

Key Clinical Message
Convulsion in diabetics is often considered as a result from fluctuation of blood glucose level. However, if a diabetic patient also presents abnormal neurological signs, mitochondrial diseases need to be considered in the differential diagnosis.
Keywords: Anticonvulsants, cerebellar ataxia, convulsion, maternally inherited diabetes and deafness
Introduction
Maternally inherited diabetes and deafness (MIDD), a mitochondrial disease first described in 1992 1, results from the mitochondrial DNA mutation m.3243A>G and accounts for up to 1% of the patients with diabetes 2. m.3243A>G is responsible for several other reported clinical syndromes and features other than MIDD, including mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS), chronic progressive external ophthalmoplegia (CPEO), isolated myopathy, cardiomyopathy, convulsions, migraine, ataxia, cognitive impairment, bowel dysmotility, and short stature. It is not clear why these various phenotypes of mitochondrial disease occur; however, it has been suggested that the level of heteroplasmy (the presence of a mixture of mutant and normal mtDNA in a cell) plays a crucial role.
We here report an unusual case of MIDD simultaneously presenting with generalized convulsion and cerebellar symptoms.
Case History
A 65-year-old Japanese woman was diagnosed with type 2 diabetes mellitus at age 53, and had experienced hearing difficulties since age 56. The family history included short stature and diabetes in her mother, maternal grandmother, and mother’s sister. The present cases were also of short stature and of low weight. Gait disturbance and dysarthria gradually progressed from age 57. Partial seizures in the right arm and foot had evolved to generalized convulsion, occurring once a month, from age 62, and these convulsions could not be suppressed with sodium valproate 400 mg/day. In addition, during the convulsions, her serum glucose level was within the normal range and the blood level of valproate was maintained at over 50 μg/mL.
Differential diagnosis, investigations, and treatment
Neurological examination at the first visit to our hospital revealed bilateral gaze-evoked horizontal nystagmus, left side dominant sensorineural deafness, coordination disorders in the extremities, cerebellar ataxic gait, and mild slurred speech; without ophthalmoplasia. The blood pressure was within normal limits. The laboratory tests revealed normal creatine kinase and renal function, high levels of lactate and pyruvate in the blood and cerebrospinal fluid. Brain magnetic resonance imaging showed atrophy of bilateral cerebellar lobes and vermis, normal-volume brainstem, and slightly atrophic cerebrum (Fig.1). Electroencephalogram (EEG) showed occasional theta slowing in the left-sided frontal, central, and parietal leads. Muscle biopsy revealed fiber size variations and scattered, ragged red fibers. Leukocyte mitochondrial gene screening showed m.3243A>G, and the patient was consequently diagnosed as MIDD with cerebellar symptoms and convulsions.
Figure 1.
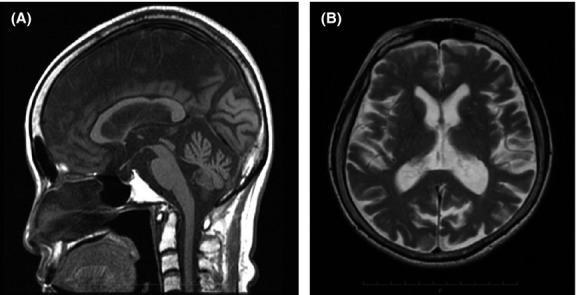
Brain magnetic resonance imaging findings. (A) T1-weighted sagittal image revealing moderate atrophy of the cerebellar vermis and no atrophy of the midbrain and pons. (B) T2-weighted axial image revealing moderate atrophy of the bilateral cerebellum hemispheres and mild atrophy of the cerebrum. No abnormal high-intensity areas are observed in the cerebrum.
Outcome and follow-up
The number of convulsions gradually decreased after clonazepam 1 mg/day was added to valproate. The patient has never experienced headaches throughout the disease duration, and no stroke-like episode has developed to date.
Discussion
We here reported a rare case of MIDD presenting with cerebellar symptoms and generalized convulsions, which were controlled effectively after adding anticonvulsants.
Recently, the involvement of the neuromuscular system in MIDD has become widely established 3–5. Several MIDD cases with concomitant cerebellar symptoms, a relatively unusual phenotype, have been reported. Kobayashi et al. suggested that involvement of the central nervous system should be considered in MIDD, as well as in other mitochondrial diseases, although the prevalence of comorbid condition was unknown 4. Fromont et al. reported that three of eleven MIDD patients (m.3243A>G:n = 9, m.14709T>C: n = 2) exhibited overt signs of cerebellar ataxia and two patients of the same group presented severe cerebellar atrophy. They also concluded that cerebellar atrophy tended to be more frequent and more severe in MIDD patients than in the patients with type 1 diabetes mellitus as control 5.
In a cohort study from 2013, 129 patients from 83 unrelated families, with m.3243A>G mtDNA mutation, were analyzed. Of these, 10% exhibited a classical MELAS phenotype, 30% had MIDD, 6% MELAS/MIDD, 2% MELAS/CPEO, and 5% MIDD/CPEO overlap syndromes. Isolated sensorineural hearing loss occurred in 3–28% of the patients 6. Although concomitant development of MIDD and MELAS was not reported, this combination may also indicate the concept that these are two phenotypic expressions of one disease 7. The studies using the proton magnetic resonance spectroscopy revealed decrease in the concentration of N–acetylaspartate, a metabolite considered as a neuroaxonal marker, in both of vermis of MIDD and cerebellum of MELAS 4,8. N–acetylaspartate is synthesized from acetyl CoA and aspartate in mitochondria 9. Thus, decrease in N–acetylaspartate suggests that MIDD and MELAS had similar pathophysiological abnormalities which were neuroaxonal loss and an impaired mitochondrial energy production in central nervous system. However, whether our case evolves to MELAS in the future remains unknown.
Convulsion is a common phenotypic feature of symptomatic, as well as nonsymptomatic mitochondrial disorders. MELAS may be associated with partial or generalized convulsions, including status epilepticus, whereas CPEO is not usually accompanied by epilepsy, except for in one previous report 10. There are only few reports on MIDD patients presenting with convulsion. Schleiffer et al. described a 27-year-old German woman with family history of MIDD for three generations associated with m.3243A>G, who experienced recurrent tonic-chronic seizures from age 6 before developing MIDD 11. Thorns et al. speculated that an increased m.3243A>G rate in oral mucosa cells from MIDD patients closely correlated with neurologic disorders such as epilepsy 12. Presumably high frequency of mitochondrial DNA mutation in the particular tissue results pronounced clinical symptom of the relevant tissue. Although the m.3243A>G rate was not quantified in our case, the convulsion and cerebellar involvement suggested that a high rate of mutation was present in cerebrum and cerebellum.
Convulsion is also a typical symptom of hypoglycemia and unilateral involuntary movements, such as hemichorea and athetosis, are known to be caused by hyperglycemia 13; however, no drastic changes in the serum glucose level were observed during the convulsive attacks. Partial convulsions with secondary generalization in our case suggest the presence of organic and/or functional disorders in the cerebral hemisphere corresponding to the unilateral slow waves in the EEG. Although the epileptogenic focuses are not always grossly evident, the cerebral cortical neuron impaired by mitochondrial dysfunction could play a more crucial role of epileptogenic focus than that of cerebellum which involvements rarely cause epilepsy. In addition to the interictal epileptiform spikes and pathological high-frequency oscillation on the EEG that have been widely investigated, the focal slow waves are common in the region of epileptic brain. Moreover, the focal delta frequency slowing has also recently been shown to be more common than interictal epileptiform spikes following febrile status epilepticus 14. Therefore, we believe that the cause of the convulsions was related to the mitochondrial disease.
Concerning the use of anticonvulsants for epilepsy in mitochondrial disorders, it is important to avoid anticonvulsants with mitochondrion-toxic side effects. They may cause severe, sometimes even fatal, side effects; may trigger or worsen epilepsy; or may make the epilepsy intractable. Valproate is the most well-known mitochondrial toxic anticonvulsant, which is known to exhibit a deleterious effect in patients carrying mitochondrial DNA polymerase gamma mutations (e.g., Alpers-Huttenlocher syndrome) and with myoclonic epilepsy with ragged red fibers syndrome 15. A common mechanism of convulsion in the patients with mitochondrial disease seems to involve cortical damage resulting from respiratory chain dysfunction 16. Inhibition and decreased activity of mitochondrial complexes I and IV, inhibition of oxygen and consumption and adenosine triphosphate synthesis are known as the mitochondrial adverse effects associated with valproate 17. Therefore, these complex factors could have affected negatively the control of convulsion in our case. In future, we are planning to taper valproate slowly over several months.
To the best our knowledge, this is the first report of MIDD associated with cerebellar symptoms and convulsion, and we believe that this report will help promote awareness about the importance of appropriate anticonvulsants use for convulsion in patients with diabetes mellitus. If a patient is suspected to have mitochondrial disorders based on the clinical course or physical examination or laboratory findings, physicians need to carefully select anticonvulsants.
Accumulation of cases is necessary to clarify the characteristics of convulsions and the long-term prognosis of patients with MIDD.
Acknowledgments
We thank Hiroshi Nemoto, MD, PhD (Hitachi Health Care Center) for the pathological diagnosis of muscle biopsy.
Conflict of Interest
None declared.
References
- Struyvenberg PA, van den Ouweland JM, Lemkes HH, Ruitenbeek W, Sandkuijl LA, Vijlder MFDe, et al. Mutation in mitochondrial tRNA(Leu)(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat. Genet. 1992;1:368–371. doi: 10.1038/ng0892-368. [DOI] [PubMed] [Google Scholar]
- Murphy R, Turnbull DM, Walker M. Hattersley AT. Clinical features, diagnosis and management of maternally inherited diabetes and deafness (MIDD) associated with the 3243A>G mitochondrial point mutation. Diabet. Med. 2008;25:383–399. doi: 10.1111/j.1464-5491.2008.02359.x. [DOI] [PubMed] [Google Scholar]
- Guillausseau PJ, Massin P, Dubois-LaForgue D, Timsit J, Virally M, Gin H, et al. Maternally inherited diabetes and deafness: a multicenter study. Ann. Intern. Med. 2001;134:721–728. doi: 10.7326/0003-4819-134-9_part_1-200105010-00008. [DOI] [PubMed] [Google Scholar]
- Kobayashi Z, Tsunemi T, Miake H, Tanaka S, Watabiki S. Morokuma Y. A mother and a child with maternally inherited diabetes and deafness (MIDD) showing atrophy of the cerebrum, cerebellum and brainstem on magnetic resonance imaging (MRI) Intern. Med. 2005;44:328–331. doi: 10.2169/internalmedicine.44.328. [DOI] [PubMed] [Google Scholar]
- Fromont I, Nicoli F, Valéro R, Felician O, Lebail B, Lefur Y, et al. Brain anomalies in maternally inherited diabetes and deafness syndrome. J. Neurol. 2009;256:1696–1704. doi: 10.1007/s00415-009-5185-4. [DOI] [PubMed] [Google Scholar]
- Nesbitt V, Pitceathly RD, Turnbull DM, Taylor RW, Sweeney MG, Mudanohwo EE, et al. The UK MRC Mitochondrial Disease Patient Cohort Study: clinical phenotypes associated with the m.3243A>G mutation—implications for diagnosis and management. J. Neurol. Neurosurg. Psychiatry. 2013;84:936–938. doi: 10.1136/jnnp-2012-303528. [DOI] [PubMed] [Google Scholar]
- Karkare K, Sinha S, Ravishankar S, Gayathri N, Yasha TC, Goyal MK, et al. Epilepsia partialis continua inmitochondrial dysfunction: interesting phenotypic and MRI observations. Ann. Indian Acad. Neurol. 2008;11:193–196. doi: 10.4103/0972-2327.42942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilichowski E, Pouwels PJ, Frahm J. Hanefeld F. Quantitative proton magnetic resonance spectroscopy of cerebralmetabolic disturbances in patients with MELAS. Neuropediatrics. 1999;30:256–263. doi: 10.1055/s-2007-973500. [DOI] [PubMed] [Google Scholar]
- Bates TE, Strangward M, Keelan J, Davey GP, Munro PM. Clark JB. Inhibition of N-acetylaspartate production: implications for 1H MRS studies in vivo. NeuroReport. 1996;7:1397–1400. [PubMed] [Google Scholar]
- de Wit HM, Westeneng HJ, van Engelen BG. Mudde AH. MIDD or MELAS: that’s not the question MIDD evolving into MELAS: a severe phenotype of the m.3243A>G mutation due to paternal co-inheritance of type 2 diabetes and a high heteroplasmy level. Neth. J. Med. 2012;70:460–462. [PubMed] [Google Scholar]
- Schleiffer T, ‘t Hart LM, Schürfeld C, Kraatz K. Riemann JF. Maternally inherited diabetes and deafness (MIDD): unusual occult exocrine pancreatic manifestation in an affected German family. Exp. Clin. Endocrinol. Diabetes. 2000;108:81–85. doi: 10.1055/s-2000-5800. [DOI] [PubMed] [Google Scholar]
- Thorns C, Widjaja A, Boeck N, Skamira C. Zühlke H. Maternally-inherited diabetes and deafness: report of two affected German families with the A3243G mitochondrial DNA mutation. Exp. Clin. Endocrinol. Diabetes. 1998;106:384–388. doi: 10.1055/s-0029-1212003. [DOI] [PubMed] [Google Scholar]
- Lin JJ, Lin GY, Shih C. Shen WC. Presentation of striatal hyperintensityon T1-weighted MRI in patients with hemiballism-hemichorea caused by non-ketotic hyperglycemia: report of seven new cases and a review of literature. J. Neurol. 2001;248:750–755. doi: 10.1007/s004150170089. [DOI] [PubMed] [Google Scholar]
- Nordli DR, Jr, Moshé SL, Shinnar S, Hesdorffer DC, Sogawa Y, Pellock JM, et al. FEBSTAT Study Team. Acute EEG findings in children with febrile status epilepticus: results of the FEBSTAT study. Neurology. 2012;79:2180–2186. doi: 10.1212/WNL.0b013e3182759766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finsterer J. Zarrouk Mahjoub S. Epilepsy in mitochondrial disorders. Seizure. 2012;21:316–321. doi: 10.1016/j.seizure.2012.03.003. [DOI] [PubMed] [Google Scholar]
- Bindoff LA. Mitochondrial function and pathology in status epilepticus. Epilepsia. 2011;52(suppl. 8):6–7. doi: 10.1111/j.1528-1167.2011.03223.x. [DOI] [PubMed] [Google Scholar]
- Finsterer J. Zarrouk Mahjoub S. Mitochondrial toxicity of antiepileptic drugs and their tolerability in mitochondrial disorders. Expert Opin. Drug Metab. Toxicol. 2012;8:71–79. doi: 10.1517/17425255.2012.644535. [DOI] [PubMed] [Google Scholar]