

Key Clinical Message
Prolonged clotting times were observed in a patient with spontaneous hemorrhage. Analysis showed severe factor X deficiency due to clearance by a noninhibitory antibody. Lymphadenopathy identified on imaging led to diagnosis of marginal B-cell lymphoma. Treatment of lymphoma with rituximab and chlorambucil resulted in complete disappearance of the bleeding disorder.
Keywords: acquired coagulation disorders, clotting factor related research, non-Hodgkin lymphoma
Introduction
Factor X, also known as Stuart-Prower factor, is a vitamin K-dependent serine protease produced in the liver. It is a key component of both intrinsic and extrinsic clotting pathways. Activated factor X cleaves prothrombin to thrombin, which facilitates the conversion of fibrinogen to fibrin which ultimately leads to the formation of a fibrin clot 1. Acquired factor X deficiency is commonly caused by the use of coumarin derivatives or due to liver disease. Both causes result in a depletion of all vitamin K-dependent coagulation factors. Liver disease additionally shows a decrease in some vitamin K-independent coagulation factors. Isolated factor X deficiency is a rare disorder and was first described in the early 1950s 2,3. Genetic factor X deficiencies are inherited in an autosomal recessive manner. Only a few families have been described, probably due to factor X’s essential role in the hemostasis 4,5. Symptoms vary from mild hemorrhage, such as bruising and mucocutaneous bleeding, to more severe bleeding, for example, gastrointestinal bleeding and muscle bleeding. Bleeding severity depends on the plasma concentration of factor X. Values between 1 and 10% are associated with minor bleeding or more serious bleeding provoked by surgical procedures or trauma, while spontaneous and severe bleeding is described with values below 1% 1,6.
Acquired isolated factor X deficiency is mostly associated with amyloid light-chain (AL)-amyloidosis 7–13. In amyloidosis, factor X binds permanently to amyloid fibrils within the vasculature, liver and spleen and is thereby scavenged from blood circulation 14. There are also a few reports of factor X deficiency concomitant with respiratory disease, antibiotic treatment, leprosy, burns or malignancy such as acute myeloid leukemia 15–28. In some cases an inhibitor was found 18,20,22–24. Rao et al. (1994), proposed that in the context of post-infectious disease, similar epitopes may be found on viral antigens leading to generation of an anti-factor X inhibitor 24. In several other cases the etiology of the acquired factor X deficiency remains unclear 17,19,21,23,27,28
In addition to earlier reports, we describe an 81-year-old patient without a bleeding history who presented with moderate to serious spontaneous bleeding symptoms due to clearance of factor X by a noninhibitory antibody. This antibody was related with an indolent malignant lymphoma. His bleeding disorder completely disappeared upon treatment of the underlying disease.
Methods
Overall standard laboratory methods were used. Coagulation assays were performed with patient plasma collected in 3.2% sodium citrate. Plasma for further testing was stored at −80°C. The prothrombin time was measured using Innovin reagent (Siemens Healthcare, Erlangen Germany) and the aPTT using Actin FSL reagent (Siemens Healthcare).
Mixing studies were done by mixing equal volumes of patients plasma and normal plasma pools (control N, Siemens Healthcare and in-house prepared pool). The aPTT was measured immediately and after 2 h of incubation at 37°C to detect a time dependent inhibitor. Heparin was neutralized in 1 mL of plasma by addition of heparinase I using Dade Hepzyme (Siemens Healthcare) according to manufacturer’s instructions. All factor assays were performed using one stage assay and factor X antigen level was determined using enzyme linked immunoassay (ELISA). For lupus anticoagulant testing a diluted Russell’s Viper Venom Time (La screen/mixing reagent, Gradipore Hawthorne NY) and a lupus sensitive aPTT (BioMerieux, Marcy l’Etoile France) were used.
Anti-factor X Bethesda assay
The Bethesda inhibitor assay for detecting inhibitory antibodies directed toward factor X was done according to the Classical Bethesda Assay described for factor VIII 29. In-house prepared pooled plasma was used as normal plasma source (a pool of more than 32 adult donors). Factor X activity was measured using the previously mentioned one stage assay.
Anti-factor X radioimmunoassay
Radioimmunoassay (RIA) for detecting factor X antibody was performed according to Wolbink et al. 30. In short, 1 mg Sepharose-immobilized protein A beads (GE Healthcare, Little Chalfont, Buckinghamshire, UK) were incubated overnight with 50 μL prediluted in phosphate-buffered saline (PBS)/0.3% bovine serum albumin (BSA; EMD Millipore, Billerica, MA) plasma of the patient, polyclonal rabbit antihuman factor X antibody or plasma from a healthy control in a total volume of 800 μL PBS-AT (PBS containing 0.3% BSA, 0.2% Tween-20 (Merck, Darmstadt, Germany) and 0.01 mol/L ethylenediamine tetraacetic acid (EDTA), thereby coupling all antibodies to the Sepharose beads. The beads were subsequently washed two times with PBS-T (PBS containing 0.005% Tween-20) and four times with tris-buffered saline (TBS)-CT (10 mmol/L Tris, 5 mmol/L CaCl2, 140 mmol/L NaCl, pH7,4 with 0.05% Tween-20). Beads were resuspended in 0.5 mL TBS-C-AT (TBS-C containing NaN3, BSA and 0.2% Tween-20). Thereafter beads were resuspended in 0.5 mL TBS-C-AT and 2 ng of biotin labeled factor X (provided kindly by M. Boon-Spijker, dept. Plasma Protein, Sanquin Research, Amsterdam) was added to the samples followed by overnight incubation and rotation using a rotor. Thereafter the beads were washed five times with TBS-CT and incubated overnight with 50 μL radioactive 125I labeled streptavidin dialyzed in TBS-CA and diluted in 500 μL TBS-C-AT. Unbound 125I was removed by washing five times with TBS-CT and samples were counted using a Wallac 1260 Multigamma II counter. Results were expressed as percentage binding of the labeled factor X.
Case history and results
An 81-year-old previously healthy Caucasian man presented on the emergency ward with a spontaneous hematoma of the skin and soft tissue in his left abdominal flank and around his left shoulder. Two weeks prior to admission, he had received a red blood cell concentrate transfusion after an episode of acute anemia caused by bleeding due to a minor trauma. Previous to this presentation, there was no history of an abnormal bleeding tendency. Complaints consisting of tiredness and slight weight loss in the past month were reported upon admission. He used hydrochlorothiazide, atenolol and lercanidipine to control hypertension. He did not use any anti-platelet drugs or anticoagulants, which was confirmed by toxicology screening.
At presentation, his vital signs were normal. Examination of the oral cavity did not show any mucosal bleeding or gingival abnormalities. The results of cardiovascular, pulmonary and neurological examinations were normal. On the left side of his abdomen there was a painful hematoma of approximately 12 × 8 inches. A painless swelling of about 8 × 8 inches was present on his left shoulder region. Computed tomography (CT) of thorax and abdomen showed intramuscular hematoma of the latissimus dorsi on the left side and enlarged inguinal, intra-abdominal and intrathoracal lymph nodes. Hepatosplenomegaly was not present.
Initial laboratory evaluation revealed anemia with a hemoglobin level of 7.1 g/dL (12.9–16.9 g/dL), with normal red blood cell indices. His white blood cell and platelet counts were normal. Plasma electrolytes and liver enzymes were within the normal ranges. However, lactate dehydrogenase (LD) was slightly elevated: 325 U/l (<248 U/l). Further analysis showed neither serum M-protein by electrophoresis nor elevation of serum-free light chains. Urine analysis was normal, including absence of Bence-Jones proteins.
Both, prothrombin time (PT) and activated partial thromboplastin time (aPTT) were prolonged 23.3 sec (PT normal range 9–12 sec) and 51 sec (aPTT normal range 24–34 sec), respectively. Fibrinogen levels were normal.
Pre-analytical contamination of the sample with heparin was ruled out using Hepzyme treatment. The in vitro addition of normal plasma to the patients plasma resulted in complete correction of PT and aPTT times, immediately after mixing as well as after 2 h of incubation at 37°C. This result is suggestive of a factor deficiency rather than an antibody neutralizing procoagulant function.
In the acute clinical situation the cause of the bleeding tendency was unknown. The patient initially received three red blood cell concentrates resulting in an increase of his hemoglobin level to 9.3 g/dL. Treatment with vitamin K, prednisone, fresh frozen plasma (FFP) and prothrombin complex concentrate (PCC) was started, which resulted in a temporary and partial response on the clotting times (Fig.1A). More detailed analysis of blood coagulation factor levels showed slightly reduced activity of factor II (54%), VII (52%), IX (56%) and XII (64%) and extremely low activity of factor X (3–4%) (Table1). The factor X activity was measured both via the intrinsic and extrinsic pathway. Additionally, a markedly reduced factor X concentration was found with the antigen assay correlating with the functional deficiency.
Figure 1.
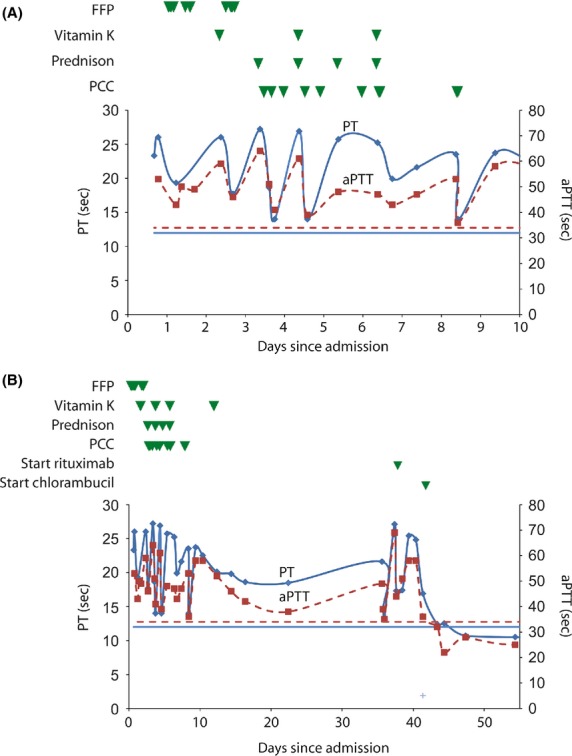
Clotting times in patient plasma in relation to treatment. (A) Effect on activated partial thromboplastin time (aPTT, dashed lines) and prothrombin time (PT, solid lines) in response to fresh frozen plasma (FFP), vitamin K and prothrombin complex concentrate (PCC) in first 10 days of hospitalization of acute setting. (B) Response during rituximab and chlorambucil treatment. Each green triangle represents one unit of FFP, 5 mg of vitamin K, 80 mg prednisone or 500 U PCC. Horizontal lines represent the upper limit of normal.
Table 1.
Blood coagulation parameters at initial laboratory screening and before treatment
| Parameter | Value | Normal range |
|---|---|---|
| PT | 23.3 | 9–12 sec |
| aPTT | 51 | 24–34 sec |
| PT mixing (1:1 normal plasma) | 11.8 | |
| PT mixing after 2 h incubation at 37°C | 11.8 | |
| aPTT mixing (1:1 normal plasma) | 29.6 | |
| aPTT mixing after 2 h incubation at 37°C | 30.4 | |
| Fibrinogen | 2.5 | 1.8–3.6 g/L |
| Factor II | 54 | 80–120% |
| Factor V | 94 | 70–140% |
| Factor VII | 52 | 65–150% |
| Factor VIII | 200 | 70–140% |
| Factor IX | 56 | 70–140% |
| Factor X (extrinsic) | 4 | 80–120% |
| Factor X (intrinsic) | 3 | 80–120% |
| Factor X antigen | 7 | 80–120% |
| Factor XI | 95 | 80–120% |
| Factor XII | 64 | 80–120% |
| Factor X inhibitor | <0.4 | <0.4 BU |
| Lupus anticoagulants | not detected | |
| D-dimer | 1042 | <500 μg/L |
PT, prothrombin time; aPTT, activated partial thromboplastin time.
The factor X deficiency in our patient was considered to be acquired, given the absence of a bleeding history. In an attempt to clarify the cause of the acquired factor X deficiency, the patient underwent a biopsy of an enlarged inguinal lymph node. The results revealed the diagnosis, a marginal-zone B-cell lymphoma. There were no amyloid fibrils seen in the biopsy of the lymph node. Furthermore, neither bone marrow biopsy examination nor the aspirate from the abdominal fat showed signs of amyloidosis.
Presence of a factor X antibody was suspected based on the lack of response to FFP and PCC. We were not able to detect an inhibitor in the mixing studies or using a Bethesda assay against functional antibodies inhibiting factor X activity. Despite the fact that there was no inhibitory antibody detectable, the probability of the presence of a noninhibitory antibody was considered and consequently the patient was first treated with prednisone as first-line treatment for an auto-immune antibody-mediated disease. Because of a new abdominal soft tissue bleeding, more rigorous treatment was initiated in an attempt to firmly tackle the marginal-zone lymphoma, considering a relation between the factor X deficiency and the lymphoma.
Treatment consisted of chemo- and immunotherapy, chlorambucil and rituximab, resulting in normalization of PT, aPTT levels (10.7 and 28 sec, respectively, Fig.1B), LDH level of 210 U/l and a factor X activity of 106% within three weeks time.
The normalization of the factor X activity after treatment of the patient strongly suggested that a noninhibitory antibody against factor X was present. Such an antibody would bind factor X without inhibiting the function but facilitating fast clearance from the circulation. Laboratory evaluation of vitamin K dependent procoagulant factor activities just before and after rituximab and chlorambucil treatment are shown in Table2. A RIA assay detecting factor X antibody binding capacity was used to demonstrate the presence of a possible factor X antibody in the patient plasma before treatment and after normalization. Indeed, the presence of a noninhibitory antibody was ultimately demonstrated in the before treatment sample of our patient and not in the sample after treatment (Table3). As a positive control rabbit antihuman factor X was taken which showed a good dose-response (Fig.2). The patient samples were measured in a 1:50 and 1:250 dilution, both in duplicate. The 1:50 dilution was within the dose–response curve of our positive control, showing a low but detectable amount of factor X binding. Plasma from a healthy person and patient plasma after treatment were used as negative controls, in this experiment. In both samples no signal above the blank was observed. In addition, the antibody found in the patient appeared to be calcium dependent. No binding of the antibody to biotinylated factor X in the absence of calcium was observed (data not shown).
Table 2.
Vitamin K dependent procoagulant clotting factor activities before and after rituximab and chlorambucil treatment
| Parameter | BT | AT | Normal range (%) |
|---|---|---|---|
| Factor II | 63 | 86 | 80–120 |
| Factor VII | 54 | 106 | 65–150 |
| Factor IX | 53 | 102 | 70–140 |
| Factor X (extrinsic) | 8 | 106 | 80–120 |
BT, before treatment; AT, after treatment.
Table 3.
Results RIA assay before and after rituximab and chlorambucil treatment
| Sample | % Binding factor X-biotin |
|---|---|
| Blank 0.3% BSA | 7.0 |
| Healthy control 1:50 | 6.7 |
| Patient BT 1:50 | 12.2 |
| Patient BT 1:250 | 8.1 |
| Patient AT 1:50 | 6.6 |
| Patient AT 1:250 | 5.3 |
BT, before treatment; AT, after treatment.
Figure 2.
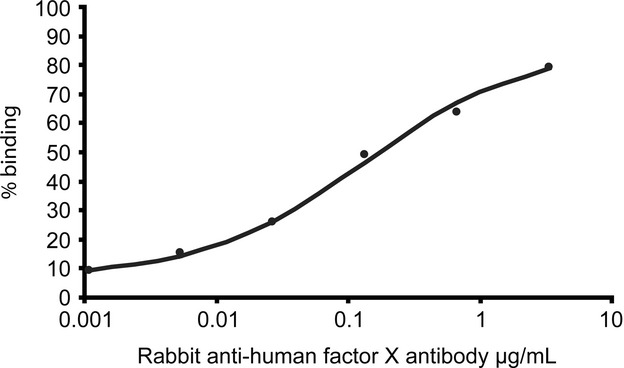
Dose response curve of rabbit anti-human factor X using radioactive immunoassay. Serial dilution of positive control rabbit anti-human factor X antibody was made and incubated overnight with Sepharose A beads to capture them. After washing, biotin labeled factor X was added and incubated with the beads overnight. Streptavidin labeled with radioactive iodine was added overnight after washing the beads again. The amount of 125I-streptavidin bound was measured after washing and calculated back to the percentage of biotin binding to the antibody.
Factor II, VII and IX were also slightly reduced in our patient, although to a much lesser extent than factor X. No binding antibody could be detected for factor II and IX using the same RIA experiment using biotinylated factor II and IX as antigen, suggesting a specific antibody against factor X only.
One year after the diagnosis, the patient remains asymptomatic with normal factor X levels. A CT-scan to evaluate effect of chlorambucil and rituximab treatment showed complete remission of the non-Hodgkin lymphoma.
Discussion
We described a previously healthy 81-year-old man with an extensive and spontaneous cutaneous- and soft tissue hematoma as well as muscle bleeding associated with a nodal marginal-zone lymphoma. The factor X activity and antigen levels were shown to be very low. Since our patient did not have a bleeding tendency in the past, we concluded that this patient suffered from an acquired factor X deficiency.
Acquired factor X deficiencies are most frequently described in relation to amyloidosis, factor X inhibitors or vitamin K deficiency 7,16. At the first instance, because all measured vitamin K-dependent clotting factors were reduced, it was thought that the patient had a mild vitamin K deficiency. However, it is rather unlikely that vitamin K deficiency was the main cause of the grossly reduced factor X activity and therefore the cause of the bleeding complication. Moreover, this assumption is in agreement with the nonresponsiveness observed after vitamin K supplementation. Also the liver function tests were normal.
Biopsy of an inguinal lymph node demonstrated a marginal-zone lymphoma. This is an indolent disease and treatment is initiated only in the presence of symptoms. Considering the physiology of the marginal zone in lymph nodes, where mature B cells can rapidly proliferate and differentiate into antibody secreting plasma cells, it was hypothesized that an antibody related factor X deficiency was present. Mixing studies and factor X Bethesda assays could not demonstrate an inhibitory antibody. Initial treatment with FFP or PCC demonstrated a temporary response in the factor X levels. The period of response did not correlate with the half-life of factor X, which is around 48–72 h 31. This result suggested the presence of an antibody causing enhanced clearance of factor X from the blood circulation. A few reports described noninhibitory autoantibody related with a factor deficiency 27,32,33.
One study describes a noninhibitory antibody in a patient with cutaneous lymphoma. The patient had combined deficiency of factor II and X. A cross-reactive antibody was found to bind to factor II, IX and X via the common metal-ion-dependent conformational epitope found on the vitamin K-dependent γ-carboxyglutamic acid (Gla) domain 27. Also, in a patient with a low grade follicular center cell non-Hodgkin lymphoma, the presence of a noninhibitory autoantibody binding via a calcium-dependent epitope of factor II was found, which accelerated its clearance 32. In an another patient also a calcium-dependent noninhibitory antibody to factor II was found, which had low affinity for factor II 33.
The presence of a noninhibitory antibody against factor X was found in our patient, which was calcium dependent. This suggests that our antibody is directed against a factor X epitope that is metal dependent, maybe also the common metal-ion-dependent conformational epitope found on the Gla domain. In the RIA assay using biotinylated factor II and IX, we could not detect binding to these clotting factors.
Due to the severity and recurrence of bleeding we had promptly started treatment with rituximab and chlorambucil according to the guidelines for treatment of nodular marginal-zone lymphoma 34. Treatment resulted in complete remission of the lymphadenopathy and normalization of factor X levels, proving that factor X deficiency was related with the non-Hodgkin lymphoma.
Acquired coagulopathies related to marginal-zone lymphomas have previously been described. Tefferi et al., reported a patient with an acquired Von Willebrand disease related to a marginal-zone lymphoma 35. The authors suggest that the von Willebrand factor (VWF) protein was absorbed by tumor cells, showing an aberrant tumor-cell expression of the platelet VWF receptor (GbIb). Another case report in which a patient was diagnosed with splenic marginal-zone lymphoma without bleeding problems, showed reduced factor activities of factor II, V, VII, VIII, IX, X, XI and XII. The authors found a specific inhibitor against factor IX and assumed presence of a non-factor specific inhibitory antibody 36.
Our case is another example of an acquired factor X deficiency associated with the presence of a lymphoproliferative disease.
This is the first patient to our knowledge diagnosed with an acquired factor X deficiency caused by a noninhibitory antibody related to nodal marginal-zone lymphoma. We advise to start treatment of the underlying disease as soon as possible when there are symptoms of hemorrhage or if there is a high risk of hemorrhage due to low-factor X levels.
Acknowledgments
The authors would to thank Prof. Dr. J. Meijers (Sanquin) and Prof. Dr. H.C.J. Eikenboom (LUMC) for helpful discussions. We thank the technicians of LabWest, Sanquin and LUMC hemostasis laboratories for technical assistance, especially A. van Leeuwen (Sanquin, Biologicals laboratory) for performing the RIA experiments. We also express our gratitude to the patient who willingly provided blood samples and biopsy material for research analysis and agreed to the publication of the results.
Conflict of Interest
None declared.
References
- Brown DL. Kouides PA. Diagnosis and treatment of inherited factor X deficiency. Haemophilia. 2008;14:1176–1182. doi: 10.1111/j.1365-2516.2008.01856.x. [DOI] [PubMed] [Google Scholar]
- Hougie C, Barrow EM. Graham JB. Stuart clotting defect. I. Segregation of an hereditary hemorrhagic state from the heterogeneous group heretofore called stable factor (SPCA, proconvertin, factor VII) deficiency. J. Clin. Invest. 1957;36:485–496. doi: 10.1172/JCI103446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telfer TP, Denson KW. Wright DR. A new coagulation defect. Br. J. Haematol. 1956;2:308–316. doi: 10.1111/j.1365-2141.1956.tb06703.x. [DOI] [PubMed] [Google Scholar]
- Dewerchin M, Liang Z, Moons L, Carmeliet P, Castellino FJ, Collen D, et al. Blood coagulation factor X deficiency causes partial embryonic lethality and fatal neonatal bleeding in mice. Thromb. Haemost. 2000;83:185–190. [PubMed] [Google Scholar]
- Peyvandi F, Menegatti M, Santagostino E, Akhavan S, Uprichard J, Perry DJ, et al. Gene mutations and three-dimensional structural analysis in 13 families with severe factor X deficiency. Br. J. Haematol. 2002;117:685–692. doi: 10.1046/j.1365-2141.2002.03486.x. [DOI] [PubMed] [Google Scholar]
- Peyvandi F, Mannucci PM, Lak M, Abdoullahi M, Zeinali S, Sharifian R, et al. Congenital factor X deficiency: spectrum of bleeding symptoms in 32 Iranian patients. Br. J. Haematol. 1998;102:626–628. doi: 10.1046/j.1365-2141.1998.00806.x. [DOI] [PubMed] [Google Scholar]
- Choufani EB, Sanchorawala V, Ernst T, Quillen K, Skinner M, Wright DG, et al. Acquired factor X deficiency in patients with amyloid light-chain amyloidosis: incidence, bleeding manifestations, and response to high-dose chemotherapy. Blood. 2001;97:1885–1887. doi: 10.1182/blood.v97.6.1885. [DOI] [PubMed] [Google Scholar]
- Glenner GG. Factor X deficiency and systemic amyloidosis. N. Engl. J. Med. 1977;297:108–109. doi: 10.1056/NEJM197707142970211. [DOI] [PubMed] [Google Scholar]
- Greipp PR, Kyle RA. Bowie EJ. Factor X deficiency in primary amyloidosis: resolution after splenectomy. N. Engl. J. Med. 1979;301:1050–1051. doi: 10.1056/NEJM197911083011907. [DOI] [PubMed] [Google Scholar]
- Korsan-Bengtsen K, Hjort PF. Ygge J. Acquired factor X deficiency in a patient with amyloidosis. Thromb Diath. Haemorrh. 1962;7:558–566. [PubMed] [Google Scholar]
- Manikkan AT. Factor X deficiency: an uncommon presentation of AL amyloidosis. Ups. J. Med. Sci. 2012;117:457–459. doi: 10.3109/03009734.2012.690457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenstein ED, Itzkowitz SH, Penziner AS, Cohen JI. Mornaghi RA. Resolution of factor X deficiency in primary amyloidosis following splenectomy. Arch. Intern. Med. 1983;143:597–599. [PubMed] [Google Scholar]
- Uprichard J. Perry DJ. Factor X deficiency. Blood Rev. 2002;16:97–110. doi: 10.1054/blre.2002.0191. [DOI] [PubMed] [Google Scholar]
- Furie B, Voo L, McAdam KP. Furie BC. Mechanism of factor X deficiency in systemic amyloidosis. N. Engl. J. Med. 1981;304:827–830. doi: 10.1056/NEJM198104023041407. [DOI] [PubMed] [Google Scholar]
- Gollard R, Rahman S. Ratnasabapathy R. Factor X inhibitor: a fulminant presentation and fatal course of a rare syndrome in a 59-year-old male. Acta Haematol. 2013;129:40–44. doi: 10.1159/000342115. [DOI] [PubMed] [Google Scholar]
- Lee G, Duan-Porter W. Metjian AD. Acquired, non-amyloid related factor X deficiency: review of the literature. Haemophilia. 2012;18:655–663. doi: 10.1111/j.1365-2516.2012.02773.x. [DOI] [PubMed] [Google Scholar]
- Mascarenhas A, Eusebio M, Freitas O. Almeida T. Acquired-transient factor X deficiency in a teenager with extensive burns. BMJ Case Rep. 2011;2011:1–3. doi: 10.1136/bcr.12.2010.3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulhare PE, Tracy PB, Golden EA, Branda RF. Bovill EG. A case of acquired factor X deficiency with in vivo and in vitro evidence of inhibitor activity directed against factor X. Am. J. Clin. Pathol. 1991;96:196–200. doi: 10.1093/ajcp/96.2.196. [DOI] [PubMed] [Google Scholar]
- Peuscher FW, van Aken WG, van Mourik JA, Swaak AJ, Sie LH. Statius van Eps LW. Acquired, transient factor X (Stuart factor) deficiency in patient with mycoplasma pneumonial infection. Scand. J. Haematol. 1979;23:257–264. doi: 10.1111/j.1600-0609.1979.tb02859.x. [DOI] [PubMed] [Google Scholar]
- Haberal M, Basaran O, Sakallioglu AE, Kesik E, Alioglu B. Ozbek N. Acquired-transient factor X deficiency associated with anticardiolipin antibodies in a child with extensive burns. J. Burn. Care Res. 2006;27:113–116. doi: 10.1097/01.bcr.0000191962.46058.bf. [DOI] [PubMed] [Google Scholar]
- Hosker JP. Jewell DP. Transient, selective factor X deficiency and acute liver failure following chest infection treated with erythromycin BP. Postgrad. Med. J. 1983;59:514–515. doi: 10.1136/pgmj.59.694.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga AT. Shafer FE. An acquired inhibitor to factor X in a pediatric patient with extensive burns. J. Pediatr. Hematol. Oncol. 1996;18:223–226. doi: 10.1097/00043426-199605000-00028. [DOI] [PubMed] [Google Scholar]
- Ness PM, Hymas PG, Gesme D. Perkins HA. An unusual factor-X inhibitor in leprosy. Am. J. Hematol. 1980;8:397–402. doi: 10.1002/ajh.2830080408. [DOI] [PubMed] [Google Scholar]
- Rao LV, Zivelin A, Iturbe I. Rapaport SI. Antibody-induced acute factor X deficiency: clinical manifestations and properties of the antibody. Thromb. Haemost. 1994;72:363–371. [PubMed] [Google Scholar]
- Caimi MT, Redaelli R, Cattaneo D, Nosari AM, Baudo F. de Cataldo F. Acquired selective factor X deficiency in acute nonlymphocytic leukemia. Am. J. Hematol. 1991;36:65–66. doi: 10.1002/ajh.2830360115. [DOI] [PubMed] [Google Scholar]
- Hsia CC, Keeney M, Bosco AA. Xenocostas A. Treatment of acquired factor X inhibitor by plasma exchange with concomitant intravenous immunoglobulin and corticosteroids. Am. J. Hematol. 2008;83:318–320. doi: 10.1002/ajh.21105. [DOI] [PubMed] [Google Scholar]
- Rochanda L, Del Zoppo GJ, Feinstein DI. Liebman HA. Approach to the treatment, characterization and diagnosis of an acquired auto-antibody directed against factors prothrombin, factor X and factor IX: a case report and review of the literature. Haemophilia. 2012;18:102–107. doi: 10.1111/j.1365-2516.2011.02553.x. [DOI] [PubMed] [Google Scholar]
- Carter C. Winfield DA. Factor X deficiency during treatment of relapsed acute myeloid leukaemia with amsacrine. Clin. Lab. Haematol. 1988;10:225–228. doi: 10.1111/j.1365-2257.1988.tb01176.x. [DOI] [PubMed] [Google Scholar]
- Peerschke EI, Castellone DD, Ledford-Kraemer M, Van Cott EM. Meijer P. Laboratory assessment of factor VIII inhibitor titer: the North American Specialized Coagulation Laboratory Association experience. Am. J. Clin. Pathol. 2009;131:552–558. doi: 10.1309/AJCPMKP94CODILWS. [DOI] [PubMed] [Google Scholar]
- Wolbink GJ, Vis M, Lems W, Voskuyl AE, de Groot E, Nurmohamed MT, et al. Development of antiinfliximab antibodies and relationship to clinical response in patients with rheumatoid arthritis. Arthritis Rheum. 2006;54:711–715. doi: 10.1002/art.21671. [DOI] [PubMed] [Google Scholar]
- Bennett SR, Lehman CM, Rodgers GM. Laboratory Hemostasis. New York: Springer; 2007. p. 244. [Google Scholar]
- Lee ES, Hibsman BK. Liebman HA. Acquired bleeding disorder in a patient with malignant lymphoma: antibody-mediated prothrombin deficiency. Cancer. 2001;91:636–641. doi: 10.1002/1097-0142(20010215)91:4<636::aid-cncr1046>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Bajaj SP, Rapaport SI, Barclay S. Herbst KD. Acquired hypoprothrombinemia due to non-neutralizing antibodies to prothrombin: mechanism and management. Blood. 1985;65:1538–1543. [PubMed] [Google Scholar]
- Reid R. Friedberg JW. Management of marginal zone lymphoma. Oncology. 2013;27:840. 842, 844. [PubMed] [Google Scholar]
- Tefferi A, Hanson CA, Kurtin PJ, Katzmann JA, Dalton RJ. Nichols WL. Acquired von Willebrand’s disease due to aberrant expression of platelet glycoprotein Ib by marginal zone lymphoma cells. Br. J. Haematol. 1997;96:850–853. doi: 10.1046/j.1365-2141.1997.d01-2088.x. [DOI] [PubMed] [Google Scholar]
- Siemens HJ, Gerke P, Steinhoff J, Roth-Isigkeit A, Wagner K. Bruckner S. A prolonged APTT in a patient with a low grade malignant NHL - a case report. Haematologica. 2002;87:ELT08. [PubMed] [Google Scholar]