

Abstract
The ADP/ATP carrier (AAC) transports ADP and ATP across the inner mitochondrial membrane. Unlike most transporters that have 2-fold direct or inverted quasi-symmetry, AAC has the apparent 3-fold rotational symmetry. Further, its transport rate is fast for transporters that carry large solutes. Here, we perform comprehensive NMR relaxation dispersion measurements for the yeast AAC carrier 3, which provide residue-specific information on the protein conformational exchange. Our data indicate that AAC is predominantly in the cytosol-facing open state and converts to a lowly populated state in an asymmetric manner despite its three-fold structural symmetry. Binding of the substrate ADP significantly increases the rate of conformational exchange, whereas the inhibitor CATR slows the exchange. These results suggest that while the transporter catalyzes the translocation of substrate, the substrate also facilitates interconversion between alternating states that may be relevant to the transport function.
A key aspect of the transport mechanism of solute carriers is the dynamic coupling between solute binding and protein conformational switch that facilitates solute translocation across the membrane1,2. With the rapid growth of crystal structures capturing various states of transporters and advances in molecular dynamics (MD) simulation, it is increasingly feasible to address transporter conformational dynamics by computational means3–5. But experimental observation of transporter dynamics is still rare and has relied mostly on spectroscopic methods such as fluorescence resonance energy transfer (FRET). The high sensitivity of FRET affords measurement at single molecule level and has been employed to gain insights into the dynamic properties of LacY, LeuT and GltPh6–10.
The use of NMR to investigate membrane protein structures and dynamics is an emerging area of research11–15. NMR has much lower sensitivity compared to FRET but holds the advantage of providing residue-specific information on conformational exchange. Since NMR experiments are sensitive to the time scale and magnitude of chemical exchange, they can be tailored to probe various dynamic processes of solute carriers. For systems that are slow on the NMR time scale, the ZZ-exchange16,17 and the chemical exchange saturation transfer (CEST)18 are sensitive methods for measuring protein conformational dynamics. For exchange processes on the μs–ms time scale, the R2 relaxation dispersion is the suitable probe19,20, which has been used successfully to detect lowly populated states (known alternatively as “invisible states”)21,22. Thus far, the only comprehensive NMR measurement of conformational exchange of a membrane transporter is on the multi-drug resistance transporter EmrE bound to the substrate TTP+13. In that study, the ZZ-exchange measurements showed that EmrE undergoes conformational exchange with a rate constant of 4.8 s−1 between an inward- and outward-facing state, and this conformational change is responsible for substrate transport through the membrane.
We sought to investigate the conformational dynamics of the mitochondrial ADP/ATP carrier (AAC), which is an essential protein in eukaryotes responsible for exchanging ADP and ATP across the inner mitochondrial membrane23,24. The AAC is a much faster transporter than EmrE; high time resolution measurements showed that the transport rates for ADP and ATP are 400 s−1 and 160 s−1, respectively25. At this rate, the ZZ-exchange is not applicable and NMR measurement of conformational exchange would require the use of the less sensitive methods for measuring R2 relaxation dispersion. The AAC belongs to the mitochondrial carrier family (MCF) of transporters that catalyzes the trafficking of metabolites and ions into and out of the matrix23,26. It is also the only MCF member for which high resolution structures have been determined27,28. The crystal structures of AAC bound to the inhibitor carboxyatractyloside (CATR) resemble an open-top barrel formed by three structurally similar domains in parallel orientation (Fig. 1a&b). Each domain consists of two transmembrane (TM) helices separated by an amphipathic (AP) helix. It has been proposed that ADP/ATP transport involves interconversion between a cytosol-facing open state (c-state) and a matrix-facing open state (m-state)24. The crystal structure of AAC is open to the cytosol side and thus represents the c-state. The m-state structure is still unknown.
Figure 1. AAC architecture and its functional reconstitution in DPC.
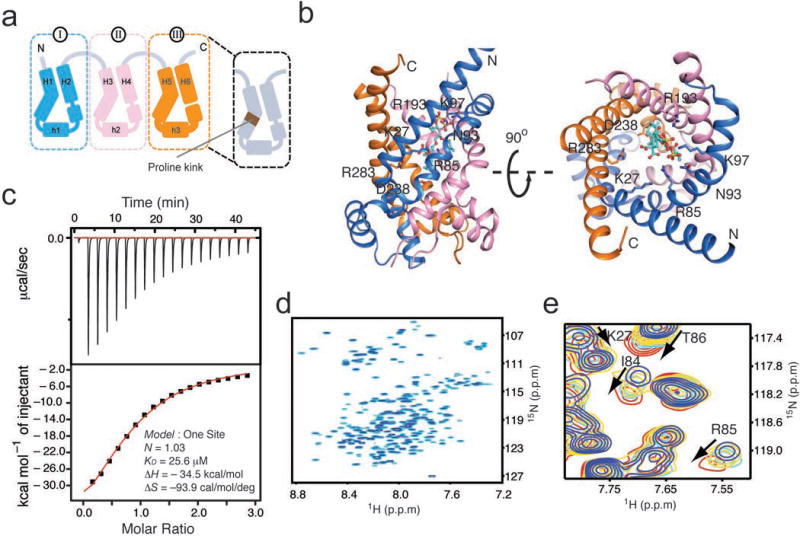
a. Schematic of the tripartite topology of AAC. AAC is composed of three domains with homologous sequence and structure. Each domain contains two long TM helices and an amphipathic helix on the mitochondrial matrix side. Conserved prolines located approximately at the middle of the odd-numbered TM helices cause large kinks in these helices.
b. Ribbon representation of the crystal structure of the yAAC3-CATR complex (PDB ID: 4C9Q28). Domains I, II, and III are colored in blue, pink and yellow, respectively, as in (a). The CATR molecule is shown as green sticks. Labeled residues are those showing close contact with CATR.
c. Isothermal titration calorimetry analysis of yAAC3 binding to CATR. The top and bottom graphs show the raw data (μcal/second vs. time) and normalized integration data (kcal/mole of injectant vs. molar ratio). Data fitting yields KD = 14.9 ± 9.6 μM (Error is given as s.d. for n=3; KD = 25.6 μM, KD = 11.0 μM, KD = 8.1 μM).
d. The 15N TROSY-HSQC spectrum of 15N/13C/2H-labelled yAAC3 reconstituted in 120 mM DPC micelles at pH 6.0, recorded at 30 °C and 1H frequency of 600 MHz.
e. A region of the 15N TROSY-HSQC showing serial perturbation of several residues of yAAC3 (0.8 mM) involved in CATR binding according to the crystal structure. The spectra were recorded at total CATR concentrations of 0, 1, 2, and 4 mM, which are shown in blue, yellow, cyan and red, respectively.
Several open questions are associated with AAC and MCF members in general, in particular, relating to their unusual structural symmetry compared to most solute carriers. First, the AAC has a 3-fold longitudinal quasi-symmetry (symmetry axis perpendicular to the membrane) instead of the more common 2-fold symmetry. It is unclear whether the AAC undergoes the “V” to inverted “V” type of conformational switch, which has been proposed as possible transport mechanism24 and has been determent as the transport mechanism for LacY10 and EmrE13. Here, we performed comprehensive measurements of R2 relaxation dispersion of the yeast AAC to address the intrinsic and substrate- and inhibitor-modulated conformational exchange of this architecturally unorthodox solute transporter.
Results
A functionally relevant NMR sample of AAC
We first established an NMR amenable and functionally relevant AAC sample. We expressed the yeast AAC carrier 3 (yAAC3) in E.coli cells and purified the carrier by Ni-NTA affinity, ion exchange, ATP affinity, and size-exclusion chromatography (Online Methods). We reconstituted the purified yAAC3 in dodeclyphosphocholine (DPC) micelles at pH 6 for NMR measurements. Since AAC has a highly specific inhibitor CATR, we used CATR-binding to examine proper folding of yAAC3 in the NMR sample. Isothermal titration calorimetry (ITC) showed that CATR binds DPC-reconstituted yAAC3 with a dissociation constant (KD) of ~15 μM (Fig. 1c; Supplementary Fig. 1a). Previously, a more native inhibitor binding study showed that atractyloside (ATR), a CATR derivative, binds yeast mitochondria over-expressing AAC carrier 2 (yAAC2) with a KD of 192 μM29. Another earlier study of ATP transport inhibition showed that CATR binds to AAC about 10 times stronger than ATR30,31. The combined results indirectly suggest the KD of CATR binding to yAAC3 is ~20 μM under native condition, which is consistent with our observed CATR binding in the NMR sample.
The sample yielded a good quality NMR spectrum (Fig. 1d) that is characteristic of α helical proteins. Chemical shift changes of a 0.8 mM yAAC3 sample titrated with CATR indicates binding saturation at around 4 mM CATR (total inhibitor concentration). Simulated binding curves for various KD values at fixed protein concentration (0.8 mM) indicates that the KD from NMR titration is ~150 μM (Supplementary Fig. 1b&c). The increased KD measured by NMR compared to ITC for CATR and ADP is most likely due to the increased detergent concentration necessary for NMR experiments. In NMR titration measurements, most of the residues expected to be involved in CATR binding showed a CATR concentration dependent chemical shift perturbation, including residues Lys27, Ile84, Arg85 and Thr86 (Fig. 1e). These residues interact with CATR according to the yeast AAC-CATR complex crystal structure determined recently28. We further showed by NMR titration that ADP addition leads to concentration dependent chemical shift perturbation for several cavity residues, including the Arg141 believed to directly interact with ADP. Analysis of the titration data suggests a KD of ~500 μM for ADP binding (Supplementary Fig. 1d&e). We further showed ADP binding with ITC (Supplementary Fig. 1f). The current and past results together indicate that the yAAC3 reconstituted in DPC is suitable for both NMR and functional measurements.
Measurement of R2 relaxation dispersion
Having a functionally relevant NMR sample of yAAC3, we conducted relaxation dispersion measurements for the transporter in three different states: the free form, in the presence of the substrate ADP, and in the presence of the inhibitor CATR. The presence of a second, less populated state in equilibrium with the major state of a protein would lead to line-broadening of the major state NMR peaks, and this effect can be modulated by applying a train of 180° radio-frequency (RF) pulses in the Carr-Purcell-Meiboom-Gill (CPMG) pulse sequence19,20. The shape of the resulting relaxation dispersion curve depends on the population of the two states (pi), the chemical shift difference (Δω), and the rate of state interconversion (kex)19,20. Since the backbone (1H, 15N) resonances of yAAC3 in a 2D 1H-15N correlation spectrum are very overlapped, we measured relaxation dispersion in the 3D mode by introducing the TROSY 15N-CPMG element32 into the 3D TROSY-HNCO pulse sequence (Supplementary Fig. 2). For peaks with lower signal-to-noise (SN) ratio, the more sensitive 2D TROSY-HSQC based experiment was used. The combined data from 3D and 2D 15N-CPMG experiments recorded at magnetic field strength of 600 and 700 MHz allowed us to derive the μs–ms motion of ~95% of the assigned residues of the free and ADP-bound yAAC3. For the CATR-bound state, a smaller number of residues could be analyzed due to decreased SN ratios.
AAC shows exchange between the c-state and an excited state
Non-flat relaxation dispersion profiles were detected for residues in the TM helices (H1-H3, H5, H6) and the AP helices h1 and h2 (examples shown in Fig. 2a). Fitting these curves individually to a numerical solution of the Bloch equation33 yields similar values of rates and populations for the free yAAC3. For the ADP- and CATR-bound forms of yAAC3, however, the rates are different between the TM and AP helices. Therefore, when performing global fitting of dispersion data to a two-state exchange model34,35 (Online Methods), TM and AP regions were treated separately for the three sample conditions, to obtain a consistent fit for all three states. For the free yAAC3, the global fit yields kex ~ 870 ± 200 s−1 for TM and 940 ± 170 s−1 for AP regions (Table 1; Supplementary Table 1). The fit also yields relative populations of 98.0 ± 0.4 % and 2.0 ± 0.4 % for the two states at equilibrium with maximum Δω being as large as 5.7 ppm in 15N. To further validate the relaxation dispersion parameter obtained from two fields, additional 2D relaxation dispersion experiments were performed at 800 MHz for free AAC and in the presence of ADP (Supplementary Fig. 3). Fitting to a two-state exchange model to data from all three fields yields very similar exchange parameters (Supplementary Table 2). Since the NMR spectrum of the free yAAC3 is very similar to that of the CATR-bound yAAC3, the major population should be in the c-state (or the ground state). The c-state transiently converts to a lowly populated excited state. Although there is no direct information on the conformation of the excite state, it is expected to be structurally distinct from the c-state based on the large Δω values.
Figure 2. Conformational exchange detected by NMR relaxation dispersion.
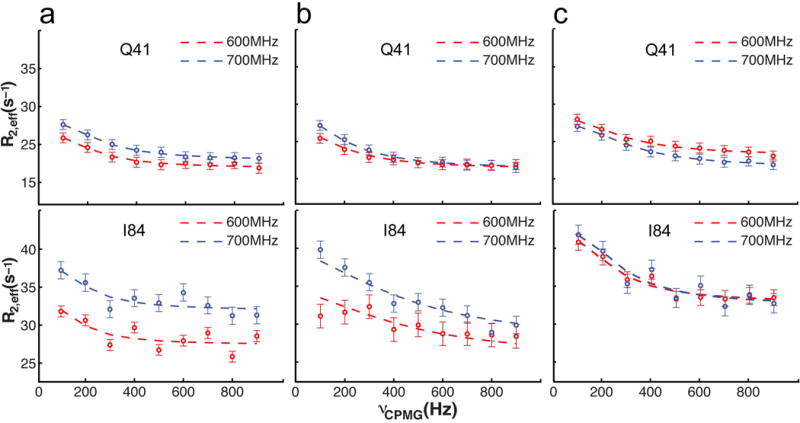
Examples of 15N relaxation dispersion profiles for Gln41 and Ile84 measured at 1H frequencies of 600 (red circles) and 700 (blue circles) MHz. Error bars are ± one s.d. values (n=4) determined from repeat experiments.
a. Relaxation dispersion of yAAC3 in the absence of ligands.
b. Relaxation dispersion of yAAC3 in the presence of 10 mM ADP.
c. Relaxation dispersion of yAAC3 in the presence of 3 mM CATR.
Table 1.
Kinetic and thermodynamic parameters of yAAC3 derived from relaxation dispersion measurements.
| State | Protein Region | kex (s−1) | pe (%) a |
|---|---|---|---|
| Free yAAC3 | TM helices | 870 ± 200 | 2.0 ± 0.4 |
| AP helices | 940 ± 170 | 2.0 ± 0.4 | |
|
| |||
| yAAC3-ADP | TM helices | 1800 ± 350 | 2.0 ± 0.6 |
| AP helices | 770 ± 450 | 2.0 ± 0.6 | |
|
| |||
| yAAC3-CATR | TM helices | 150 ± 110 | 2.0 ± 1.5 |
| AP helices | 1040 ± 300 | 2.0 ± 1.0 | |
The symbol pe denotes the population of the excited state.
Modulation of AAC dynamics by inhibitor and substrate
The addition of 3 mM CATR decreased the exchange rate for the TM region by more than 5-fold (kex = 150 ± 110 s−1) (Fig. 2c; Table 1), which is expected from the action of the inhibitor to bind the cavity of the transporter. But remarkably, the presence of 10 mM ADP, which is believed to also bind the cavity, increased the exchange rate of the TM region by more than 2-fold (kex = 1800 ± 350 s−1) (Fig. 2b; Table 1). This observation indicates that ADP is facilitating the conversion between the c-state and the excited state. For both inhibitor and substrate binding, the exchange rate of the AP helices is not substantially affected, suggesting that the intrinsic motion of the AP helices is subject to a different mode than the TM region. This is consistent with the AAC structure in which the loops connecting the AP helices to the TM helices are mostly flexible27,28. In both cases of CATR and ADP binding, the relative populations of the two states also do not appear to be affected, although the uncertainties are substantially larger than those of fitting results for the free yAAC3 (Table 1; Supplementary Table 1).
AAC conformational exchange is asymmetric
Despite the 3-fold quasi-symmetry of the AAC structure, the residues that show significant exchange are asymmetrically distributed (Fig. 3a). The large exchanges are concentrated in Domain I, the kink region of H1 shows the strongest chemical exchange, e.g., Δω of Ala26 and Ser31 are 5.7 ± 0.9 and 5.5 ± 1.4 ppm, respectively (Fig. 3b). Moreover, the binding of ADP or CATR does not substantially alter the pattern of affected residues (Supplementary Fig. 4). The large chemical shift differences between the ground and excited states are indicative of a major conformational rearrangement in that region. The peaks corresponding to the H3 kink region are too broad and weak to be analyzed, however, the strong line-broadening is likely also due to μs–ms exchange. Several residues on the matrix side of H1, H3, and H5 show exchange as well, including residues that belong to the AAC signature sequence, e.g., Gln41, Ala146 and Met246. These residues are parts of helical segments that pack with each other to close the matrix side of the cavity. Chemical exchange in this region is likely associated with opening of the cavity to the matrix side.
Figure 3. The intrinsic chemical exchange map of yAAC3.
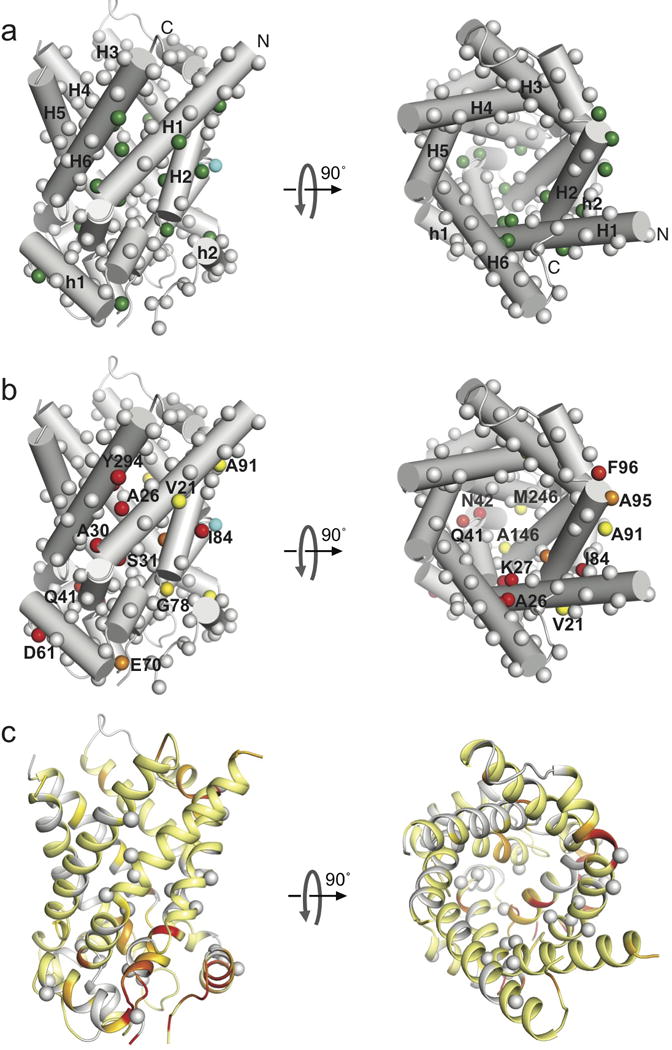
a. Cylinder representation of yAAC3 (PDB ID: 4C9Q28) showing backbone amide 15N nuclei of free yAAC3 with significant chemical exchange (green spheres). These residues all show kex ~ 870 ± 200 s−1. Grey spheres represent residues with flat relaxation dispersion curves. Spheres in cyan indicate severely exchange-broadened residues.
b. The same representation as in (a) showing residue-specific chemical shift differences between the ground and excited states, Δω. The spheres are colored according to the Δω value using a linear gradient from yellow (0.5 ppm) to red (4 ppm).
c. Residue-specific chemical shift changes (ΔΩ) induced by the addition of 10 mM ADP mapped to the yAAC3 structure in the same orientation as in (a) and (b). The ΔΩ are shown with a linear color spectrum scale from 0.01 ppm (yellow) to 0.03 ppm (red). The residues having strong relaxation dispersion are indicated as grey spheres, for showing the difference between chemical exchange and ADP-induced chemical shift changes.
Discussion
Our relaxation dispersion data show that yAAC3, both free and in the presence of substrate, is predominantly in the c-state, which is the low-energy state. The high-energy state only accounts for about 2% of the population at equilibrium. The c-state being the ground state is consistent with previous findings that AAC in isolated mitochondria adopts the c-state conformation23. We believe that the excited state is likely to have a similar conformation as the m-state because the large Δω values are associated with residues of the proline-kink (Fig. 1a, 3b) and conformational rearrangements of the proline kinks are required for switching from the c-state to the m-state23,24. Furthermore, residues involved in closing the matrix side of the AAC cavity, namely Gln41, Ala146 and Met246, showed large conformational exchange, which is most likely due to the opening of the matrix side when transitioning from the c- to the m-state23,24. The striking asymmetry in state distribution is in contrast with many other solute transporters that have more equal populations of the alternating access states such as LacY10, LeuT7 and EmrE13. We acknowledge that the AP helices are positively charged and thus the presence of membrane potential in vivo might affect the population distribution.
The Δω mapping in Fig. 3b indicates that the switch from the c-state to the excited state involves structural rearrangement in the kink regions and the matrix side region of the odd numbered helices as well as the N-terminal half of H2. The asymmetric pattern of exchange appears to be an intrinsic property of yAAC3 because neither substrate nor inhibitor significantly alters the Δω distribution. Ligand binding only modulats the rate of exchange between the two states. We emphasize that the modulation of relaxation dispersion by the ligands are not a consequence of chemical shift perturbation induced by ligand binding because the chemical shift changes induced by ADP binding are on average ~50-fold smaller than the Δω values of conformational exchange and are associated with a different set of residues from those showing relaxation dispersion (Fig. 3c). The observation that CATR binding slows down conformational exchange by more than 5-fold is consistent with the experience that successful crystallization of AAC thus far can be achieved only in the presence of CATR27,28, which may facilitate crystal formation by making the AAC less dynamic. In contrast to CATR, ADP increases the exchange rate from 800 to 1800 s−1, and the fast rate is compatible with AAC being a fast transporter (rate ~400 s−1 observed in liposome assays25). Earlier studies using NMR and FRET on EmrE27,28 and LeuT27,28 have shown that substrates are capable of modulating the transition rate between inward-open and inward-closed states without changing the population distribution between the two states27,28.
A conceptual model of ligand-modulated dynamics of AAC can be proposed based on our NMR measurements and previous functional data. The rate of chemical exchange as derived from relaxation dispersion data is determined by the energy barrier separating the two alternating access states required for the transport (Fig. 4a). Binding of substrate lowers the activation energy either by destabilizing the c-state or by stabilizing the transition state, which results in greater than 2-fold increase in the exchange rate. Conversely, CATR binding raises the activation energy and this can be explained by two possible mechanisms. First, CATR binding stabilizes the c-state, which would lower the energy of the c-state and thus alter the population distribution between the c-state and the excited state. However, since a 5-fold decrease in rate results in ~1.5% decrease in excited population, which is the same as the uncertainty in pe (Table 1), we cannot conclude whether or not CATR binding lowers the energy of the c-state. Another possible mechanism is that CATR binding does not necessarily lower the energy of the c-state, but instead functions as a molecular wedge that attenuates the transition from the c-state to the excited state. In this case, the energy barrier between the two states would be raised without changing the relative populations of the states. This is a more likely scenario because CATR binds mainly to the side of the AAC cavity that shows strong exchange.
Figure 4. A model for explaining ligand modulated conformational exchange of AAC.
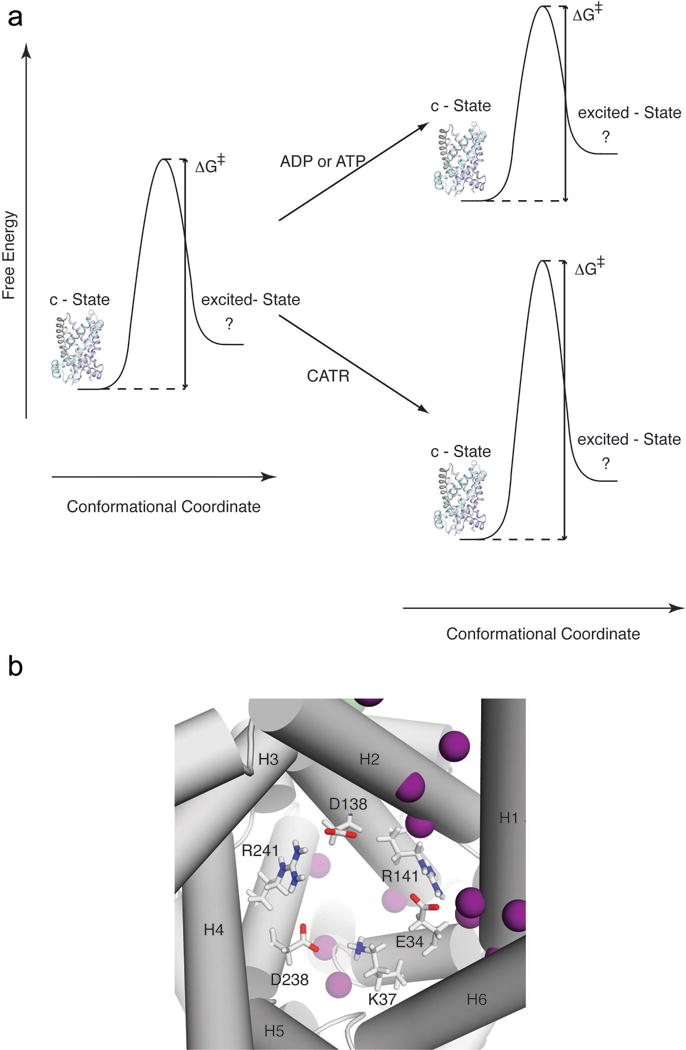
a. Energy diagrams of AAC in the absence and presence of ligands. The low energy state of the free AAC adopts the c-state conformation, which is represented by the crystal structure. The c-state transiently switches to a high energy m-state, for which the structure is unknown. The binding of the inhibitor CATR raises the activation energy of the conformational switch whereas the binding of the substrate ADP lowers the activation energy.
b. A deep view into the yAAC3 cavity showing three acidic and three basic residues forming a network of inter-helical salt bridges (red lines) that “locks” the proline-kink segments in a conformation that closes the matrix side of the cavity. ADP pyrophosphate is expected to transiently form salt bridges with the basic residues, which disrupt the inter-helical salt bridges and thus facilitate the conformational switch to the m-state. This mechanism is consistent with the large chemical exchange observed for this region of the yAAC3 cavity in the presence of ADP (purple spheres; see also Supplementary Fig. 4).
Transporters have been optimized to selectively transport substrates. It is conceivable that they adopt structures for which the interconversion between two alternating access states can be selectively facilitated by the designated substrate. For yAAC3, the ADP facilitated switch from the c-state to the excited state can be explained based on the structure of the c-state and the proposed ADP binding site5,36. In the c-state crystal structure, the closed matrix side of the cavity is stabilized by a network of inter-helical salt bridges that hold the odd numbered TM helices together (Fig. 4b). These salt bridges are formed between Glu34 and Arg141, Lys37 and Asp238, and Asp138 and Arg2415,27,28. It has been predicted based on a molecular dynamics (MD) simulation and functional data that upon binding to the bovine AAC, the phosphate groups of ADP form multiple salt bridges with the three basic residues and that the adenine ring interacts with a hydrophobic pocket formed by Gly188, Ile189 and Tyr192 of H45,36. The majority of interactions of ADP with AAC are located in domains I and II, which could explain the asymmetry of conformational exchange detected by the relaxation dispersion experiments. Furthermore, the MD simulation suggests that ADP directly interacts with Arg141 and Lys37 and this has the consequence of disrupting the inter-helical salt bridges, thus facilitate the matrix side to open5.
ONLINE METHODS
Sample preparation
A gene encoding residues 1–307 of the yeast ADP/ATP carrier 3 (yAAC3) with a N-terminal 6XHis-tag was synthesized by Genscript and subsequently cloned into pET21a expression vector. The plasmid containing the yAAC3 insertion was transformed into E.coli BL21(DE3) (New England Biolabs) expression strain. The cells were grown at 37 °C in M9 minimal media to O.D. ~0.8. After induction with 0.5 mM IPTG, the protein was expressed for 4 hours at 37 °C. The yAAC3 was expressed in inclusion bodies and refolded following a previously established protocol that generated folded protein for functional assays37. The cells were lysed in the Lysis Buffer containing 20 mM HEPES (pH 7.4), 150 mM NaCl, 30 mM lysozyme and 1 mM β-mercaptoethanol (BME). After centrifugation at 20,000 × g for 30 minutes, the pellet containing the inclusion body was collected and solubilized with the Solubilizing Buffer containing 1.67% (w/v) N-lauroylsarcosine (sarkosyl), 20 mM HEPES (pH 7.4), 150 mM NaCl, 1 mM BME and 0.05% polyethylene glycol 4000 (PEG). After solubilization at 4 °C for 16 hours, the solution was centrifuged at 20,000 x g for 30 minutes to remove the insoluble debris. The supernatant containing the solubilized yAAC3 was rapidly diluted 3-fold with the Dilution Buffer containing 6 mM dodeclyphosphocholine (DPC), 20 mM HEPES (pH 7.4), 150 mM NaCl and 200 mM imidazole, and then passed through Ni-NTA resin (Thermo Scientific) pre-equilibrated with the dilution buffer, and eluted with the Elution Buffer containing 3 mM DPC, 20 mM HEPES (pH 7.4), 150 mM NaCl and 200 mM imidazole. To remove the misfolded protion, the yAAC3 elution was dialyzed against the Dialysis Buffer containing 20 mM HEPES (pH 7.4) and 30 mM NaCl for 4 hours and purified with Hi-trap Q FF column (GE Lifesciences). To further fractionize the active form, the yAAC3 purified from the Q column was loaded onto resins crosslinked with ATP (Jena Bioscience). After extensive wash with Wash Buffer 20 mM HEPES (pH 7.4), 3 mM DPC and 50 mM NaCl, yAAC3 was eluted with 20 mM HEPES (pH 7.4), 3 mM DPC and 500 mM NaCl and finally passed through a Superdex 200 size exclusion column (GE lifesciences) in the FPLC Buffer containing 30 mM MES (pH 6.0), 20 mM NaCl, 5 mM BME and 3 mM DPC. The homogenous yAAC3 fractions were pooled and concentrated to be the final NMR sample containing 0.8 mM yAAC3, 120 mM DPC, 5 mM BME, 30 mM MES (pH 6.0) and 20 mM NaCl.
ITC experiment
The calorimetric titrations were carried out at 30 °C on a MicroCal ITC200 instrument. A solution of 50 μM yAAC3 in buffer containing 30 mM MES (pH 6.0) and 20 mM NaCl with 3 mM DPC was loaded into the sample cell. Carboxyatractyloside (CATR) was dissolved in the same buffer to 1 mM concentration and loaded to the injection syringe. Titration curves were generated by 18 successive injections of 2 μL CATR spaced at 160 s intervals. The ITC experiments were performed in triplicate and the data was analyzed using the ORIGIN 7 software. The dissociation constant (KD) and the stoichiometry (N) were calculated by fitting the thermograms to one set of binding sites.
NMR resonance assignment
Sequence-specific assignment of backbone 1HN, 15N, 13Cα, 13Cβ and 13C’ resonances was first carried out for the apo AAC. The triple resonance experiments used for this purpose include TROSY-based HNCA, HN(CO)CA, HNCACB, HN(CO)CACB, HN(CA)CO and HNCO38,39. Additionally, the assignments were validated using a 3D (HN, HN)-HSQC-NOESY-TROSY spectrum with 15N, 15N and 1HN evolution in the t1, t2 and t3 dimensions, respectively, recorded with NOE mixing time of 250 ms. These experiments were performed using a 0.8 mM 15N/13C/2H-labeled apo AAC sample at 30°C on a 600 MHz Bruker spectrometer equipped with a cryogenic TXI probe. Resonance assignments were obtained for ~80% of all backbone and 13Cβ resonances. Resonance assignment of the ADP or CATR bound state was achieved by tracing the assigned resonances of the apo AAC when the AAC sample was titrated with increasing amount of ADP or CATR. The NMR experiments used for the titration are 2D 1H-15N TROSY-HSQC and 3D TROSY-HNCO recorded under the same condition. Data were processed using NMRPipe40 and analyzed using CcpNmr41.
NMR relaxation dispersion measurements
Backbone 15N single-quantum Carr-Purcell-Meiboom-Gill (CPMG) relaxation dispersion experiments were performed on a 600 MHz, a 700 MHz, and a 800 MHz Agilent DD2 spectrometer equipped with a third generation cryogenic probe. The CPMG dispersion experiment measures the modulation of the transverse relaxation rate (R2(1/τcp)) as a function of the delay (τcp) between 180º radio frequency pulses. As shown in the data analysis, the R2 vs. τcp data can be fitted to determine equilibrium populations (pa, pb), kinetics (kex = k1 + k-1), and chemical shift differences (Δω) between the two interconverting conformations.
Residue-specific R2 dispersion was measured using the 2D 15N TROSY-CPMG 42 and the 3D TROSY-CPMG-HNCO (Fig. S3) experiments. Since 35% of the AAC resonances are overlapped in the 2D 1H-15N TROSY-HSQC spectrum, it was essential to record the 3D TROSY-CPMG-HNCO spectra, which provided the large majority of the relaxation dispersion measurements. The CPMG experiments use a 20 ms constant-time 15N relaxation delay with τcp spacing of 236.778 μs, 271.3 μs, 314.143 μs, 375.667 μs, 459.0 μs, 584.0 μs, 792.333 μs, 1.209 μs, and 2.459 μs, which correspond to CPMG-based radio frequency field strengths, νCPMG, ranging from 100 to 900 Hz43. The spectra with different τcp were recorded in an interleaved fashion and duplicate data sets were recorded at two τcp values for error analysis. All spectra were recorded at 30 °C using 0.8 mM 15N/13C/2H-labeled free yAAC3 and the same sample in the presence of 10 mM ADP or 3 mM CATR. For the 2D 15N TROSY-CPMG 128 data points were recorded in the indirect dimension with 24 scans per point for the free AAC and 64 scans for the substrate and inhibitor bound states. For the 3D TROSY-CPMG-HNCO experiment, 60 and 25 data points were recorded for the 15N and 13C indirect dimension, respectively, using 8 scans per point.
NMR relaxation dispersion data analysis
Conformational exchanges of free AAC, AAC in the presence ADP, and AAC bound to CATR are in the intermediate to slow exchange regime (150 – 1800 s−1). To avoid systematic errors fitting different time regimes, all dispersion curves were fit to an exact numerical solution of the Bloch equation using a model of two-site exchange to obtain the calculated relaxation rates 43,44. The parameters of the two-site exchange model, rate of conformational exchange, kex, the populations of the two involved states, (pa, pb), and the 15N chemical shift difference between the two states, Δω were obtained by performing least-square fits to minimize , where and are the experimental and calculated relaxation rates, respectively, and the summation is over all residues included in the fit. The uncertainties in relaxation rates are given by and were calculated from repeat experiments as described previously44.
First, relaxation dispersion profiles with R2 exchange contributions > 4 s−1 were fit using individual exchange parameters to a two-site exchange model to yield residue-specific values for the rate of exchange kex, the chemical shift difference between the two states, Δω, and the populations of the states, pi. In a second step, residues with similar kex and pi were fit in global fits assuming uniform rate constants and populations, but site-specific values for Δω, as the use of a global fit and the use of multiple magnetic fields lead to a higher certainty in the obtained parameters45,46. In a third step, residues with R2 exchange contributions between 2 s−1 and 4 s−1 were fit individually with kex and pi constrained to the values obtained from the global fit to determine their Δω.
Supplementary Material
Acknowledgments
We thank R. Sounier, M. Berardi and K. Oxenoid for insightful discussion and Z. Liu for collecting CPMG data on the Agilent 800 MHz spectrometer at the National Center for Protein Science at Shanghai, China. S.B. is supported as a recipient of an Erwin Schrödinger postdoctoral fellowship of the Austrian Science Fund (FWF, J3251). This work was supported by the NIH grant GM094608 (to J.J.C.). The main NMR facility used for this study was supported by a US National Institutes of Health grant P41 EB-002026.
Footnotes
Author Contributions S.B., Q.Y., and J.J.C. conceived of the study; Q.Y., S.B., and C.R. prepared samples and performed ligand binding assays; S.B. collected and analyzed NMR data; S.B., Q.Y., and J.J.C wrote the paper.
References
- 1.Forrest LR, Kramer R, Ziegler C. The structural basis of secondary active transport mechanisms. Biochim Biophys Acta. 2011;1807:167–88. doi: 10.1016/j.bbabio.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 2.Shi Y. Common folds and transport mechanisms of secondary active transporters. Annu Rev Biophys. 2013;42:51–72. doi: 10.1146/annurev-biophys-083012-130429. [DOI] [PubMed] [Google Scholar]
- 3.Khalili-Araghi F, et al. Molecular dynamics simulations of membrane channels and transporters. Curr Opin Struct Biol. 2009;19:128–37. doi: 10.1016/j.sbi.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wisedchaisri G, Park MS, Iadanza MG, Zheng H, Gonen T. Proton-coupled sugar transport in the prototypical major facilitator superfamily protein XylE. Nat Commun. 2014;5:4521. doi: 10.1038/ncomms5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Y, Tajkhorshid E. Electrostatic funneling of substrate in mitochondrial inner membrane carriers. Proc Natl Acad Sci U S A. 2008;105:9598–603. doi: 10.1073/pnas.0801786105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akyuz N, Altman RB, Blanchard SC, Boudker O. Transport dynamics in a glutamate transporter homologue. Nature. 2013;502:114–8. doi: 10.1038/nature12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Erkens GB, Hanelt I, Goudsmits JM, Slotboom DJ, van Oijen AM. Unsynchronised subunit motion in single trimeric sodium-coupled aspartate transporters. Nature. 2013;502:119–23. doi: 10.1038/nature12538. [DOI] [PubMed] [Google Scholar]
- 8.Zhao Y, et al. Single-molecule dynamics of gating in a neurotransmitter transporter homologue. Nature. 2010;465:188–93. doi: 10.1038/nature09057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao Y, et al. Substrate-modulated gating dynamics in a Na+-coupled neurotransmitter transporter homologue. Nature. 2011;474:109–13. doi: 10.1038/nature09971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Majumdar DS, et al. Single-molecule FRET reveals sugar-induced conformational dynamics in LacY. Proc Natl Acad Sci U S A. 2007;104:12640–5. doi: 10.1073/pnas.0700969104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hwang PM, Bishop RE, Kay LE. The integral membrane enzyme PagP alternates between two dynamically distinct states. Proc Natl Acad Sci U S A. 2004;101:9618–23. doi: 10.1073/pnas.0402324101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu JJ, Horst R, Katritch V, Stevens RC, Wuthrich K. Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science. 2012;335:1106–10. doi: 10.1126/science.1215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morrison EA, et al. Antiparallel EmrE exports drugs by exchanging between asymmetric structures. Nature. 2011;481:45–50. doi: 10.1038/nature10703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nygaard R, et al. The dynamic process of beta(2)-adrenergic receptor activation. Cell. 2013;152:532–42. doi: 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oxenoid K, Chou JJ. The present and future of solution NMR in investigating the structure and dynamics of channels and transporters. Curr Opin Struct Biol. 2013;23:547–54. doi: 10.1016/j.sbi.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morrison EA, Henzler-Wildman KA. Transported substrate determines exchange rate in the multidrug resistance transporter EmrE. J Biol Chem. 2014;289:6825–36. doi: 10.1074/jbc.M113.535328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Imai S, et al. Functional equilibrium of the KcsA structure revealed by NMR. J Biol Chem. 2012;287:39634–41. doi: 10.1074/jbc.M112.401265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vallurupalli P, Bouvignies G, Kay LE. Studying "invisible" excited protein states in slow exchange with a major state conformation. J Am Chem Soc. 2012;134:8148–61. doi: 10.1021/ja3001419. [DOI] [PubMed] [Google Scholar]
- 19.Palmer AG, 3rd, Kroenke CD, Loria JP. Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Methods Enzymol. 2001;339:204–38. doi: 10.1016/s0076-6879(01)39315-1. [DOI] [PubMed] [Google Scholar]
- 20.Mittermaier A, Kay LE. New tools provide new insights in NMR studies of protein dynamics. Science. 2006;312:224–8. doi: 10.1126/science.1124964. [DOI] [PubMed] [Google Scholar]
- 21.Neudecker P, et al. Structure of an intermediate state in protein folding and aggregation. Science. 2012;336:362–6. doi: 10.1126/science.1214203. [DOI] [PubMed] [Google Scholar]
- 22.Whittier SK, Hengge AC, Loria JP. Conformational motions regulate phosphoryl transfer in related protein tyrosine phosphatases. Science. 2013;341:899–903. doi: 10.1126/science.1241735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klingenberg M. The ADP and ATP transport in mitochondria and its carrier. Biochim Biophys Acta. 2008;1778:1978–2021. doi: 10.1016/j.bbamem.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 24.Kunji ER, Robinson AJ. Coupling of proton and substrate translocation in the transport cycle of mitochondrial carriers. Curr Opin Struct Biol. 2010;20:440–7. doi: 10.1016/j.sbi.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 25.Gropp T, et al. Kinetics of electrogenic transport by the ADP/ATP carrier. Biophys J. 1999;77:714–26. doi: 10.1016/S0006-3495(99)76926-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Monne M, Palmieri F. Antiporters of the mitochondrial carrier family. Curr Top Membr. 2014;73:289–320. doi: 10.1016/B978-0-12-800223-0.00008-6. [DOI] [PubMed] [Google Scholar]
- 27.Pebay-Peyroula E, et al. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature. 2003;426:39–44. doi: 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]
- 28.Ruprecht JJ, et al. Structures of yeast mitochondrial ADP/ATP carriers support a domain-based alternating-access transport mechanism. Proc Natl Acad Sci U S A. 2014;111:E426–34. doi: 10.1073/pnas.1320692111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Babot M, Blancard C, Zeman I, Lauquin GJ, Trezeguet V. Mitochondrial ADP/ATP carrier: preventing conformational changes by point mutations inactivates nucleotide transport activity. Biochemistry. 2012;51:7348–56. doi: 10.1021/bi300978z. [DOI] [PubMed] [Google Scholar]
- 30.Riccio P, Aquila H, Klingenberg M. Purification of the carboxy-atractylate binding protein from mitochondria. FEBS Lett. 1975;56:133–8. doi: 10.1016/0014-5793(75)80127-x. [DOI] [PubMed] [Google Scholar]
- 31.Bamber L, Harding M, Butler PJ, Kunji ER. Yeast mitochondrial ADP/ATP carriers are monomeric in detergents. Proc Natl Acad Sci U S A. 2006;103:16224–9. doi: 10.1073/pnas.0607640103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loria JP, Rance M, Palmer AG., 3rd A TROSY CPMG sequence for characterizing chemical exchange in large proteins. J Biomol NMR. 1999;15:151–5. doi: 10.1023/a:1008355631073. [DOI] [PubMed] [Google Scholar]
- 33.Korzhnev DM, Neudecker P, Mittermaier A, Orekhov VY, Kay LE. Multiple-site exchange in proteins studied with a suite of six NMR relaxation dispersion experiments: an application to the folding of a Fyn SH3 domain mutant. J Am Chem Soc. 2005;127:15602–11. doi: 10.1021/ja054550e. [DOI] [PubMed] [Google Scholar]
- 34.Tollinger M, Skrynnikov NR, Mulder FA, Forman-Kay JD, Kay LE. Slow dynamics in folded and unfolded states of an SH3 domain. J Am Chem Soc. 2001;123:11341–52. doi: 10.1021/ja011300z. [DOI] [PubMed] [Google Scholar]
- 35.Korzhnev DM, et al. Low-populated folding intermediates of Fyn SH3 characterized by relaxation dispersion NMR. Nature. 2004;430:586–90. doi: 10.1038/nature02655. [DOI] [PubMed] [Google Scholar]
- 36.Robinson AJ, Kunji ER. Mitochondrial carriers in the cytoplasmic state have a common substrate binding site. Proc Natl Acad Sci U S A. 2006;103:2617–22. doi: 10.1073/pnas.0509994103. [DOI] [PMC free article] [PubMed] [Google Scholar]
REFERENCES for ONLINE METHODS
- 37.Heimpel S, Basset G, Odoy S, Klingenberg M. Expression of the mitochondrial ADP/ATP carrier in Escherichia coli. Renaturation, reconstitution, and the effect of mutations on 10 positive residues. J Biol Chem. 2001;276:11499–506. doi: 10.1074/jbc.M010586200. [DOI] [PubMed] [Google Scholar]
- 38.Kay LE, Torchia DA, Bax A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry. 1989;28:8972–9. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- 39.Pervushin K, Riek R, Wider G, Wuthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci U S A. 1997;94:12366–71. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delaglio F, et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–93. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 41.Vranken WF, et al. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–96. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 42.Loria JP, Rance M, Palmer AG., 3rd A TROSY CPMG sequence for characterizing chemical exchange in large proteins. J Biomol NMR. 1999;15:151–5. doi: 10.1023/a:1008355631073. [DOI] [PubMed] [Google Scholar]
- 43.Tollinger M, Skrynnikov NR, Mulder FA, Forman-Kay JD, Kay LE. Slow dynamics in folded and unfolded states of an SH3 domain. J Am Chem Soc. 2001;123:11341–52. doi: 10.1021/ja011300z. [DOI] [PubMed] [Google Scholar]
- 44.Korzhnev DM, Neudecker P, Mittermaier A, Orekhov VY, Kay LE. Multiple-site exchange in proteins studied with a suite of six NMR relaxation dispersion experiments: an application to the folding of a Fyn SH3 domain mutant. J Am Chem Soc. 2005;127:15602–11. doi: 10.1021/ja054550e. [DOI] [PubMed] [Google Scholar]
- 45.Ishima R, Torchia DA. Error estimation and global fitting in transverse-relaxation dispersion experiments to determine chemical-exchange parameters. J Biomol NMR. 2005;32:41–54. doi: 10.1007/s10858-005-3593-z. [DOI] [PubMed] [Google Scholar]
- 46.Kovrigin EL, Kempf JG, Grey MJ, Loria JP. Faithful estimation of dynamics parameters from CPMG relaxation dispersion measurements. J Magn Reson. 2006;180:93–104. doi: 10.1016/j.jmr.2006.01.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.