

Abstract
Recently, a number of promising approaches have been developed using synthetic chemistry, materials science, and bioengineering-based strategies to address challenges in the design of more effective cancer vaccines. At the stage of initial priming, potency can be improved by maximizing vaccine delivery to lymph nodes. Because lymphatic uptake from peripheral tissues is strongly size-dependent, antigens and adjuvants packaged into optimally-sized nanoparticles access the lymph node with much greater efficiency than unformulated vaccines. Once primed, T cells must home to the tumor site. Because T cells acquire the necessary surface receptors in the local lymph node draining the tissue of interest, vaccines must be engineered that reach organs such as the lung and gut, which are common sites of tumor lesions but inaccessible by traditional vaccination routes. Particulate vaccine-carriers can improve antigen exposure in these organs, resulting in greater lymphocyte priming. Immunomodulatory agents can also be injected directly into the tumor site to stimulate a systemic response capable of clearing even distal lesions; materials have been designed that entrap or slowly release immunomodulators at the tumor site, reducing systemic exposure and improving therapeutic efficacy. Finally, lessons learned from the design of biomaterial-based scaffolds in regenerative medicine have led to the development of implantable vaccines that recruit and activate antigen presenting cells to drive anti-tumor immunity. Overall, these engineering strategies represent an expanding toolkit to create safe and effective cancer vaccines.
Motivation for cancer vaccine engineering
Therapeutic vaccination is one of the oldest and most studied concepts in cancer immunotherapy. Yet, in contrast to prophylactic vaccines against infectious disease, which have had a major impact on public health, therapeutic vaccines against cancer have generally been much less successful, and only a single cancer vaccine has been FDA approved to date (1, 2). This is likely due to a variety of factors, including a paucity of truly foreign antigens expressed by tumor cells, lack of infection-associated inflammatory cues that drive productive immunity, chronic antigen exposure, the presence of a highly immunosuppressive microenvironment in solid tumors, and our as yet still poor understanding of how to induce strong and sustained T-cell-mediated immune responses in humans. However, there are at least three reasons why cancer vaccines should see renewed interest as part of the cancer immunotherapy armamentarium, based on recent rapid advances in the field: First, the advent of clinical-stage therapeutics that can directly influence the immunological status of the tumor microenvironment, such as checkpoint blockade antibodies (3), regulatory T-cell-modulating chemotherapy (4), and IDO inhibitors (5) (to name a few), now provide a number of ways to overcome immunosuppressive pathways in patients. Secondly, the availability of an ever-growing array of targeted drugs that can dramatically (but transiently) lower tumor burden provides a window of opportunity for vaccines to act in a setting of minimal disease, and some of these drugs may act synergistically with the immune response (6). Lastly, powerful genomic sequencing capabilities are enabling the possibility of patient-specific vaccines targeting defined neoantigens, which have the potential for alleviating the safety and efficacy challenges of targeting unmutated self antigens (7–9). Altogether, these recent developments in cancer therapy strongly motivate renewed efforts to develop effective therapeutic cancer vaccine approaches.
How might we enhance the vaccines themselves to enable therapeutic immunization to reach its full potential in this new era of cancer immunotherapy? First is the issue of vaccine potency, as measured by the number, functionality, and avidity of antigen-specific T-cells induced by cancer vaccines. A number of experimental and licensed infectious disease vaccines induce robust multifunctional CD4+ and CD8+ T-cell responses in humans that can be detected directly ex vivo and measured even by relatively low-sensitivity methods like peptide-MHC tetramer staining (10, 11). By contrast, with a few exceptions (12, 13), the response to cancer vaccines is often only robustly detected by expanding/stimulating patient T-cells over 1–2 weeks ex vivo (14–16) – a direct indicator of the low frequency of responding cells. These results may be partly due to issues of tolerance to self-antigens and systemic immunosuppression in cancer patients, but also may reflect the common use of minimal-epitope peptide vaccines and weak adjuvants which have known immunological shortcomings (17). Equally important is for vaccines to be capable of promoting T-cell responses enriched in high-avidity, polyfunctional T-cells with high proliferative capacity that avoid induction of an exhausted/terminally-differentiated phenotype. Finally, devising vaccine strategies that prime effective trafficking of effector cells to tumor sites is critical, which in cases such as mucosal tumors, could be directly influenced by vaccines that program expression of appropriate tissue homing receptors (18). Thus, a number of strategies exist to enhance current vaccine approaches to increase the efficacy of therapeutic anti-tumor immune responses.
There are many ways to improve therapeutic vaccines rooted in traditional vaccinology principles such as microbial vector development, molecular biology, and adjuvant design, but in this brief perspective, we will review promising recent preclinical and early clinical developments derived from approaches based in immune engineering–bringing methods from chemistry, chemical engineering, materials science, and biological engineering to bear on the problem of therapeutic vaccine design. Such approaches are particularly well suited to augmenting vaccines based on subunit antigen (defined protein, peptide, or polysaccharide epitopes) and tumor cell lysate-based vaccines, and we focus on these two ubiquitous classes of cancer vaccine antigens.
Targeting vaccines to lymph nodes
A very basic issue in generating robust immunity with cancer vaccines is efficient delivery of vaccine components to lymphoid tissues, the sites of immune response orchestration. Nearly all vaccines are administered parenterally, either intramuscularly or subcutaneously. Following injection of soluble protein/peptide vaccines, antigen arrives in draining lymph nodes in two phases: first, lymph node-resident dendritic cells (DCs) directly access antigen as it drains through afferent ducts, and present antigen to T cells to initiate an immune response. This response is sustained during the second phase, when migratory DCs or monocytes that have phagocytosed additional antigen at the site of injection arrive in the lymph node (19). In some settings, however, only the first phase may be necessary: In mice vaccinated with protein antigens fused to an anti-CD205 antibody to target cross-presenting lymph node-resident DCs, migratory DC depletion actually enhanced T cell priming (20). Migratory DCs were shown to contain expression signatures enriched in genes associated with immune suppression, compared to cross-presenting lymph node-resident DCs. That lymph node delivery is key to vaccine potency is shown by studies of intra-lymph node injections, which demonstrated that peptide or DNA vaccines injected directly into LNs are 100-fold more potent than the same vaccine administered subcutaneously (21, 22).
Although the fate of injected vaccines is a complex interplay of numerous parameters, the physical size of vaccine components– whether they be particulates or individual molecules– plays a significant role in determining the outcome, as shown in Figure 1A (23). Molecules or particles injected in tissue can be cleared by either entering the blood or the lymph. In classic studies in sheep comparing the biodistribution of a series of molecules of varying molecular weight from tissues following injection, Supersaxo and colleagues showed that large proteins preferentially convected to the lymph node rather than being lost to systemic circulation (24). This is because the lymphatic endothelium has valve-like openings enabling the entry of large particles, while the capillary endothelium is lined by an uninterrupted basement membrane that blocks the transit of large macromolecules. A linear correlation is observed between molecular weight and the fraction of lymph node (LN) uptake up to a threshold of 45 kDa (corresponding to a size of approximately 4–5 nm in diameter for a globular protein), at which point nearly 100% of protein is delivered to the lymph and downstream draining lymph node (25). Consistent with this finding, unformulated peptides (26), molecular adjuvants (26–28), and small protein antigens show very poor uptake in lymph nodes, and soluble small-molecule adjuvants often show significant systemic inflammatory toxicity (26–28). While both preclinical and clinical studies have often sought to solve this problem by administering vaccines in “depot”-based adjuvants such as incomplete Freund’s adjuvant, it has been shown that passive, non-inflammatory depots of antigen at the injection site become a decoy for effector cells that leads to deletion of the very T-cells that are meant to be primed by the vaccine (29).
Figure 1.
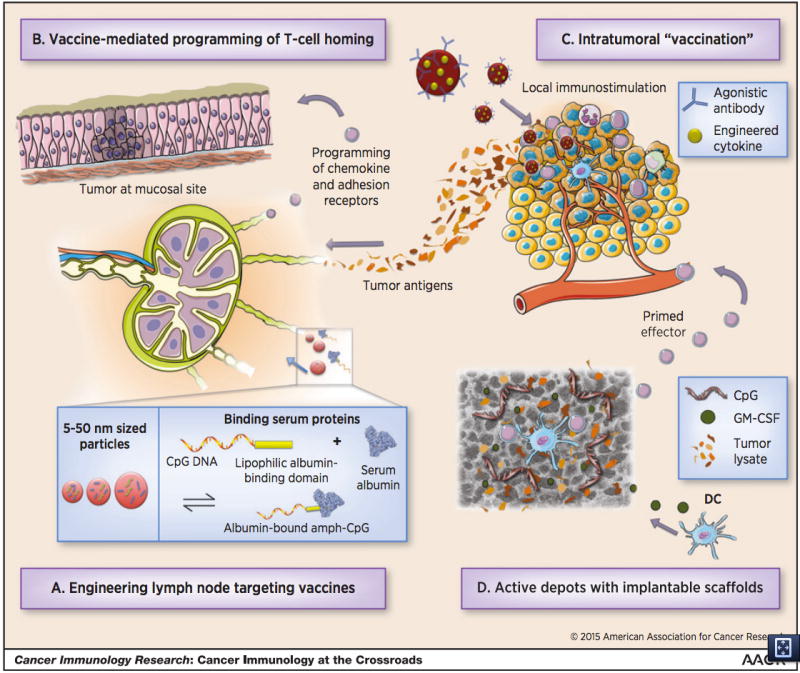
A schematic showing four strategies for engineering more effective cancer vaccines. In (A), vaccines are engineered to drain efficiently to lymph nodes. Particles between 5–50nm drain more effectively than particles of other sizes, and molecular vaccines can be engineered to bind serum proteins to meet this size criteria for effective lymphatic drainage. In (B), T-cell homing to specific sites can be directed by the route of administration. For sites like the lung and the gut, engineering of biomaterial carriers may facilitate delivery. Immunomodulatory therapies can be introduced directly into the tumor to generate anti-tumor responses, shown in (C). Implantable biomaterial scaffolds can be loaded with tumor antigens and inflammatory signals to create and in-situ dendritic cell vaccine, shown in (D).
The size-dependent physiology of lymphatic trafficking has motivated studies of synthetic nanoparticles larger than individual proteins as carriers to efficiently deliver small antigens/molecular adjuvants to the lymph node. Maximal lymph node targeting is a size optimization problem as particles that are larger than the average pore size in the extracellular matrix may become entrapped in the tissue rather than convecting to lymphatics. A series of studies by three different groups demonstrated that nanoparticles with diameters under ~50 nm target lymph nodes much more efficiently than larger particles. Reddy et al. injected labeled 20 nm, 45 nm, and 100 nm poly(propylene sulfide) nanoparticles intradermally and sampled the draining lymph nodes up to 120 hours later; while the 20 nm and 45 nm ultra-small nanoparticles were present in the lymph node throughout the time points sampled, 100 nm particles could not be detected (30). Manolova, et al. (31) and Fifis et al. (32) reached similar conclusions using virus-like particles or synthetic polystyrene nanoparticles of different sizes as carriers for vaccines, demonstrating that particles in the 20–40 nm size range were much more effective for lymph node delivery and subsequent vaccine responses than larger particles. Nanoparticles can also be used to deliver potent molecular adjuvants to lymph nodes, and promising data in both mice (28, 33–38) and non-human primates (34, 39) suggests this is an approach that should be moved toward clinical testing, especially for small molecule adjuvants where such approaches can increase both the safety and potency. Finally, on arrival in lymph nodes, nanoparticles have the potential to impact multiple aspects of antigen processing and presentation by enabling antigen and adjuvant to be co-delivered into recipient antigen presenting cells (41), promoting cross presentation of antigen (33, 42), and acting as intracellular/extracellular vaccine depots (42, 43). An exciting recent study demonstrated a method to coat polymer nanoparticles with native tumor cell-derived plasma membranes, leading to cross presentation of tumor membrane-associated antigens, providing a means to combine complex tumor-derived antigen mixtures with particle-based lymph node targeting (44).
A second strategy to target vaccines to lymph nodes is to exploit reversible binding to proteins naturally meeting the size-dependent criteria for effective lymphatic uptake. For example, albumin, the most prevalent protein in blood and interstitial fluid, is a 66 KDa globular protein with a hydrodynamic diameter of approximately 5 nm– and thus traffics one way from the blood to the lymph in the interstitial space. Liu et al. conjugated peptide antigens and CpG adjuvant to saturated hydrocarbon lipid tails chosen to promote binding to fatty acid-binding pockets of albumin (26). Importantly, these conjugates were comprised of an albumin-binding tail linked to the antigen via a highly water soluble poly(ethylene glycol) (PEG) chain. This PEG spacer solubilized the conjugates and prevented the conjugate from stably inserting in cell membranes, an important distinction from traditional lipopeptide vaccines. Upon injection, these vaccine amphiphiles bound to albumin, leading to >10-fold increases in lymph node accumulation relative to the parent vaccine molecules. In therapeutic melanoma and cervical cancer tumor models, lipid-conjugated vaccines were able to significantly delay growth of established tumors at doses where traditional peptide/adjuvant vaccines were completely ineffective.
Vaccine-Mediated programming of T-cell homing to tumor sites
It has been observed in many clinical studies that the presence of circulating tumor antigen-specific T-cells does not correlate with clinical outcome, and this is consistent with the expectation that activated T-cells must home to tumor sites to impact disseminated disease. Cancer vaccines can impact this phase of the immune response by ensuring induction of appropriate tissue-homing receptor profiles on newly primed tumor-specific lymphocytes, as seen in Figure 1B. A key strategy to control tissue-specific effector cell trafficking is via choice of vaccination site, because dendritic cells in lymph nodes draining different tissue sites express factors regulating the expression of tissue homing receptors on T-cells primed in these sites. Thus, T-cells primed in mediastinal lymph nodes express α4β1 integrins and home to the lungs; DCs in skin-draining lymph nodes induce T-cells to express cutaneous lymphocyte antigen (CLA) and CCR4 to home to the skin; and DCs of the gut-associated lymphoid tissues secrete retinoic acid, programming expression of α4β7 and CCR9 on T-cells for homing to the gut lamina propria (45). While these tissue-specific homing patterns have all been defined in the setting of T-cell trafficking to normal tissues, they are also critical in the therapeutic setting of effector T-cells homing to tumor sites. For example, in orthotopic tumor models of head and neck and lung cancer, Sandoval, et al. demonstrated that mucosal, but not intramuscular, delivery of vaccines can promote CD8-mediated rejection of mucosal tumors (46). Human papillomavirus 16 E7-expressing TC-1 cells were engrafted in the submucosal lining of the tongue or in the lung as model mucosal tumors. Shiga toxin 1 subunit B (STxB) E7 fusions in combination with αGalCer adjuvant were administered intranasally or intramuscularly, and while both vaccines generated systemic CD8+ T-cell responses, intranasal delivery resulted in more efficient tumor clearance in both models. Mechanistically, this was traced to mucosal imprinting of activated antigen-specific T cells, as measured by CD49a and CD103 expression, which allowed for effective homing and infiltration at the tumor site. Thus, strategies to enhance local tissue immunization may have a significant impact on the efficacy of cancer vaccines.
To this end, nanoparticle formulations discussed above for parenteral immunization have also been shown to enhance vaccine antigen/adjuvant uptake across pulmonary and nasal mucosa, which could promote tumor-homing T-cell responses in the setting of lung carcinoma, head and neck cancer, and treatment of lung metastases in a variety of other cancers. Nanoparticles can co-deliver antigen and molecular adjuvants to dendritic cells in the airway mucosa and promote uptake by DCs prior to mucociliary clearance (36, 47, 48). For example, exploiting the high density of dendritic cells lining alveoli in the lungs, pulmonary vaccination with lipid nanocapsules carrying a protein antigen and Toll-like receptor agonists led to increased persistence of antigen in the lungs 24 hours after administration, and subsequently greatly increased trafficking of antigen to lung-draining lymph nodes several days later. This enhanced antigen delivery translated to >10-fold increases in T-cell priming compared to soluble forms of the same antigen and adjuvant, and enabled pulmonary nanocapsule vaccination to be 100% protective in a lung metastasis model, compared to only 20% protection in the equivalent soluble vaccine (49). In a similar vein, intranasal vaccination with antigen-carrying poly(γ-glutamic acid) nanoparticles enhanced therapeutic protection against melanoma lung metastases (50). Pulmonary vaccination with PEGylated poly(propylene sulfide) nanoparticles conjugated to antigens using a reduction-sensitive linker combined with soluble CpG has been shown to enhance antigen uptake in lung-draining lymph nodes and subsequent lung-homing antigen specific T-cell populations (51, 52). Thus, several types of nanoparticle formulations have shown efficacy in enhancing mucosa-homing T-cell responses and mucosal anti-tumor immunity.
Analogous to pulmonary vaccination for protection of airway mucosal tissues, oral vaccination may facilitate anti-tumor immunity in the gastrointestinal tract, and could thus help protect against cancers of the throat, stomach, intestine, and colon. A non-obvious benefit of vaccinating the gastrointestinal tract is that T cell priming in the lymphoid organs of the large intestine can induce protection of rectal and vaginal mucosa, which are difficult to vaccinate directly (53). The design of effective oral vaccines that reach the large intestine has been challenging largely because of the low pH and destructive enzymatic activity characteristic of the gut. To solve this problem, Zhu et al. developed PLGA nanoparticles that encapsulated peptide antigen and three TLR agonists: MALP-2, poly(I:C), and CpG (53). These nanoparticles were subsequently encapsulated within anionic pH-responsive polymer capsules. The capsules were designed to have mean diameters > 10 μm to prevent non-specific phagocytosis and uptake by Peyer’s patches in the small intestine, and to dissolve at pH values greater than 7, characteristic of the terminal ileum of the large intestine, to allow for vaccine release only in this localized region of interest. Significantly stronger T cell responses in the large intestine were generated when capsules of appropriate pH responsiveness were used as a coating rather than an alternative polymer that dissolved at more acidic pH. This general delivery strategy may thus hold potential in the treatment of colorectal tumors and establishes a paradigm for targeting other regions of the GI tract. Together, these studies demonstrate that physically programmable properties of particulate vaccine carriers can be used to specifically target different organs to target the immune response to the required site of protection.
Exploiting the tumor site as an antigen source
A seminal observation in cancer immunology was Dr. William Coley’s discovery in 1893 that repeated intratumoral injection of bacteria could induce tumor rejection. Nearly one hundred years later, trials in humans found that intratumoral administration of BCG in metastatic melanoma lesions resulted not only in the regression of 90% of the injected lesions, but also 17% of distal tumors (54). Although intratumoral injections of immunomodulators are intended to be local treatments, in many cases they can generate a systemic immune response capable of targeting distal tumors in a vaccine-like manner, turning the tumor itself into an in situ vaccine, as depicted in Figure 1C. Importantly, this strategy does not depend on the discovery of tumor-specific antigens, and instead exploits the tumor itself as a source of antigen.
Local administration of diverse immunostimulatory agents to an accessible lesion has been effective at promoting systemic tumor rejection in animal models and humans. For example, intratumoral injection of CpG, anti-OX40, and anti-CTLA4 in mouse lymphoma models can eradicate regulatory T-cells from tumors (55). In humans, topical application of a cream prepared with 5% imiquimod, a TLR7 agonist, has been shown to induce 80% histological clearance in human patients with superficial basal cell carcinoma (56). In some cases, local administration of immunomodulators increases susceptibility to subsequent systemic therapy. In mouse melanoma models with tumors on both the right and left flank, Zamarin et al. showed that oncolytic virus injections into one tumor increased lymphocytic infiltration even in the contralateral tumor, improving the efficacy of systemically administered anti-CTLA4 therapy (57). Analogously in humans, early clinical trials suggest that stereotactic body radiotherapy, where radiation is precisely delivered to tumor sites to enhance local inflammation, can improve responses to IL-2 therapy in patients with metastatic lesions (58). Despite the promise of intratumoral injections in promoting anti-tumor immunity, one deficiency in intratumoral administration of soluble therapeutics is that locally-applied drugs can still rapidly leak into the systemic circulation. This has been observed in many studies in small animals (60–62) and with immunotherapy in humans, where intratumorally-injected cytokines have been measured in the systemic circulation within minutes (63). Such systemic dissemination both weakens the potency of the therapy by clearing the drug from the tumor and gives rise to systemic inflammatory toxicity.
To promote such “in situ vaccination”, biomaterials have been designed to trap immunomodulatory molecules in the tumor environment. For example, slow-release particles or hydrogels have been injected peritumorally or intratumorally, to allow local permeation of tumors with immunostimulatory drugs released from localized depots. This has been demonstrated with biodegradable microspheres releasing IL-12 (62) and hydrogel matrices releasing an IL-15 superagonist (64), both of which led to non-toxic but potent induction of CD8+ T-cell responses against treated tumors. Such approaches can enable otherwise toxic treatments to be safely administered while eliciting systemic anti-tumor immunity. For example, anti-CD137 and IL-2 administered directly into solid melanoma tumors disseminated into the systemic circulation, inducing systemic inflammation, including IL-6 and TNF-α in serum and major weight loss in mice (60). However, intratumoral injection of these same immunomodulators covalently anchored to liposomes prevented their dissemination outside of the local microenvironment (60, 61), eliminating their toxicity and enabling the drugs to remain concentrated at the tumor site for 96 hours post injection. These intratumoral immunoliposomes acted as vaccines and elicited systemic T-cell responses; mice that rejected treated tumors on one flank could also substantially delay the growth of an untreated tumor on the contralateral flank, in the complete absence of supporting systemic therapy. Thus, even relatively simple strategies can be employed to significantly alter the efficacy and safety of immunotherapeutic drugs in this setting.
Active depots with implantable vaccine scaffolds
The only FDA-approved cancer vaccine to date is Provenge, an autologous cell-based therapeutic vaccine against castration-resistant metastatic prostate cancer (65). Although this vaccine was shown to extend survival in prostate cancer patients by 4 months, its implementation is clinically complex, since peripheral blood is first collected from patients, shipped to a cell preparation facility, treated with antigen ex vivo, shipped back to the clinical site, and subsequently re-infused into the patient. Clinical trials of related processes based on the isolation of precursor cells, differentiation of these cells into dendritic cells in vitro, activation and antigen loading of the resulting DCs, and injection as cellular vaccines have also shown promise (66, 67) but with the same logistical concerns. In an attempt to harness the power of DC vaccines without the practical limitations of cell therapy, several strategies have been developed to create implantable or injectable implants that would mimic this series of ex vivo treatment steps directly in patients. The common premise of these approaches is to employ a synthetic matrix or scaffold that when placed in vivo (e.g., following a minor subcutaneous implantation procedure) would release/present cues in the local tissue that enable the processes of attracting, differentiating, activating, and antigen loading dendritic cells, which would subsequently traffic to local draining lymph nodes to initiate an anti-tumor immune response. This concept leverages a large body of experience from the tissue engineering and regenerative medicine field, where biomaterial scaffolds designed to attract and program cell fate have been studied for more than 20 years (68). These biodegradable scaffolds may release immunomodulatory agents with defined spatial and temporal profiles that can be engineered by manipulating the material properties of the implant. Multiple agents can be loaded into a single immunomodulatory scaffold, including antigen, adjuvant, and cytokine support, and they can be designed to promote cell recruitment and modulation within the scaffold itself.
One of the first reports of a DC-programming vaccine system utilized millimeter scale polymer rods that released the chemoattractant CCL19 along with tumor lysate as an antigen preparation (69). These attractant-releasing implants recruited antigen presenting cells to the vaccine site, which correlated with enhanced tumor regression in a therapeutic lung carcinoma model. More recently, Ali et al. designed centimeter-scale porous polymer disks composed of the same polymer used in resorbable sutures (poly(lactide-co-gyolide) or PLGA); these disks were loaded with 3 components: GM-CSF, CpG DNA, and tumor lysate, as shown in Figure 1D (70). These scaffolds released GM-CSF to recruit and differentiate dendritic cells into the structure and CpG as a danger signal to activate DCs internalizing antigens in the tumor lysate. These scaffolds were capable of protecting mice from B16F10 melanoma challenge in a prophylactic setting. In a follow up study, this scaffold vaccine, in combination with vaccination using irradiated tumor cells transduced to express GM-CSF, was shown to also greatly enhance protection relative to non-scaffolded vaccines or GM-CSF-producing tumor cell-based vaccination alone in the therapeutic setting, results which correlated with enhanced recruitment of plasmacytoid DCs, cross-presenting CD8+ DCs, and elevated IL-12 production in the scaffold implants (71). In a rat glioma model, PLGA scaffold vaccines implanted after partial tumor resection resulted in significantly enhanced survival over control blank PLGA matrices (72). Efficacy in this model was only seen when scaffolds were placed next to the resection site but not within the resection site, highlighting the importance of implantation site for these implantable scaffold-based vaccines. Based on these encouraging preclinical results, this promising PLGA scaffold vaccine system was recently moved into a first-in-human phase I trial in patients with melanoma (73).
A number of strategies have sought to generate an in situ-forming immunomodulatory depot that does not require surgical implantation like the PLGA-based scaffolds described above. In a recent report, antigen and adjuvant were mixed with chitosan and hydroxyapatite and co-injected with crosslinking agent tripolyphosphate and chondroitin sulphate via a two needle aligned injection (74). These two aqueous solutions crosslinked in vivo to form a biodegradable hydrogel vaccine that was capable of inducing humoral responses durable for more than a year after implantation following a single injection; this type of sustained release implant may also be of interest for driving anti-tumor T-cell responses. In a second example, Kim et al. demonstrated that biodegradable mesoporous silica rods could nonspecifically coalesce to form a scaffold-like structure following subcutaneous injection (75). When formulated with GM-CSF, CpG, and antigen, these injectable scaffolds were capable of recruiting DCs and priming T-cell responses that were capable of delaying the outgrowth of OVA-expressing tumors in a prophylactic setting. While still in early stages of preclinical development, these “injectable scaffold” approaches may provide a facile strategy to repeatedly prime and boost anti-tumor immunity. Importantly, both implanted and injectable matrix-based vaccines are powerful technological platforms for examining the importance of timing, dosing, and physical localization of immunostimulatory cues on the output immune response, making these systems both potential therapeutics and valuable tools for determining how these factors quantitatively influence the immune response.
Conclusions
Although traditional techniques inspired by prophylactic vaccines activate immune responses, new vaccine concepts are of interest to overcome tumor antigen tolerance and tumor-induced immunosuppression in the setting of advanced cancer and to drive immune responses of the appropriate magnitude and quality to treat large metastastic tumor burdens. Approaches grounded in engineering methods for creating synthetic materials and synthesizing new molecules offer a number of strategies to enhance cancer immunotherapy and cancer vaccines in particular, including improving the delivery of vaccine components to lymphoid organs, optimally programming activated T-cells to home to tumor sites, prolonging immunomodulation of lesions following intratumoral injection, and programming sequential events in immunization from a single injectable or implantable device. Overall, such engineering-based approaches have shown great promise in pre-clinical models, and the next few years should see a number of these approaches moved into clinical testing in patients.
Acknowledgments
This work was supported by the NIH (CA174795 and CA172164), the V Foundation, and the Bridge Project of the Koch Institute and the Dana Farber Harvard Cancer Center. KDM is supported by a graduate fellowship from the Hertz Foundation. KDM and NKM are supported by graduate research fellowships from NSF.
References
- 1.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10(9):909–15. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, Thatcher N, Wagstaff J, Zielinski C, Faulkner I, Mellstedt H. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol. 2014;11(9):509–24. doi: 10.1038/nrclinonc.2014.111. [DOI] [PubMed] [Google Scholar]
- 3.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 4.Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, Solary E, Le Cesne A, Zitvogel L, Chauffert B. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. 2007;56(5):641–8. doi: 10.1007/s00262-006-0225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vacchelli E, Aranda F, Eggermont A, Sautes-Fridman C, Tartour E, Kennedy EP, Platten M, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology. 2014;3(10):ARTN e957994. doi: 10.4161/21624011.2014.957994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12(4):237–51. doi: 10.1038/nrc3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fritsch EF, Hacohen N, Wu CJ. Personal neoantigen cancer vaccines: The momentum builds. Oncoimmunology. 2014;3:e29311. doi: 10.4161/onci.29311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, Mulder GE, Toebes M, Vesely MD, Lam SS, Korman AJ, Allison JP, Freeman GJ, Sharpe AH, Pearce EL, Schumacher TN, Aebersold R, Rammensee HG, Melief CJ, Mardis ER, Gillanders WE, Artyomov MN, Schreiber RD. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515(7528):577–81. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, Modrusan Z, Mellman I, Lill JR, Delamarre L. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515(7528):572–6. doi: 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
- 10.Barnes E, Folgori A, Capone S, Swadling L, Aston S, Kurioka A, Meyer J, Huddart R, Smith K, Townsend R, Brown A, Antrobus R, Ammendola V, Naddeo M, O’Hara G, Willberg C, Harrison A, Grazioli F, Esposito ML, Siani L, Traboni C, Oo Y, Adams D, Hill A, Colloca S, Nicosia A, Cortese R, Klenerman P. Novel adenovirus-based vaccines induce broad and sustained T cell responses to HCV in man. Sci Transl Med. 2012;4(115):115ra1. doi: 10.1126/scitranslmed.3003155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller JD, van der Most RG, Akondy RS, Glidewell JT, Albott S, Masopust D, Murali-Krishna K, Mahar PL, Edupuganti S, Lalor S, Germon S, Del Rio C, Mulligan MJ, Staprans SI, Altman JD, Feinberg MB, Ahmed R. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity. 2008;28(5):710–22. doi: 10.1016/j.immuni.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 12.Speiser DE, Lienard D, Rufer N, Rubio-Godoy V, Rimoldi D, Lejeune F, Krieg AM, Cerottini JC, Romero P. Rapid and strong human CD8+ T cell responses to vaccination with peptide, IFA, and CpG oligodeoxynucleotide 7909. J Clin Invest. 2005;115(3):739–46. doi: 10.1172/JCI23373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenberg SA, Sherry RM, Morton KE, Scharfman WJ, Yang JC, Topalian SL, Royal RE, Kammula U, Restifo NP, Hughes MS, Schwartzentruber D, Berman DM, Schwarz SL, Ngo LT, Mavroukakis SA, White DE, Steinberg SM. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol. 2005;175(9):6169–76. doi: 10.4049/jimmunol.175.9.6169. [DOI] [PubMed] [Google Scholar]
- 14.Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, Staehler M, Brugger W, Dietrich P-Y, Mendrzyk R, Hilf N, Schoor O, Fritsche J, Mahr A, Maurer D, Vass V, Trautwein C, Lewandrowski P, Flohr C, Pohla H, Stanczak JJ, Bronte V, Mandruzzato S, Biedermann T, Pawelec G, Derhovanessian E, Yamagishi H, Miki T, Hongo F, Takaha N, Hirakawa K, Tanaka H, Stevanović S, Frisch J, Mayer-Mokler A, Kirner A, Rammensee H-G, Reinhardt C, Singh-Jasuja H. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nature Medicine. 2012 doi: 10.1038/nm.2883. [DOI] [PubMed] [Google Scholar]
- 15.Odunsi K, Matsuzaki J, Karbach J, Neumann A, Mhawech-Fauceglia P, Miller A, Beck A, Morrison CD, Ritter G, Godoy H, Lele S, Dupont N, Edwards R, Shrikant P, Old LJ, Gnjatic S, Jäger E. Efficacy of vaccination with recombinant vaccinia and fowlpox vectors expressing NY-ESO-1 antigen in ovarian cancer and melanoma patients. Proceedings of the National Academy of Sciences. 2012 doi: 10.1073/pnas.1117208109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, Gailani F, Riley L, Conlon K, Pockaj B, Kendra KL, White RL, Gonzalez R, Kuzel TM, Curti B, Leming PD, Whitman ED, Balkissoon J, Reintgen DS, Kaufman H, Marincola FM, Merino MJ, Rosenberg SA, Choyke P, Vena D, Hwu P. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. The New England journal of medicine. 2011;364(22):2119–27. doi: 10.1056/NEJMoa1012863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Melief CJM, van der Burg SH. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nature reviews Cancer. 2008;8(5):351–60. doi: 10.1038/nrc2373. [DOI] [PubMed] [Google Scholar]
- 18.Sandoval F, Terme M, Nizard M, Badoual C, Bureau MF, Freyburger L, Clement O, Marcheteau E, Gey A, Fraisse G, Bouguin C, Merillon N, Dransart E, Tran T, Quintin-Colonna F, Autret G, Thiebaud M, Suleman M, Riffault S, Wu TC, Launay O, Danel C, Taieb J, Richardson J, Zitvogel L, Fridman WH, Johannes L, Tartour E. Mucosal imprinting of vaccine-induced CD8(+) T cells is crucial to inhibit the growth of mucosal tumors. Sci Transl Med. 2013;5(172):172ra20. doi: 10.1126/scitranslmed.3004888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Itano AA, McSorley SJ, Reinhardt RL, Ehst BD, Ingulli E, Rudensky AY, Jenkins MK. Distinct Dendritic Cell Populations Sequentially Present Antigen to CD4 T Cells and Stimulate Different Aspects of Cell-Mediated Immunity. Immunity. 2003;19(1):47–57. doi: 10.1016/s1074-7613(03)00175-4. [DOI] [PubMed] [Google Scholar]
- 20.Anandasabapathy N, Feder R, Mollah S, Tse SW, Longhi MP, Mehandru S, Matos I, Cheong C, Ruane D, Brane L, Teixeira A, Dobrin J, Mizenina O, Park CG, Meredith M, Clausen BE, Nussenzweig MC, Steinman RM. Classical Flt3L-dependent dendritic cells control immunity to protein vaccine. Journal of Experimental Medicine. 2014;211(9):1875–91. doi: 10.1084/jem.20131397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johansen P, Haffner AC, Koch F, Zepter K, Erdmann I, Maloy K, Simard JJ, Storni T, Senti G, Bot A, Wuthrich B, Kundig TM. Direct intralymphatic injection of peptide vaccines enhances immunogenicity. Eur J Immunol. 2005;35(2):568–74. doi: 10.1002/eji.200425599. [DOI] [PubMed] [Google Scholar]
- 22.Maloy KJ, Erdmann I, Basch V, Sierro S, Kramps TA, Zinkernagel RM, Oehen S, Kundig TM. Intralymphatic immunization enhances DNA vaccination. Proc Natl Acad Sci U S A. 2001;98(6):3299–303. doi: 10.1073/pnas.051630798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nature Reviews Immunology. 2010;10(11):787–96. doi: 10.1038/nri2868. [DOI] [PubMed] [Google Scholar]
- 24.Supersaxo A, Hein WR, Steffen H. Effect of Molecular Weight on the Lymphatic Absorption of Water-Soluble Compounds Following Subcutaneous Administration. Pharm Res. 1990;7(2):167–9. doi: 10.1023/A:1015880819328. [DOI] [PubMed] [Google Scholar]
- 25.McLennan DN, Porter CJH, Charman SA. Subcutaneous drug delivery and the role of the lymphatics. Drug Discovery Today: Technologies. 2005;2(1):89–96. doi: 10.1016/j.ddtec.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 26.Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B, Van Egeren DS, Park C, Irvine DJ. Structure-based programming of lymph-node targeting in molecular vaccines. Nature. 2014;507(7493):519–22. doi: 10.1038/nature12978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smirnov D, Schmidt JJ, Capecchi JT, Wightman PD. Vaccine adjuvant activity of 3M-052: an imidazoquinoline designed for local activity without systemic cytokine induction. Vaccine. 2011;29(33):5434–42. doi: 10.1016/j.vaccine.2011.05.061. [DOI] [PubMed] [Google Scholar]
- 28.Hanson MC, Crespo MP, Abraham W, Moynihan KD, Szeto GL, Chen SH, Melo MB, Mueller S, Irvine DJ. Nanoparticulate STING agonists are potent lymph node-targeted vaccine adjuvants. J Clin Invest. doi: 10.1172/JCI79915. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hailemichael Y, Dai Z, Jaffarzad N, Ye Y, Medina MA, Huang XF, Dorta-Estremera SM, Greeley NR, Nitti G, Peng W, Liu C, Lou Y, Wang Z, Ma W, Rabinovich B, Sowell RT, Schluns KS, Davis RE, Hwu P, Overwijk WW. Persistent antigen at vaccination sites induces tumor-specific CD8(+) T cell sequestration, dysfunction and deletion. Nat Med. 2013;19(4):465–72. doi: 10.1038/nm.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reddy ST, Rehor A, Schmoekel HG, Hubbell JA, Swartz MA. In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. Journal of Controlled Release. 2006;112(1):26–34. doi: 10.1016/j.jconrel.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 31.Manolova V, Flace A, Bauer M, Schwarz K, Saudan P, Bachmann MF. Nanoparticles target distinct dendritic cell populations according to their size. European Journal of Immunology. 2008;38(Lcmv):1404–13. doi: 10.1002/eji.200737984. [DOI] [PubMed] [Google Scholar]
- 32.Fifis T, Gamvrellis A, Crimeen-Irwin B, Pietersz GA, Li J, Mottram PL, McKenzie IF, Plebanski M. Size-dependent immunogenicity: therapeutic and protective properties of nano-vaccines against tumors. J Immunol. 2004;173(5):3148–54. doi: 10.4049/jimmunol.173.5.3148. [DOI] [PubMed] [Google Scholar]
- 33.Moon JJ, Suh H, Bershteyn A, Stephan MT, Liu H, Huang B, Sohail M, Luo S, Um SH, Khant H, Goodwin JT, Ramos J, Chiu W, Irvine DJ. Interbilayer-crosslinked multilamellar vesicles as synthetic vaccines for potent humoral and cellular immune responses. Nat Mater. 2011;10(3):243–51. doi: 10.1038/nmat2960. Epub 2011/02/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kasturi SP, Skountzou I, Albrecht RA, Koutsonanos D, Hua T, Nakaya HI, Ravindran R, Stewart S, Alam M, Kwissa M, Villinger F, Murthy N, Steel J, Jacob J, Hogan RJ, Garcia-Sastre A, Compans R, Pulendran B. Programming the magnitude and persistence of antibody responses with innate immunity. Nature. 2011;470(7335):543–7. doi: 10.1038/nature09737. Epub 2011/02/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.St John AL, Chan CY, Staats HF, Leong KW, Abraham SN. Synthetic mast-cell granules as adjuvants to promote and polarize immunity in lymph nodes. Nature Materials. 2012 doi: 10.1038/nmat3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Titta A, Ballester M, Julier Z, Nembrini C, Jeanbart L, van der Vlies AJ, Swartz MA, Hubbell JA. Nanoparticle conjugation of CpG enhances adjuvancy for cellular immunity and memory recall at low dose. Proc Natl Acad Sci U S A. 2013;110(49):19902–7. doi: 10.1073/pnas.1313152110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O’Neil CP, Lee LK, Swartz MA, Hubbell JA. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat Biotechnol. 2007;25(10):1159–64. doi: 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- 38.Thomas SN, Vokali E, Lund AW, Hubbell JA, Swartz MA. Targeting the tumor-draining lymph node with adjuvanted nanoparticles reshapes the anti-tumor immune response. Biomaterials. 2014;35(2):814–24. doi: 10.1016/j.biomaterials.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 39.Fraser CC, Altreuter DH, Ilyinskii P, Pittet L, LaMothe Ra, Keegan M, Johnston L, Kishimoto TK. Generation of a universal CD4 memory T cell recall peptide effective in humans, mice and non-human primates. Vaccine. 2014;32(24):2896–903. doi: 10.1016/j.vaccine.2014.02.024. [DOI] [PubMed] [Google Scholar]
- 40.Wu TYH, Singh M, Miller AT, De Gregorio E, Doro F, D’Oro U, Skibinski DAG, Mbow ML, Bufali S, Herman AE, Cortez A, Li Y, Nayak BP, Tritto E, Filippi CM, Otten GR, Brito LA, Monaci E, Li C, Aprea S, Valentini S, Calabro S, Laera D, Brunelli B, Caproni E, Malyala P, Panchal RG, Warren TK, Bavari S, O’Hagan DT, Cooke MP, Valiante NM. Rational design of small molecules as vaccine adjuvants. Science translational medicine. 2014;6(263):12. doi: 10.1126/scitranslmed.3009980. [DOI] [PubMed] [Google Scholar]
- 41.Kourtis IC, Hirosue S, de Titta A, Kontos S, Stegmann T, Hubbell JA, Swartz MA. Peripherally administered nanoparticles target monocytic myeloid cells, secondary lymphoid organs and tumors in mice. PloS one. 2013;8(4):e61646. doi: 10.1371/journal.pone.0061646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shen H, Ackerman AL, Cody V, Giodini A, Hinson ER, Cresswell P, Edelson RL, Saltzman WM, Hanlon DJ. Enhanced and prolonged cross-presentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles. Immunology. 2006;117(1):78–88. doi: 10.1111/j.1365-2567.2005.02268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krishnamachari Y, Geary SM, Lemke CD, Salem AK. Nanoparticle delivery systems in cancer vaccines. Pharm Res. 2011;28(2):215–36. doi: 10.1007/s11095-010-0241-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fang RH, Hu CM, Luk BT, Gao W, Copp JA, Tai Y, O’Connor DE, Zhang L. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014;14(4):2181–8. doi: 10.1021/nl500618u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.von Andrian UH, Mackay CR. T-Cell Function and Migration: Two Sides of the Same Coin. The New England Journal of Medicine. 2000;343(14):15. doi: 10.1056/NEJM200010053431407. [DOI] [PubMed] [Google Scholar]
- 46.Sandoval F, Terme M, Nizard M, Badoual C, Bureau M-F, Freyburger L, Clement O, Marcheteau E, Gey A, Fraisse G, et al. Mucosal imprinting of vaccine-induced CD8+ T cells is crucial to inhibit the growth of mucosal tumors. Science translational medicine. 2013;5(172):172ra20–ra20. doi: 10.1126/scitranslmed.3004888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nochi T, Yuki Y, Takahashi H, Sawada S, Mejima M, Kohda T, Harada N, Kong IG, Sato A, Kataoka N, Tokuhara D, Kurokawa S, Takahashi Y, Tsukada H, Kozaki S, Akiyoshi K, Kiyono H. Nanogel antigenic protein-delivery system for adjuvant-free intranasal vaccines. Nat Mater. 2010;9(7):572–8. doi: 10.1038/nmat2784. Epub 2010/06/22 nmat2784 [pii] [DOI] [PubMed] [Google Scholar]
- 48.Li AV, Moon JJ, Abraham W, Suh H, Elkhader J, Seidman MA, Yen M, Im EJ, Foley MH, Barouch DH, Irvine DJ. Generation of effector memory T cell-based mucosal and systemic immunity with pulmonary nanoparticle vaccination. Sci Transl Med. 2013;5(204):204ra130. doi: 10.1126/scitranslmed.3006516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li AV, Moon JJ, Abraham W, Suh H, Elkhader J, Seidman MA, Yen M, Im E-J, Foley MH, Barouch DH, et al. Generation of Effector Memory T Cell–Based Mucosal and Systemic Immunity with Pulmonary Nanoparticle Vaccination. Science translational medicine. 2013;5(204):204ra130–204ra130. doi: 10.1126/scitranslmed.3006516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matsuo K, Koizumi H, Akashi M, Nakagawa S, Fujita T, Yamamoto A, Okada N. Intranasal immunization with poly(γ-glutamic acid) nanoparticles entrapping antigenic proteins can induce potent tumor immunity. Journal of Controlled Release. 2011;152(2):310–6. doi: 10.1016/j.jconrel.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 51.Stano A, Nembrini C, Swartz MA, Hubbell JA, Simeoni E. Nanoparticle size influences the magnitude and quality of mucosal immune responses after intranasal immunization. Vaccine. 2012 doi: 10.1016/j.vaccine.2012.10.050. Epub 2012/10/30 S0264-410X(12)01496-X [pii] [DOI] [PubMed] [Google Scholar]
- 52.Nembrini C, Stano A, Dane KY, Ballester M, van der Vlies AJ, Marsland BJ, Swartz MA, Hubbell JA. Nanoparticle conjugation of antigen enhances cytotoxic T-cell responses in pulmonary vaccination. Proceedings of the National Academy of Sciences. 2011;108(44):E989–E97. doi: 10.1073/pnas.1104264108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu Q, Talton J, Zhang G, Cunningham T, Wang Z, Waters RC, Kirk J, Eppler B, Klinman DM, Sui Y, Gagnon S, Belyakov IM, Mumper RJ, Berzofsky JA. Large intestine-targeted, nanoparticle-releasing oral vaccine to control genitorectal viral infection. Nature medicine. 2012;18(8):1291–6. doi: 10.1038/nm.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morton DL, Eilber FR, Holmes EC, Hunt JS, Ketcham AS, Silverstein MJ, Sparks FC. BCG Immunotherapy of Malignant Melanoma: Summary of a Seven-year Experience. Annals of Surgery. 1974;180(4):635–41. doi: 10.1097/00000658-197410000-00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G, Rajapaksa R, Green MR, Torchia J, Brody J, Luong R, Rosenblum MD, Steinman L, Levitsky HI, Tse V, Levy R. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. Journal of Clinical Investigation. 2013;123(6):2447–63. doi: 10.1172/JCI64859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schulze HJ, Cribier B, Requena L, Reifenberger J, Ferrandiz C, Garcia Diez A, Tebbs V, McRae S. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: results from a randomized vehicle-controlled phase III study in Europe. British Journal of Dermatology. 2005;152(5):939–47. doi: 10.1111/j.1365-2133.2005.06486.x. [DOI] [PubMed] [Google Scholar]
- 57.Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, Merghoub T, Wolchok JD, Allison JP. Localized Oncolytic Virotherapy Overcomes Systemic Tumor Resistance to Immune Checkpoint Blockade Immunotherapy. Science Translational Medicine. 2014;6(226):12. doi: 10.1126/scitranslmed.3008095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seung SK, Curti BD, Crittenden M, Walker E, Coffey T, Siebert JC, Miller W, Payne R, Glenn L, Bageac A, Urba WJ. Phase 1 Study of Stereotactic Body Radiotherapy and Interleukin-2: Tumor and Immunological Responses. Science Translational Medicine. 2012;4(137):7. doi: 10.1126/scitranslmed.3003649. [DOI] [PubMed] [Google Scholar]
- 59.Le Mercier I, Poujol D, Sanlaville A, Sisirak V, Gobert M, Durand I, Dubois B, Treilleux I, Marvel J, Vlach J, Blay JY, Bendriss-Vermare N, Caux C, Puisieux I, Goutagny N. Tumor Promotion by Intratumoral Plasmacytoid Dendritic Cells Is Reversed by TLR7 Ligand Treatment. Cancer Research. 2013;73(15):4629–40. doi: 10.1158/0008-5472.CAN-12-3058. [DOI] [PubMed] [Google Scholar]
- 60.Kwong B, Gai SA, Elkhader J, Wittrup KD, Irvine DJ. Localized immunotherapy via liposome-anchored Anti-CD137 + IL-2 prevents lethal toxicity and elicits local and systemic antitumor immunity. Cancer Res. 2013;73(5):1547–58. doi: 10.1158/0008-5472.CAN-12-3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kwong B, Liu H, Irvine DJ. Induction of potent anti-tumor responses while eliminating systemic side effects via liposome-anchored combinatorial immunotherapy. Biomaterials. 2011;32(22):5134–47. doi: 10.1016/j.biomaterials.2011.03.067. Epub 2011/04/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Egilmez NK, Jong YS, Sabel MS, Jacob JS, Mathiowitz E, Bankert RB. In situ tumor vaccination with interleukin-12-encapsulated biodegradable microspheres: induction of tumor regression and potent antitumor immunity. Cancer Res. 2000;60(14):3832–7. [PubMed] [Google Scholar]
- 63.Herpen V, Huijbens R, Looman M, de Vries J. Pharmacokinetics and Immunological Aspects of a Phase Ib Study with Intratumoral Administration of Recombinant Human Interleukin-12 in Patients with …. Clinical Cancer …. 2003 [PubMed] [Google Scholar]
- 64.Hori Y, Stern PJ, Hynes RO, Irvine DJ. Engulfing tumors with synthetic extracellular matrices for cancer immunotherapy. Biomaterials. 2009;30(35):6757–67. doi: 10.1016/j.biomaterials.2009.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pasquié J-l, Ph D, Scavée C, Bordachar P, Clémenty J, Haïssaguerre M. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. New England Journal of Medicine. 2010:2373–83. doi: 10.1056/NEJMoa1407764. [DOI] [Google Scholar]
- 66.Mitchell DA, Batich KA, Gunn MD, Huang MN, Sanchez-Perez L, Nair SK, Congdon KL, Reap EA, Archer GE, Desjardins A, Friedman AH, Friedman HS, Herndon JE, 2nd, Coan A, McLendon RE, Reardon DA, Vredenburgh JJ, Bigner DD, Sampson JH. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature. 2015;519(7543):366–9. doi: 10.1038/nature14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Palucka K, Banchereau J. Dendritic-Cell-Based Therapeutic Cancer Vaccines. Immunity. 2013;39(1):38–48. doi: 10.1016/j.immuni.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Langer R, Vacanti JP. Tissue engineering. Science. 1993;260(5110):920–6. doi: 10.1126/science.8493529. [DOI] [PubMed] [Google Scholar]
- 69.Kumamoto T, Huang EK, Paek HJ, Morita A, Matsue H, Valentini RF, Takashima A. Induction of tumor-specific protective immunity by in situ Langerhans cell vaccine. Nature biotechnology. 2002;20(January):64–9. doi: 10.1038/nbt0102-64. [DOI] [PubMed] [Google Scholar]
- 70.Ali Oa, Huebsch N, Cao L, Dranoff G, Mooney DJ. Infection-mimicking materials to program dendritic cells in situ. Nature materials. 2009;8(2):151–8. doi: 10.1038/nmat2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ali Oa, Emerich D, Dranoff G, Mooney DJ. In situ regulation of DC subsets and T cells mediates tumor regression in mice. Science translational medicine. 2009;1(8):8ra19-8ra. doi: 10.1126/scitranslmed.3000359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ali OA, Doherty E, Bell WJ, Fradet T, Hudak J, Laliberte M-T, Mooney DJ, Emerich DF. The efficacy of intracranial PLG-based vaccines is dependent on direct implantation into brain tissue. Journal of Controlled Release. 2011;154(3):249–57. doi: 10.1016/j.jconrel.2011.06.021. [DOI] [PubMed] [Google Scholar]
- 73.Stephen Hodi FM Dana-Farber Cancer Institute. clinicaltrials.gov. 2014. Dendritic Cell Activating Scaffold in Melanoma; p. NCT01753089. [Google Scholar]
- 74.Chua BY, Sekiya T, Al Kobaisi M, Short KR, Mainwaring DE, Jackson DC. A single dose biodegradable vaccine depot that induces persistently high levels of antibody over a year. Biomaterials. 2015;53:50–7. doi: 10.1016/j.biomaterials.2015.02.066. [DOI] [PubMed] [Google Scholar]
- 75.Kim J, Li WA, Choi Y, Lewin Sa, Verbeke CS, Dranoff G, Mooney DJ. Injectable, spontaneously assembling, inorganic scaffolds modulate immune cells in vivo and increase vaccine efficacy. Nature Biotechnology. 2014;(December) doi: 10.1038/nbt.3071. [DOI] [PMC free article] [PubMed] [Google Scholar]