 1994;205:73–78. doi: 10.1006/bbrc.1994.2631. [DOI] [PubMed] [Google Scholar]
- 57.Kanai Y, Ochiai A, Shibata T, Oyama T, Ushijima S, Akimoto S, et al. c-erbB-2 gene product directly associates with β-catenin and plakoglobin. Biochem Biophys Res Commun. 1995;208:1067–1072. doi: 10.1006/bbrc.1995.1443. [DOI] [PubMed] [Google Scholar]
- 58.Shibata T, Ochiai A, Kanai Y, Akimoto S, Gotoh M, Yasui N, et al. Dominant negative inhibition of the association between β-catenin and c-erbB-2 by N-terminally deleted β-catenin suppresses the invasion and metastasis of cancer cells. Oncogene. 1996;13:883–889. [PubMed] [Google Scholar]
- 59.Fujii K, Furukawa F, Matsuyoshi N. Ligand activation of overexpressed epidermal growth factor receptor results in colony dissociation and disturbed E-cadherin function in HSC-1 human cutaneous squamous carcinoma cells. Exp Cell Res. 1996;223:50–62. doi: 10.1006/excr.1996.0057. [DOI] [PubMed] [Google Scholar]
- 60.Takahashi K, Suzuki K, Tsukatani Y. Induction of tyrosine phosphorylation and association of β-catenin with EGF receptor upon tryptic digestion of quiescent cells at confluence. Oncogene. 1997;15:71–78. doi: 10.1038/sj.onc.1201160. [DOI] [PubMed] [Google Scholar]
- 61.Bonvini P, An WG, Rosolen A, Nguyen P, Trepel J, Garcia de Herreros A, et al. Geldanamycin abrogates ErbB2 association with proteasome-resistant β-catenin in melanoma cells, increases β-catenin-E-cadherin association, and decreases β-catenin-sensitive transcription. Cancer Res. 2001;61:1671–1677. [PubMed] [Google Scholar]
- 62.Schroeder JA, Adriance MC, McConnell EJ, Thompson MC, Pockaj B, Gendler SJ. ErbB-β-catenin complexes are associated with human infiltrating ductal breast and murine mammary tumor virus (MMTV) -Wnt-1 and MMTV-c-Neu transgenic carcinomas. J Biol Chem. 2002;277:22692–22698. doi: 10.1074/jbc.M201975200. [DOI] [PubMed] [Google Scholar]
- 63.Hiscox S, Jiang WG. Association of the HGF/SF receptor, c-met, with the cell-surface adhesion molecule, E-cadherin, and catenins in human tumor cells. Biochem Biophys Res Commun. 1999;261:406–411. doi: 10.1006/bbrc.1999.1002. [DOI] [PubMed] [Google Scholar]
- 64.Nakopoulou L, Gakiopoulou H, Keramopoulos A, Giannopoulou I, Athanassiadou P, Mavrommatis J, et al. c-met tyrosine kinase receptor expression is associated with abnormal β-catenin expression and favourable prognostic factors in invasive breast carcinoma. Histopathology. 2000;36:313–325. doi: 10.1046/j.1365-2559.2000.00847.x. [DOI] [PubMed] [Google Scholar]
- 65.Davies G, Jiang WG, Mason MD. HGF/SF modifies the interaction between its receptor c-Met, and the E-cadherin/catenin complex in prostate cancer cells. Int J Mol Med. 2001;7:385–388. doi: 10.3892/ijmm.7.4.385. [DOI] [PubMed] [Google Scholar]
- 66.Brady-Kalnay SM, Rimm DL, Tonks NK. Receptor protein tyrosine phosphatase PTPmu associates with cadherins and catenins in vivo. J Cell Biol. 1995;130:977–986. doi: 10.1083/jcb.130.4.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Muller T, Choidas A, Reichmann E, Ullrich A. Phosphorylation and free pool of β-catenin are regulated by tyrosine kinases and tyrosine phosphatases during epithelial cell migration. J Biol Chem. 1999;274:10173–10183. doi: 10.1074/jbc.274.15.10173. [DOI] [PubMed] [Google Scholar]
- 68.Meng K, Rodriguez-Pena A, Dimitrov T, Chen W, Yamin M, Noda M, et al. Pleiotrophin signals increased tyrosine phosphorylation of β-catenin through inactivation of the intrinsic catalytic activity of the receptor-type protein tyrosine phosphatase β/ξ. Proc Natl Acad Sci USA. 2000;97:2603–2608. doi: 10.1073/pnas.020487997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Behrens J, Vakaet L, Friis R, Winterhager E, Van Roy F, Mareel MM, et al. Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/β-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J Cell Biol. 1993;120:757–766. doi: 10.1083/jcb.120.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roura S, Miravet S, Piedra J, Garcia de Herreros A, Dunach M. Regulation of E-cadherin/catenin association by tyrosine phosphorylation. J Biol Chem. 1999;274:36734–36740. doi: 10.1074/jbc.274.51.36734. [DOI] [PubMed] [Google Scholar]
- 71.Piedra J, Martinez D, Castano J, Miravet S, Dunach M, de Herreros AG. Regulation of β-catenin structure and activity by tyrosine phosphorylation. J Biol Chem. 2001;276:20436–20443. doi: 10.1074/jbc.M100194200. [DOI] [PubMed] [Google Scholar]
- 72.O’Dwyer ME, Druker BJ. The role of the tyrosine kinase inhibitor STI571 in the treatment of cancer. Curr Cancer Drug Targets. 2001;1:49–57. doi: 10.2174/1568009013334250. [DOI] [PubMed] [Google Scholar]
- 73.Torrance CJ, Jackson PE, Montgomery E, Kinzler KW, Vogelstein B, Wissner A, et al. Combinatorial chemo-prevention of intestinal neoplasia. Nat Med. 2000;6:1024–1028. doi: 10.1038/79534. [DOI] [PubMed] [Google Scholar]