

Abstract
Purpose
To describe chronological electrographic features of the interictal EEG background observed in two patients with MMPEI from neonatal to early infantile period.
Methods
EEGs of two patients who fulfilled diagnostic criteria for MMPEI were acquired over the period of 6 months to monitor treatment efficacy and characterize seizures and other paroxysmal events.
Results
Both patients followed a similar sequential pattern. A distinctive evolution from a dysmature term neonatal EEG pattern to an asynchronous suppression burst pattern was observed prior to the interictal background becoming continuous.
Conclusions
Physicians providing care to infants with intractable epilepsy and burst suppression EEG pattern should be alert to the possibility of MMPEI. An earlier diagnosis of MMPEI would help guide diagnostic workup including genetic testing.
Keywords: Suppression burst, interictal EEG, Malignant Migrating Partial Epilepsy in Infancy (MMPEI), EEG evolution, infantile seizures
Introduction
Malignant migrating partial epilepsy in infancy (MMPEI) is a rare early infantile epileptic encephalopathy associated with intractable epilepsy and poor neurodevelopmental outcome (Coppola et al, 1995). The diagnosis of MMPEI is based on ictal electroencephalographic characteristics of the EEG showing the discharges randomly involving multiple independent cerebral sites, moving from one cortical area to another in consecutive seizures (Coppola et al, 1995).
A number of genetic causes of MMPEI have been identified, including mutations in KCNT1 (Barcia et al, 2012), PLCB1 (Poduri et al, 2012), SCN1A (Freilich et al, 2011), SLC25A22 (Poduri et al, 2013), and TBC1D24 (Milh et al, 2013). Most patients with MMPEI have intractable epilepsy and significant neurodevelopmental outcomes (Coppola, 2013). There is limited literature describing interictal EEG characteristics in these patients preceding the ictal electrographic trademark of MMPEI (Coppola et al, 1995 and 2009). We present chronological observations of the interictal and ictal EEG findings of two patients fulfilling diagnostic criteria for MMPEI.
Methods
Subjects were recruited as part of the Genetic Studies of Developmental Brain Disorders protocol approved by the Research Subjects Review Board of the University of Rochester Medical Center. Informed consent was obtained in all cases. Retrospective clinical histories, primary EEG studies, and brain MRIs were reviewed.
The interictal and ictal EEG recordings were performed in a Level 4 Epilepsy Center in accordance to the Minimum Technical Standards for Pediatric Electroencephalography (ACNS, 2006) using an XLTEK vEEG telemetry monitoring system with 21 channels of electrodes placed in accordance with the international 10–20 system of electrode placement. Supplementary ECG and respiratory channels were also used as part of the monitoring including. The tracings were reviewed by JB and OS. There was no disagreement in the interpretation.
Case Reports
Subject DB13-002 was a male born full-term via spontaneous vaginal delivery. He initially had movements concerning for seizures on the second day of life, but was not confirmed to have seizures until 9 days of life. Subject DB12-014 was a female born full-term via repeat cesarean section with no pregnancy or delivery complications. She initially had seizures at six days of life. Extensive work up for inborn errors of metabolism was unrevealing. MRI of the brain at 10 days for DB13-002 and 17 days for DB12-014 were normal.
Each child was treated with vitamin B6 without improvement. Both patients were treated with multiple antiepileptic medications in various combinations as well as the ketogenic diet without significant improvement in seizure control. Sequential EEGs were utilized to monitor treatment efficacy allowing surveillance of the chronological electroencephalographic progression from birth to 6 months of age. Subject DB13-002 was identified by research whole exome sequencing to have a de novo c.1420C>T, p.Arg474Cys mutation in KCNT1. Subject DB12-014 was found by clinical gene sequencing to have a de novo c.4718T>C, p.Leu1573Pro mutation in SCN2A.
Subject DB13-002 died at the age of 18 months after complications from recurrent respiratory illnesses. Subject DB12-014 at last follow-up at 16 months had medically refractory epilepsy, and has global developmental delay.
EEG findings
Each child had the first EEG at 39 weeks of conceptional age (CA). Subsequent EEGs were repeated at 1 to 7 week intervals. Few conspicuous electrographic similarities of the interictal background were observed. At no point was the EEG considered normal in either subject. The first EEG tracing at 39 weeks CA was characterized by the excessive degree of interhemispheric asynchrony with interburst intervals varying between 2 and 5 seconds. Moreover, there was excessive suppression of interburst background activity, particularly during sleep (Fig. 1). The first electro-clinical seizure was recorded at the age of 39 weeks CA in DB13-002 and 40 weeks CA in DB12-014. In each patient the seizures had polymorphic electrographic patterns and were multifocal in onset.
Figure 1.
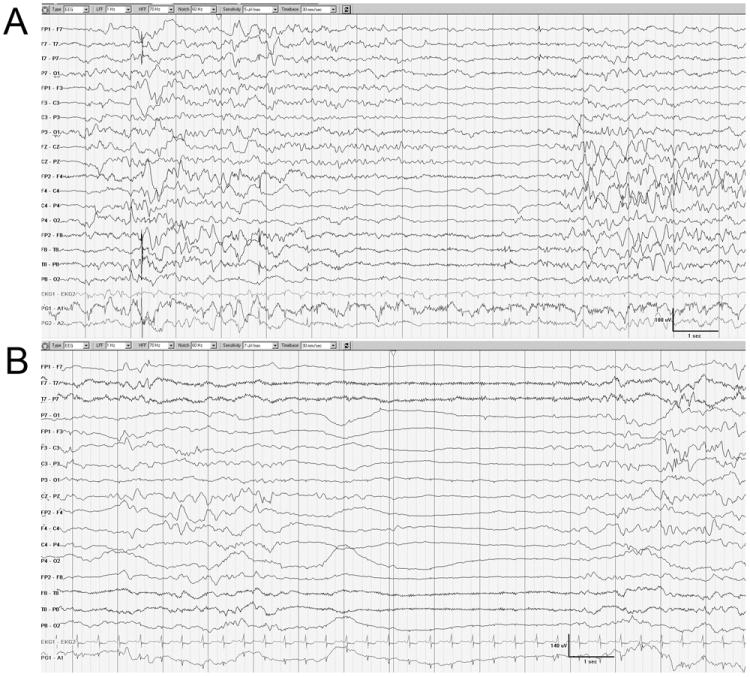
Similar interictal patterns of the first recording at 39 weeks of CA. The background is discontinuous and asynchronous between hemispheres in DB13-002 (A; waking state) and DB12-014 (B; sleeping state).
Subsequently, a distinguishing pattern of persistent and invariable asynchronous suppression burst ensued first at the age of 46 days in DB13-002 (there was longer periods of time between first and second EEG for this case), and 13 days in DB12-014. This pattern became distinguishing and the dominant feature of the interictal electrographic tracing. The bursts of cerebral activity contained morphologically variable high voltage (ranging from 50 to over 100 microvolts) spikes, spike-wave complexes, and polyspikes. The bursts lasted between 5-10 seconds in each hemisphere and alternated with irregular 2-10 second periods of background suppression with background being lower than 25, and frequently below 10 microvolts. Although these bursts were similar in morphology and amplitude in each hemisphere, they occurred independently creating notable checkerboard appearance. This pattern was invariable and persisted during clinical waking and sleeping states (Fig. 2). There was no normal sleep architecture during clinical sleep. At this chronological stage the patients continued having frequent electrographically polymorphic and multifocal seizures.
Figure 2.
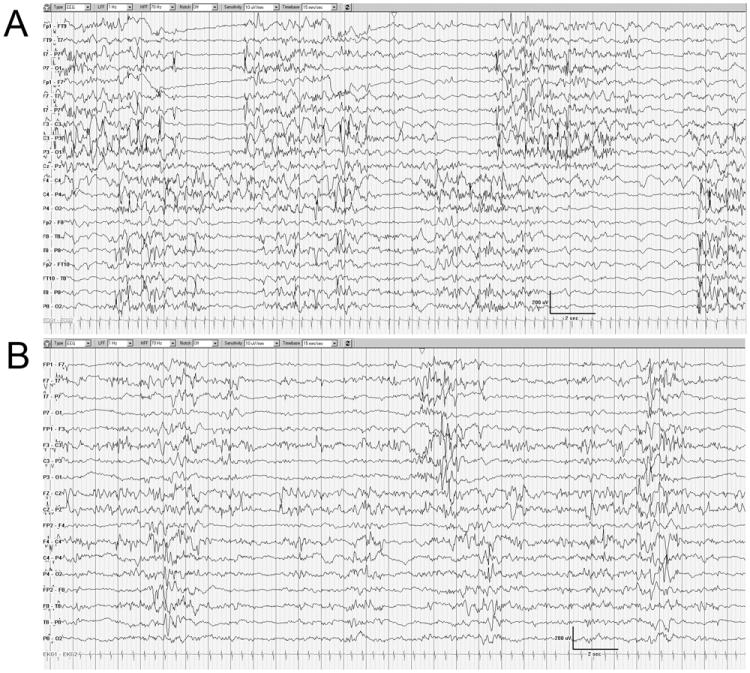
The EEG background is characterized by the asynchronous and discontinuous suppression pattern in DB13-002 (A) and DB12-014 (B). The state is indeterminate sleep.
The asynchronous burst suppression pattern was subsequently replaced by a continuous background with intermixed multifocal epileptiform potentials. The continuous EEG background was first noted at 107 and 67 postnatal days in DB13-002 and DB12-014 respectively (Fig. 3). As the background becomes continuous the ictal patterns were relatively monomorphic and migrating resembling those reported in patients with MMPEI (Coppola et al, 1995), first observed at 107 postnatal days in DB13-002 and 102 postnatal days in DB12-014 (Fig. 4).
Figure 3.
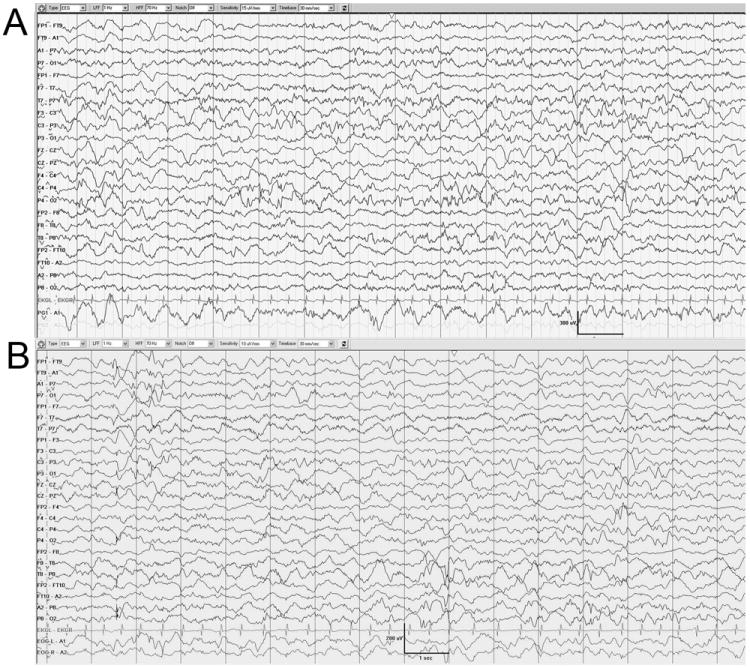
The background is continuous and contains multifocal epileptiform discharges. The periods of suppression are no longer evident. DB13-002 (A) chronological age is 107 days. DB12-014 (B) chronological age is 67 days.
Figure 4.
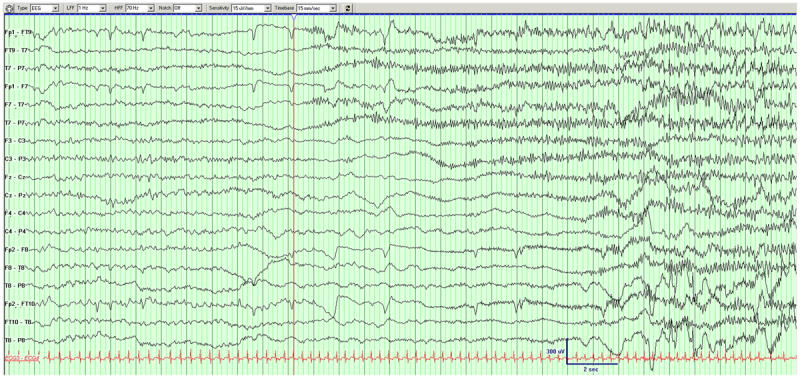
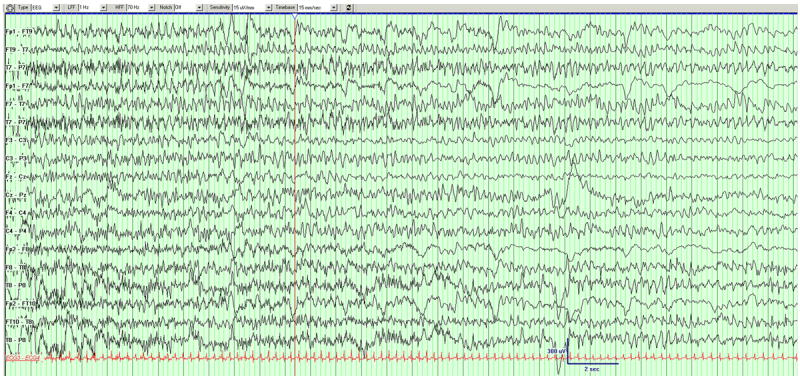
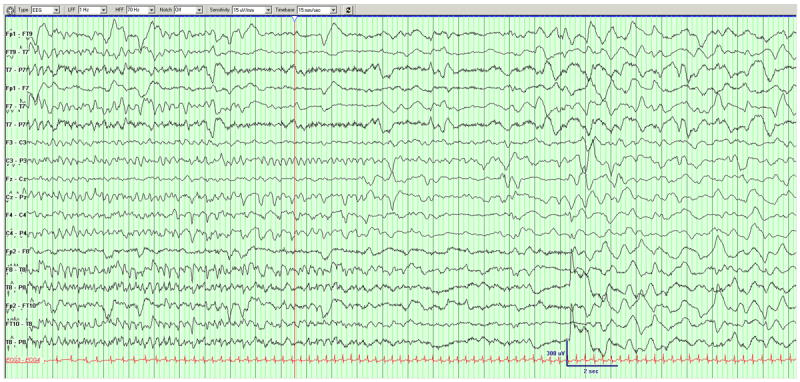
Example of a seizure in DB12-014. Left hemispheric seizure migrates to the right hemisphere.
Discussion
The diagnostic criteria for MMPEI are based on the characteristic ictal electrographic patterns in neonates with the onset of seizures in the first 6 months of life with progressive deterioration of psychomotor development (Coppola, 2013). The focus of our report are the sequential interictal EEG features observed over a period of time in two patients fulfilling diagnostic criteria for MMPEI with seizures first manifesting in the neonatal period. We found consistent abnormalities in the interictal background continuity and interhemispheric synchrony. In addition, a transient suppression burst pattern was noted in both of our subjects with MMPEI. These observations broaden the described EEG findings in MMPEI patients.
The evolution from discontinuous to continuous cerebral pattern is a normal expected physiologic process observed in preterm infants without neurological abnormalities (Vecchierini et al, 2007). The EEG of the full term newborn is expected to be continuous during waking and active sleep with trace alternant present only during quiet sleep. (Clancy et al, 2002). In our patients the degree of asynchrony and discontinuity at 39 weeks of CA was excessive, suggesting cortical neuronal dysfunction. Continuity was achieved only months later in both subjects, past the neonatal period.
Interhemispheric synchrony is likewise age-dependent and defined as a temporal delay no longer than 1.5 to 2 seconds between the bursts of identical waveforms between hemispheres (Andre et al, 2010.) The degree of synchrony should approach 100% in normal term neonates (Tsuchida et al, 2012). Interhemispheric asynchrony has been previously described in patients with corpus callosum dysgenesis (Shany, 2011). In addition, it is postulated that thalamic afferent input on cortical generators and rapid dendritic spine development and synaptogenesis are responsible for the interhemispheric synchrony that is expected to ensue close to full-term gestational age (Silvestri-Hobson, 2012). Striking interhemispheric asynchrony in our patients, particularly in the setting of normal corpus callosum morphology, raises the possibility of abnormalities in thalamocortical circuitry or aberrant synaptogenesis.
The next striking chronological feature of the interictal background included suppression burst (SB) pattern with alternating periods of relative of background inactivity between hemispheres. The SB pattern is described in the states of hypoxia, drug-related intoxication, hypothermia, and anesthesia (Amzica, 2009). The SB EEG patterns in neonates without these cofactors have traditionally been associated with either Ohtahara or early myoclonic epilepsy (EME) syndromes (Ohtahara, 2003). Both syndromes are included under the common designation of early infantile (neonatal) epileptic syndromes or encephalopathies with suppression-burst (EIEE) (Ohtahara, 2003). The key electrographic difference between these syndromes is persistent periodic appearing suppression-burst pattern appearing continuously in both waking and sleeping states in OS (Panayiotopoulos, 2010) versus presence of SB primarily in sleeping states in EME (Ohtahara, 2003 and 2006). The SB pattern in OS is typically bilaterally synchronous between hemispheres (Panayiotopoulos, 2010) but may show some asymmetry (presumably due to underlying structural lesion) (Ohtahara, 2003). In EME, SB often shows some asynchrony and irregular burst-to-burst intervals (Ohtahara, 2003). The SB EEG pattern in our patients was a transient phenomenon. Characteristically, SB pattern was noted in both waking and sleeping states and was associated with marked degree of interhemispheric asynchrony, sharing characteristics of both OS and EME syndromes, constituting a continuum of epileptic encephalopathies with suppression burst patterns.
The physiological basis of the SB state has been attributed to various mechanisms resulting in transient cortical neuronal inactivity, including those induced by deep level of anesthesia (Ching et al, 2011). These mechanisms mainly explain SB pattern that is synchronous between the two hemispheres. The strikingly disjointed inter-hemispheric SB pattern noted in our patients, particularly in the absence of callosal defects, remains difficult to explain.
Despite the EEG becoming continuous the multifocal spikes persisted and were considered abnormal as neonatal sharp transients were expected to disappear past 46 weeks of conceptional age (Fisch, 1999).
Electro-clinical seizures were noted in both subjects in the neonatal period. Initially the seizures were polymorphous and multifocal remaining confined to a single hemisphere without a characteristic migrating phenomenon. The migrating nature of the seizures in our subjects became apparent only when the background became continuous. Further description of the ictal patterns is beyond the focus of this report.
Several genetic etiologies of MMPEI and other developmental epilepsies have recently been identified. De novo gain-of-function mutations affecting the C-terminal domain of the KCNT1 potassium channel have been reported in patients with MMPEI (Barcia et al, 2012). KCNT1 is a calcium-activated potassium channel that regulates the rate of bursting and enhances the accuracy with which action potentials lock to incoming stimuli (Barcia et al, 2012). One of our patients, DB13-002, was found to have a KCNT1 mutation. DB12-014 was found to have a mutation in SCN2A, which is a known sodium channel gene implicated in infant onset epilepsy (Baasch et al, 2014; Matalon et al, 2014; Hackenburg et al, 2014), including infantile spasms (Sundaram et al, 2013) and Ohtahara syndrome (Nakamura et al, 2013).
The interictal EEGs of these patients underwent rapid sequential metamorphosis from abnormal neonatal pattern to a transient alternating burst suppression state prior to becoming continuous. This transient developmental phenomenon was recognized solely due to frequent electrographic assessments which allowed us to observe conspicuous patterns of early interictal electrographic evolution not previously reported in MMPEI. Replication of these findings will be essential in establishing a characteristic electrographic developmental signature for MMPEI to improve recognition of this rare disorder. This in turn could provide further insight into the developmental process of cerebral malfunction in patients with MMPEI.
Although these EEG patterns cannot be rendered specific for MMPEI, the physicians providing care to the infants with medically intractable epilepsy and asynchronous burst suppression EEG pattern need to be alert to the possibility of MMPEI and consider expansion of available genetic testing to include known mutations associated with this disease. This report is a contribution to greater awareness of the chronological changes in the interictal EEG of patients with manifestations of MMPEI in the neonatal period which may facilitate an early diagnosis of this rare condition.
Acknowledgments
We wish to thank our research families for their participation. Research reported in this work was supported by the National Institute of Neurologic Disorders and Stroke (NINDS) and the National Institutes of Health (NIH) under award numbers K12NS066098 (to LES) and K08NS078054 (to ARP).
Contributor Information
Olga Selioutski, Department of Neurology, University of Rochester Medical Center, Rochester, NY; Strong Epilepsy Center, University of Rochester Medical Center, Rochester NY.
Laurie Seltzer, Department of Neurology, University of Rochester Medical Center, Rochester, NY; Strong Epilepsy Center, University of Rochester Medical Center, Rochester NY.
James Burchfiel, Department of Neurology, University of Rochester Medical Center, Rochester, NY; Strong Epilepsy Center, University of Rochester Medical Center, Rochester NY.
Alex Paciorkowski, Department of Neurology, University of Rochester Medical Center, Rochester, NY; Departments of Pediatrics and Biomedical Genetics, Center for Neural Development & Disease, University of Rochester Medical Center, Rochester, NY.
Giuseppe Erba, Department of Neurology, University of Rochester Medical Center, Rochester, NY; Strong Epilepsy Center, University of Rochester Medical Center, Rochester NY.
References
- American Clinical Neurophysiology Society (ACNS) Guideline Two: Minimum Technical Standards for Pediatric Electroencephalography. American Clinical Neurophysiology Society; 2006. [Google Scholar]
- Amzica F. Basic physiology of burst-suppression. Epilepsia. 2009;50(Suppl. 12):38–39. doi: 10.1111/j.1528-1167.2009.02345.x. [DOI] [PubMed] [Google Scholar]; Andréa AM, Lamblinb M-D, d’Allest AM, Curzi-Dascalovad L, Moussalli-Salefranquee F, NguyenThe Tichf S, Vecchierini-Blineaug M-F, Wallois F, Walls-Esquivel E, Plouin P. EEG in premature and full term infants. Developmental features and glossary. Plouine Neurophysiologie Clinique/Clinical Neurophysiology. 2010;40:59–124. doi: 10.1016/j.neucli.2010.02.002. [DOI] [PubMed] [Google Scholar]
- Baasch A-L, Huning I, Gilissen C, Klepper J, Veltman J, Gillessen-Kaesbach G, Hoischen A, Lohmann K. Exome sequencing identifies a de novo SCN2A mutation in a patient with intractable seizures, severe intellectual disability, optic atrophy, muscular hypotonia, and brain abnormalities. Epilepsia. 2014;55(4):e25–e29. doi: 10.1111/epi.12554. [DOI] [PubMed] [Google Scholar]
- Barcia G, Fleming MR, Deligniere A, Gazula V, Brown MR, Langouet M, Chen H, Kronengold J, Abhyankar A, Cilio R, Nitschke P, Kaminska A, Boddaert N, Casanova JL, Desguerre I, Munnich A, Dulac O, Kaczmarek LK, Colleaux L, Nabbout R. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nature Genetics. 2012 Nov;44(11):1255–9. doi: 10.1038/ng.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy R, Bergqvist C, Dlugos D. Neonatal Electrencephalography. Current practice of clinical electroencephalography Chapter 6 [Google Scholar]; Ebersol J, Pedley T. LWW. 2002 [Google Scholar]; Coppola G, Plouin P, Chiron C, Robain O, Dulac O. Migraing partial seizures in infancy: a malignant disorder with developmental arrest. Epilepsia. 1995;36(10):1017–1024. doi: 10.1111/j.1528-1157.1995.tb00961.x. [DOI] [PubMed] [Google Scholar]
- Coppola G. Malignant migrating partial seizures in infancy: An epilepsy syndrome of unknown etiology. Epilepsia. 2009;50(Suppl. 5):49–51. doi: 10.1111/j.1528-1167.2009.02121.x. [DOI] [PubMed] [Google Scholar]
- Coppola G. Handb Clin Neurol. Vol. 111. Elsevier B.V; 2013. Malignant migrating partial seizures in infancy; pp. 605–9. [DOI] [PubMed] [Google Scholar]
- EMEDICINE. Silvestri-Hobson RC, Benbadis SR. Abnormal Neonatal EEG. Updated Jul 27, 2012 < http://emedicine.medscape.com/article/1139692-overview#showall>.
- Fisch BJ. Fisch and Spehlmann’s EEG primer. Elsevier; 1999. [Google Scholar]
- Freilich ER, Jones JM, Gaillard WD, Conry JA, Tsuchida TN, Reyes C, Dib-Hajj S, Waxman SG, Meisler MH, Pearl PL. Novel SCN1A mutation in a proband with malignant migrating partial seizures of infancy. Arch Neurol. 2011 May;68(5):665–71. doi: 10.1001/archneurol.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackenberg A, Baumer A, Sticht H, Schmitt B, Kroell-Seger J, Wille D, Joset P, Papuc S, Rauch A, Plecko B. Infantile Epileptic Encephalopathy, Transient Choreoathetotic Movements, and Hypersomnia due to a De Novo Missense Mutation in the SCN2A Gene. Neuropediatrics. 2014 Apr 7; doi: 10.1055/s-0034-1372302. [DOI] [PubMed] [Google Scholar]; Matalon D, Goldberg E, Medne L, Marsh E. Confirming an expanded spectrum of SCN2A mutations: a case series. Epileptic disorders : international epilepsy journal with videotape. 2014 Mar; doi: 10.1684/epd.2014.0641. [DOI] [PubMed] [Google Scholar]
- Milh M, Falace A, Villeneuve N, Vanni N, Cacciagli P, Assereto S, Nabbout R, Benfenati F, Zara F, Chabrol B, Villard L, Fassio A. Novel compound heterozygous mutations in TBC1D24 cause familial malignant migrating partial seizures of infancy. Hum Mutat. 2013 Jun;34(6):869–72. doi: 10.1002/humu.22318. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Kato M, Osaka H, Yamashita S, Nakagawa E, Haginoya K, Tohyama J, Okuda M, Wada T, Shimakawa S, Imai K, Takeshita S, Ishiwata H, Lev D, Lerman-Sagie T, Cervantes-Barragán DE, Villarroel CE, Ohfu M, Writzl K, Gnidovec Strazisar B, Hirabayashi S, Chitayat D, Myles Reid D, Nishiyama K, Kodera H, Nakashima M, Tsurusaki Y, Miyake N, Hayasaka K, Matsumoto N, Saitsu H. Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology. 2013 Sep 10;81(11):992–8. doi: 10.1212/WNL.0b013e3182a43e57. [DOI] [PubMed] [Google Scholar]
- Ohtahara S, Yamatogi Y. Epileptic Encephalopathies in Early Infancy With Suppression-Burst. Journal of Clinical Neurophysiology. 2003;20(6):398–407. doi: 10.1097/00004691-200311000-00003. [DOI] [PubMed] [Google Scholar]
- Ohtahara S, Yamatogi Y. Ohtahara syndrome: With special reference to its developmental aspects for differentiating from early myoclonic. Epilepsy Research. 2006;70S:S58–S67. doi: 10.1016/j.eplepsyres.2005.11.021. [DOI] [PubMed] [Google Scholar]
- Panayiotopoulos CP. Atlas of Epilepsies. Springer-Verlag London limited; 2010. pp. 847–8. [Google Scholar]; Poduri A, Chopra SS, Neilan EG, Elhosary PC, Kurian MA, Meyer E, Barry BJ, Khwaja OS, Salih MA, Stödberg T, Scheffer IE, Maher ER, Sahin M, Wu BL, Berry GT, Walsh CA, Picker J, Kothare SV. Homozygous. PLCB1 deletion associated with malignant migrating partial seizures in infancy. Epilepsia. 2012 Aug;53(8):e146–50. doi: 10.1111/j.1528-1167.2012.03538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poduri A, Heinzen EL, Chitsazzadeh V, Lasorsa FM, Elhosary PC, LaCoursiere CM, Martin E, Yuskaitis CJ, Hill RS, Atabay KD, Barry B, Partlow JN, Bashiri FA, Zeidan RM, Elmalik SA, Kabiraj MM, Kothare S, Stödberg T, McTague A, Kurian MA, Scheffer IE, Barkovich AJ, Palmieri F, Salih MA, Walsh CA. SLC25A22 is a novel gene for migrating partial seizures in infancy. Ann Neurol. 2013 Dec;74(6):873–82. doi: 10.1002/ana.23998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shany E, Berger I. Neonatal Electroencephalography: Review of a Practical. Approach J Child Neurol. 2011;26:341. doi: 10.1177/0883073810384866. [DOI] [PubMed] [Google Scholar]
- Sundaram SK, Chugani HT, Tiwari VN, Huq AH. SCN2A mutation is associated with infantile spasms and bitemporal glucose hypometabolism. Pediatr Neurol. 2013 Jul;49(1):46–9. doi: 10.1016/j.pediatrneurol.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchida T, Wusthoff C, Shellhaas R, Abend N, Hahn C, Sullivan J, Nguyen S, Weinstein S, Scher M, Riviello J, Clancy R American Clinical Neurophysiology Society (ACNS) Critical Care Monitoring Committee. American Clinical Neurophysiology Society; 2012. Guideline Two: ACNS Standardized EEG Terminology and Categorization for the Description of Continuous EEG Monitoring in Neonates. Report of the American Clinical Neurophysiology Society. [DOI] [PubMed] [Google Scholar]
- Tsurusaki Y, Miyake N, Hayasaka K, Matsumoto N, Saitsu H. Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology. 2013 Sep 10;81(11):992–8. doi: 10.1212/WNL.0b013e3182a43e57. [DOI] [PubMed] [Google Scholar]
- Vecchierinia MF, Andréb M, d’Allest AM. Normal EEG of premature infants born between 24 and 30 weeks gestational age: Terminology, definitions and maturation aspects. Neurophysiologie Clinique/Clinical Neurophysiology. 2007;37:311–323. doi: 10.1016/j.neucli.2007.10.008. [DOI] [PubMed] [Google Scholar]