

Abstract
Dietary carcinogens, such as 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), and chronic inflammation have each been implicated as etiological agents in prostate cancer. We hypothesized that bacterial prostatitis would accelerate PhIP-induced pre-invasive lesions in the rat prostate. Male Fischer 344 rats were assigned into 4 groups: Control (untreated), PhIP (200 ppm in the diet for 20 weeks), E. coli (prostatic inoculation in week 10), or PhIP+E. coli. Study animals were monitored for a total of 52 weeks and were euthanized as necessary based on strict criteria for health status and tumor burden. Animals treated with E. coli initially developed acute and chronic inflammation in all lobes of the prostate, whereas inflammation was observed predominantly in the ventral lobe at time of death. PhIP+E. coli-treated animals exhibited a marked decrease in survival compared to PhIP-alone treated animals as a result of an increase in the number of invasive cancers that developed at multiple sites including the skin, small intestine, and Zymbal’s gland. Despite their earlier mortality, PhIP+E. coli-treated animals developed an increased average number of precancerous lesions within the prostate compared to PhIP-treated animals, with a significantly increased Ki-67 index. Multiplexed serum cytokine analysis indicated an increase in the level of circulating IL-6 and IL-12 in PhIP+E. coli-treated animals. Elevated serum IL-6 levels correlated with the development of precancerous lesions within the prostate. These results suggest that bacterial infections and dietary carcinogens - two conceivably preventable cancer risk factors – may synergistically promote tumorigenesis.
Keywords: Prostate cancer, PhIP, infection, inflammation
Introduction
An emerging concept in cancer epidemiology is the coupling of genetic and genomic studies with “exposomics”, or the collective study of life-course environmental exposures (i.e., the “exposome”), in relation to human disease (1). For prostate cancer, one such “environmental exposure”, the Western diet, is a potential source of carcinogens that afflict the prostate. Dietary carcinogens may stand as one of the major means to acquire the genetic mutation(s) required for tumor initiation (2). In this regard, much attention has been given to a class of dietary mutagens called heterocyclic amines (HCAs) that are generated in meats cooked under high temperature conditions and are linked to cancer of multiple organs including prostate, colon and breast (3, 4). In prostate cancer, a particular focus has been given to the cancer-inducing capabilities of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), which is the most abundant of the HCAs produced by high temperature cooking of meats. Although not all studies are positive, a number of epidemiological studies have linked the consumption of meat and very well cooked meat with risk of overall prostate cancer and/or aggressive prostate cancer (4–9). In rodents, dietary exposure to PhIP represents one of the examples in which carcinogen exposure results in neoplastic lesions of the prostate (10), providing further evidence of a potential role for dietary intake of PhIP in prostate cancer development.
Likewise, over the last 10–15 years it has become clear that inflammatory cells and chronic inflammation, in addition to at times attacking cancer by immune surveillance, can lead to increased cancer occurrence and disease progression (11). In fact, chronic inflammation is now regarded as an additional “enabling characteristic” of cancer (12). A number of infectious agents that generally produce long standing chronic inflammation are linked to cancer, and it is currently estimated that at least 1 in 6 cancers worldwide are caused by preventable infections (13). In the case of prostate cancer, a potential role for infectious agents has long been suspected (14) and this has been bolstered by the frequent observation of unexplained acute and chronic inflammation and inflammation-associated lesions in radical prostatectomy specimens, autopsy prostate tissues, transurethral resections of the prostate, and prostate needle biopsies (15).
Interestingly, numerous microorganisms have been identified in association with prostate cancer using both microbial culture and culture-independent molecular techniques. Many are pro-inflammatory bacterial species connected with conditions including bacterial prostatitis and/or urinary tract infections (reviewed in (14)), including Escherichia coli. E. coli is frequently identified as a causative agent in bacterial prostatitis and has also been detected in both benign prostatic hyperplasia (BPH) and prostate cancer tissues (16, 17). E. coli DNA has also been found in corpora amylacea (18) which are tiny concretions that are very common in the prostate of the adult male and are now considered to be remnants of past acute inflammatory events in the prostate (19). Infection of the mouse prostate with uropathogenic strains of E. coli (UPEC) has been reported to induce epithelial proliferation and reactive hyperplasia (20), dysplasia and oxidative DNA damage (21) and marked reduction of the potential prostate cancer tumor suppressor, NKX3.1 (22). A prostate-derived UPEC strain has also been shown to induce inflammation in the prostate microenvironment and accelerate tumorigenesis in the Hi-MYC transgenic mouse model of prostate cancer (23).
When rats consume PhIP, pre-invasive prostate cancer lesions develop exclusively in the ventral lobe of the prostate, and not in the dorsal, lateral or anterior lobes (10). This is strikingly analogous to the “lobe” specificity observed in human prostate cancer, where cancer development predominantly occurs in the peripheral zone. Interestingly, inflammation and atrophy were shown to precede prostatic neoplasia in a previous study of PhIP consumption in Fischer 344 rats (24). This is also analogous to the suspected early role for prostatic inflammation in the development of a putative risk factor/precursor lesion to human prostate cancer development, namely proliferative inflammatory atrophy (PIA) (15). In another study of PhIP-induced prostate carcinogenesis in transgenic Fischer 344 Big Blue® rats, it was determined that all lobes of the prostate (ventral, dorsal, lateral, anterior) are subjected to markedly elevated mutation frequencies after PhIP consumption (25). Therefore, mutation frequency alone cannot account for the distinct ventral lobe specificity to cancer development in rats treated with PhIP. Intriguingly, the study by Nakai et al. (25) demonstrated a PhIP-induced increase in the Ki-67 (proliferative) index, the apoptotic index, and in the number of stromal mast cells and macrophages in the ventral lobe of the rat prostate that did not occur in the other prostatic lobes. These results indicate that PhIP can act as both a prostate cancer “initiator” as well as a lobe-specific cancer “promoter”. Furthermore, the finding that infiltration of inflammatory cells (chiefly mast cells and to a lesser degree macrophages) was restricted to the ventral lobe of the prostate in the Nakai et al. (25) study indicated that inflammation may play a mechanistic role in PhIP-induced carcinogenesis. Since chronic inflammation is thought to be a driver of cancer initiation and progression, we reasoned that carcinogen exposure (e.g. PhIP) might synergize with chronic inflammation induced by an infectious agent (beyond an increase in macrophage and mast cells with PhIP alone) and would accelerate prostatic carcinogenesis in the rat prostate.
Materials and Methods
Animals
All studies were approved by the Johns Hopkins Animal Care and Use Committee (ACUC). For the 52 week study, rats were randomly divided into the following groups: control (n=15), PhIP (n=14), E. coli (n=11), and PhIP+E. coli (n=10). The control and PhIP groups were shared with a recent study on the chemopreventative effects of tomato+broccoli on PhIP-induced carcinogenesis (26). Additional information on the study animals and the rodent diets can be found in the Supplementary Methods.
E. coli Infections
The strain of E. coli (CP1) used to elicit prostatitis in this model is a clinically relevant UPEC strain previously studied in the context of chronic pelvic pain as well as the Hi-MYC transgenic model of prostate carcinogenesis in mice (23, 27). The CP1 strain, though technically a UPEC strain, is from Group B1, whose members normally lack extraintestinal virulence factors (28) but when selected for in clinical disease states can exhibit high levels of virulence (29). CP1 E. coli was cultured as previously described (27), washed and resuspended in sterile 1X PBS. A volume of 100 µl at a dose of approximately 107 CFU/100 µl was introduced into the rat prostate via urethral catheterization using sterilized polyethylene tubing in week 10 of the study.
Image Analysis
Slides were scanned using the Aperio ScanScope (CS model, Aperio, Vista, CA) and viewed using the freeware ImageScope Viewer Software (Aperio version 10.2.2.2353). The mean percentage area of cribriform PIN/CIS lesions was determined as previously described (26). Inflammation analyses are described in the Supplementary Methods (see Supplementary Fig. S1). Ventral prostate mast cell counts were obtained by Toluidine Blue staining as previously described (25), followed by slide scanning, blinding in terms of treatment group, and manual counting of positively staining cells per total ventral prostate area as assessed using the Aperio ImageScope Viewer Software. For calculation of the Ki-67 index, scanned slides were blinded in terms of treatment group, and analyzed with Infinity Analyze (Lumenera Corp., Ottawa, Ontario) counting software. Ki-67 index was calculated as the percentage of brown staining nuclei of all nuclei present in cribriform PIN/CIS lesions.
Immunohistochemistry (IHC)
Slides containing sections of formalin fixed, paraffin embedded (FFPE) rat prostate tissue were steamed for 25 min (E. coli IHC) or 40 min (Ki-67, CD68) in high temperature target retrieval (HTTR) solution for antigen retrieval (Dako). Slides were then incubated with an anti-E. coli antibody (Virostat, Inc., product #1001) at a dilution of 1:2000, anti-CD68 antibody (Abcam ab125212) at 1:1000, or anti-Ki-67 antibody (Novocastra) at 1:1000 for 45 min at room temperature, followed by incubation with secondary antibody (PowerVision, Leica Microsystems) for 30 min at room temperature. Staining was visualized using 3,3’-Diamino-benzidine (Sigma FAST 3,3’-Diamino-benzidine tablets), and slides were counterstained with hematoxylin.
Chromogenic in situ Hybridization (CISH)
IL-6 CISH was performed on FFPE ventral prostate tissues using the RNAscope 2.0 FFPE Brown Reagent Kit (Advanced Cell Diagnostics, Inc.) with probe Mm-Il6 and the manufacturer’s recommended protocol.
Serum Cytokine Analysis
Blood was collected into BD Vacutainer serum tubes (product number 366441) at time of sacrifice by cardiac puncture and serum was stored at −80 °C until the time of cytokine analysis. Multiplexed serum cytokine analysis was performed using the Luminex platform at the Immunology Core Laboratory at the Johns Hopkins Sidney Kimmel Comprehensive Cancer Center. A panel of 11 cytokines (GM-CSF, IFNγ, IL-1a, IL-1b, IL-4, IL-6, IL-10, IL-17, IL-12 (p70), Leptin, and TNFα) were analyzed in duplicate using a custom MILLIPLEX MAP Rat Cytokine/Chemokine Magnetic Bead Panel (EMD Millipore).
Statistics
Unless otherwise noted, data were compared among treatments by unpaired two-tailed t tests using GraphPad Prism Software (version 5.0; GraphPad Software, Inc., La Jolla, CA), and values were considered significantly different at p < 0.05.
Results
Induction of bacterial prostatitis in Fischer 344 rats
An 18 week pilot study was conducted to determine if CP1 E. coli could induce inflammation in the rat prostate. Early infection (2–4 days post-inoculation) of the rat prostate was characterized by dense infiltrates of neutrophils into glandular lumens that were identified in all lobes of the prostate, as well as chronic inflammation to a lesser extent in the stroma (Supplementary Fig. S2). At 18 weeks post-inoculation, chronic inflammation persisted as evidenced by dense accumulations of lymphocytes and macrophages in the stroma (Supplementary Fig. S2). We did not observe evidence of infection at other sites in the genitourinary tract, such as kidney or bladder inflammation.
The combinatorial effects of PhIP and E. coli in the F344 rat model
The study design for the 52 week study is shown in Fig. 1A. At the start of the study, animals were randomized into groups with approximately equal body weight averages: control 131.5 ± 2.1 g (± standard error), E. coli 135.6 ± 3.0 g, PhIP 134.1 ± 2.5 g, and PhIP+E. coli 133.0 ± 2.3 g. After two weeks of consumption of PhIP-containing diets, the body weights in the PhIP and PhIP+E. coli groups significantly diverged from control rats and E. coli treated rats, p < 0.0001 (Supplementary Fig. S3A). The trend for PhIP fed groups having the smallest body weights was maintained until the completion of the study at 52 weeks (478.1 ± 14.5 g, p < 0.0001 for PhIP treated rats and 466.9 ± 14.8 g, p < 0.0001 for PhIP + E. coli treated rats) compared to control (556.2 ± 8.3 g) or E. coli treated rats (557.4 ± 10.3 g). Decreased food intake and body weight has previously been reported for rats consuming PhIP (24). Importantly, there was no overall difference in PhIP-containing diet consumption between the PhIP and PhIP+E. coli groups. In weeks 1–20, PhIP treated animals ate an average of 14.9 ± 0.2 g per day and PhIP+E. coli treated animals ate an average of 15.2 ± 0.5 g per day, p = 0.62. There was a significant decrease in the amount of PhIP-containing diet consumption in the PhIP+E. coli group compared to the PhIP group during week 10 following E. coli inoculations, followed by a significant increase in PhIP diet consumption in the PhIP+E. coli group compared to the PhIP group in week 12 (Supplementary Fig. S3B).
Figure 1.
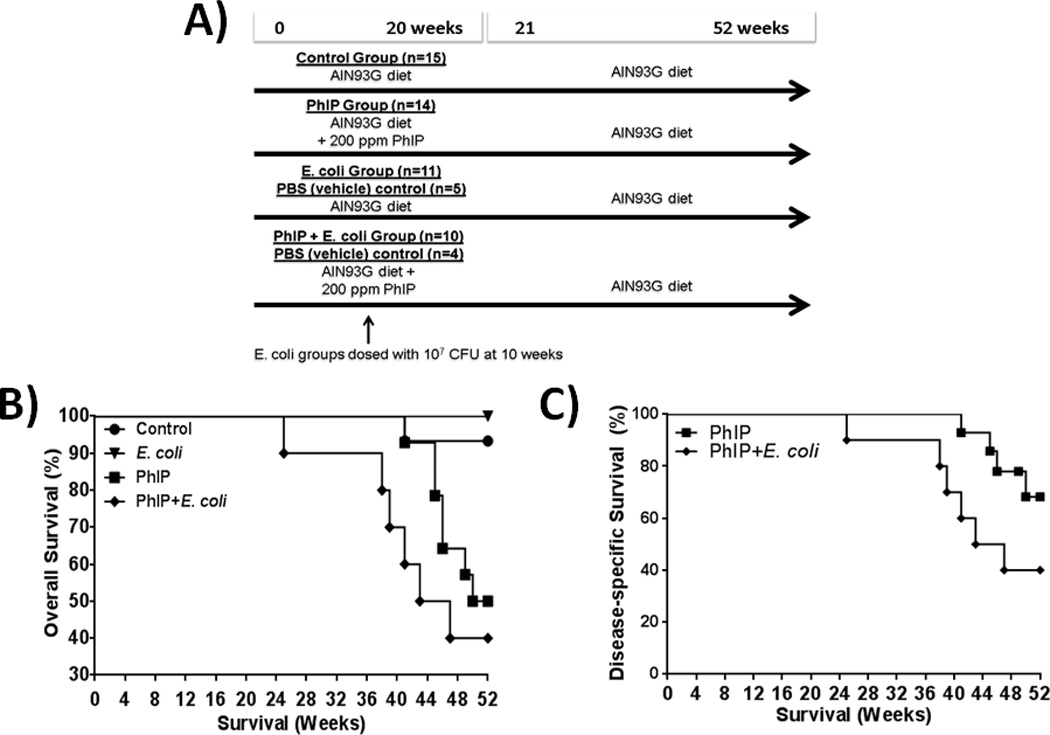
Decreased survival due to the combinatorial effects of PhIP and E. coli. Study design (A) overall survival (B) and disease-specific (e.g., invasive cancer) survival (C).
PhIP+E. coli treatment leads to decreased survival due to malignancy compared to PhIP treatment alone
As shown in Fig. 1B, by the end of 52 weeks, one animal in the control group and no animals in the E. coli group died or had to be euthanized. The control animal was found dead of unknown causes. The animals consuming PhIP-alone exhibited decreased survival beginning at 41 weeks. This decrease in survival was due to the development of PhIP-induced tumors at sites other than the prostate, such as small intestine and skin. In this regard, PhIP-treated male F344 rats are known to develop tumors at sites in addition to the prostate including the skin, small intestines, and colon (26). Three of the PhIP-alone treated animals (45, 46 and 49 weeks at death) had to be euthanized per ACUC criteria for non-invasive benign skin tumors. These animals were censored at the respective weeks of death in cause-specific survival analyses since death was not due to invasive cancer (Fig. 1C). Surprisingly, PhIP+E. coli treated animals exhibited a significant decrease in survival due to malignancy compared to PhIP-alone treated animals (p=0.049, Gehan-Breslow-Wilcoxon test, Fig. 1C).
Ventral prostate tumor incidence
The ventral prostates of PhIP consuming animals develop neoplastic lesions resembling cribriform prostatic intraepithelial neoplasia (PIN) in humans. These lesions often end up distending the ducts/acini, which resembles intraductal carcinoma in the human prostate. Rather than referring to these lesions as intraductal, we consider them as carcinoma in situ (CIS) because intraductal cancer in the human prostate is generally assumed to be the result of retrograde spread of invasive adenocarcinoma back into ducts/acini; and, in the PhIP treated rats, there are no invasive carcinomas so these lesions cannot be from retrograde spread. Together we refer to the pathological lesions of cribriform PIN and CIS as cribriform PIN/CIS lesions and group them together for analysis. One of our initial aims for this study was to compare ventral prostate tumor incidence, number of lesions, and tumor volume in PhIP versus PhIP+E.coli treated animals. Since PhIP-induced prostate lesions increase in frequency and size over time, the unexpected decrease in survival in the PhIP+E. coli group (mean cause-specific survival of 6 weeks less than PhIP-alone treated animals) precluded the conduction of direct, age-matched, comparisons of PhIP-induced prostate cribriform PIN/CIS lesions between the two treatment groups. Therefore, we characterized the lesions that were present at time of death. Note that this will serve to attenuate any effect of E. coli on PhIP-induced PIN/CIS lesions since animals treated with both E. coli and PhIP died at a younger age than those treated with PhIP or E. coli alone.
One animal in the control diet group developed a spontaneous ventral prostate cribriform PIN/CIS lesion and no lesions were observed in the E. coli group. PhIP and PhIP+E. coli treated rats developed cribriform PIN/CIS lesions in 92.9% and 90% of animals, respectively (Fig. 2A,B). None of the prostate tumors reached the strict survival cutoff of 2 cm and survival of the animals was not impacted by the induced PIN/CIS lesions. Even with the discrepancy in survival between the PhIP and PhIP+E. coli groups, PhIP+E. coli animals developed a slightly higher average number of cribriform PIN/CIS lesions (an average of 1.6 lesions/rat in the PhIP group versus 2.1 lesions/rat in the PhIP+E. coli group, Fig. 2C). Area analysis was also performed to quantify the percentage of the ventral prostate that was comprised of cribriform PIN/CIS lesions. The average percentage of ventral prostate comprised of cribriform PIN/CIS was slightly higher for the PhIP group (2.0%) compared to the PhIP+E. coli group (1.7%, Fig. 2D), although again, the PhIP+E. coli animals were on average younger at time of death. Of interest, the Ki-67 index of cribriform PIN/CIS lesions in PhIP+E. coli treated animals was roughly twice that of PhIP-alone treated rats (p=0.0036, Fig. 2E-G).
Figure 2.
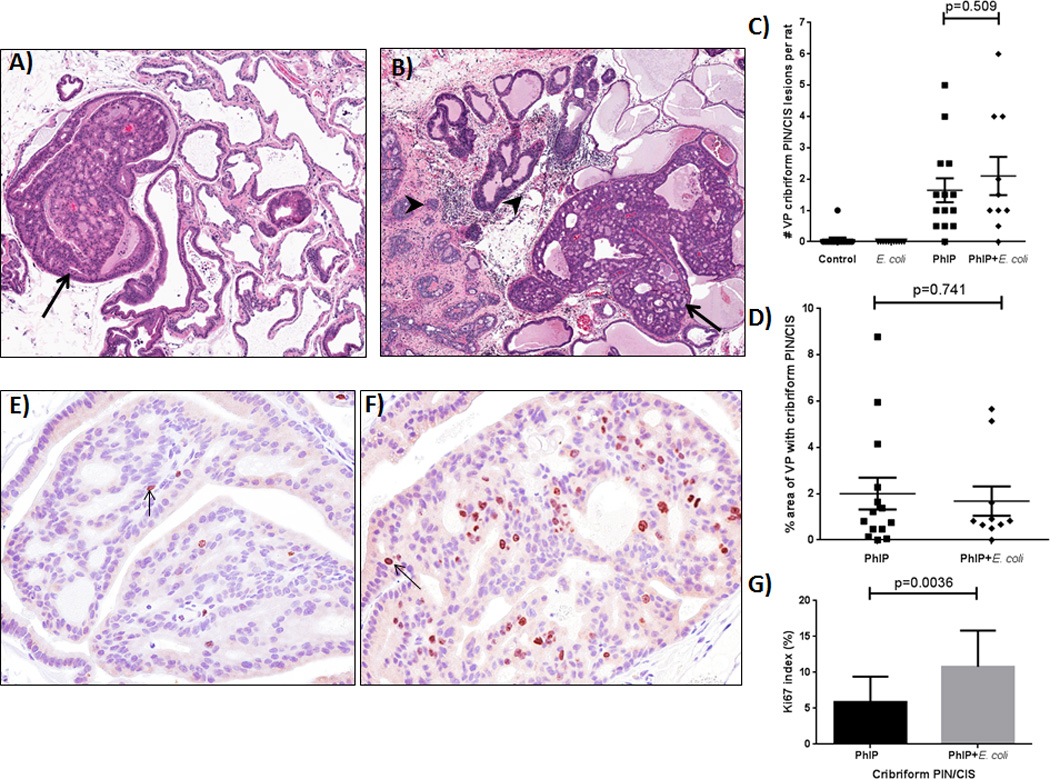
Development of prostatic cribriform PIN/CIS lesions in PhIP treated animals. Examples of cribriform PIN/CIS lesions (arrows) in the ventral prostate (VP) lobe of PhIP (A) and PhIP+E. coli (B) treated animals. Note chronic inflammation (arrowheads) in PhIP+E. coli treated animal. (C) Number of VP cribriform PIN/CIS lesions observed in study animals. (D) Size of cribriform PIN/CIS lesions as percent area of VP. Two H&E step sections were analyzed per animal and average values are reported. Representative images of Ki-67 IHC in the VP of PhIP (E) and PhIP+E. coli (F) treated animals and Ki-67 index (G).
Ventral lobe specificity of prostatic inflammation
We evaluated the presence of E. coli-induced acute and/or chronic inflammation as characterized by dense inflammatory cell infiltrates that were observed on H&E stained slides at time of death (see Supplementary Fig. S1). Acute and chronic inflammation was rarely observed in the prostate lobes of PhIP-alone treated animals and never observed in control animals at time of death (Fig. 3A). Likewise, acute and chronic inflammation was never observed in any of the PBS (vehicle) control animals (see Supplementary Fig. S2). Acute and chronic inflammation was present in the greatest percentage of animals in the ventral prostate lobe of E. coli and PhIP+E. coli treated animals, but inflammation was also present to a lesser extent in the dorsal and anterior lobes (Fig. 3A). The incidence of chronic inflammation was significantly higher in the ventral lobe of PhIP+E. coli treated animals versus the dorsal and anterior lobes (p = 0.023) and higher in the ventral lobe of E. coli-alone treated animals compared to the dorsal and anterior lobes but did not reach statistical significance (Fig. 3A). Lateral prostate lobes could not be reliably evaluated for the presence of acute or chronic inflammation since they were uniformly acutely inflamed in all animals at each time point (see Discussion). Acute inflammation was observed in many of the E. coli-alone treated animals at end of study (42 weeks post-inoculation). In three cases, the prostate was found to be abscessed at end of study in E. coli-alone treated animals. In contrast, the inflammation observed in PhIP+E. coli treated animals was predominantly chronic inflammation at time of death/end of study (Fig. 3A). No evidence of intact E. coli cells (as determined by E. coli-specific IHC) was seen in PhIP+E. coli treated animals at time of death, and it is possible that PhIP could have been toxic to the E. coli. In contrast, two of the E. coli-alone treated animals (both of which had abscesses observed in the prostate) had evidence of intact E. coli cells and active infections at end of study (Supplementary Fig. S4).
Figure 3.
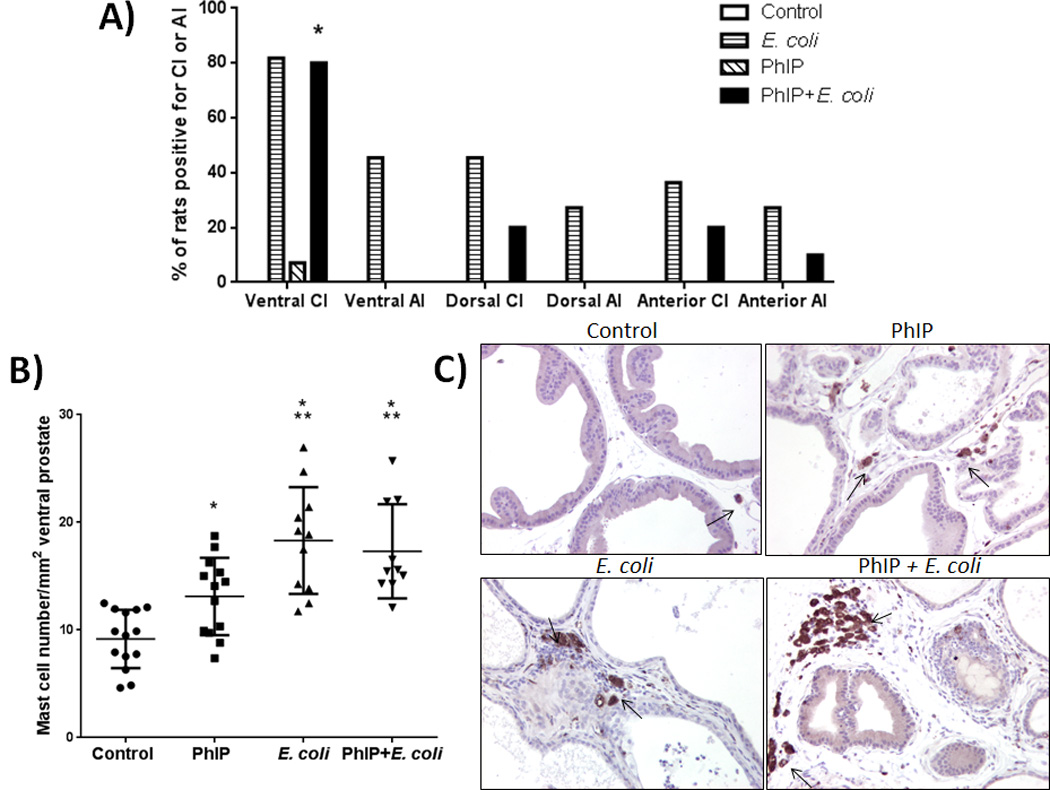
Ventral lobe specificity of chronic inflammation and increased mast cell numbers. Inflammation analysis of prostate lobes at time of death / end of study (A). AI = acute inflammation, CI = chronic inflammation. * Significantly different from dorsal and anterior CI (p = 0.023) by chi-square test. No inflammation was observed in the control group or in PBS (vehicle) treated animals. Mast cell quantification in ventral prostates at time of death/end of study (B). * p < 0.005 versus control, ** p < 0.05 versus PhIP. Representative macrophage (CD68) IHC images (C) from all groups at time of death/end of study. Arrows point to prostate-infiltrating CD68+ macrophages.
The predilection of the ventral lobe for both PhIP-induced pre-cancerous lesions as well as chronic inflammation is of keen interest. Nakai et al. (25) quantified mast cells and macrophages in the prostate immediately after 8 weeks of dietary exposure to PhIP. Although the level of “inflammation” in the Nakai et al. study was not analogous to the severe infection-induced chronic inflammation that we describe in the present study, there was a quantifiable and significant increase in the numbers of mast cells and macrophages specifically in the ventral prostate (25). In the present study, we confirmed this finding of elevated mast cell numbers in the ventral prostate lobe while rats consume PhIP in animals that were sacrificed directly after 20 weeks of PhIP treatment (Supplementary Fig. S5). Intriguingly, quantification of mast cells in the ventral prostate at time of death in the present study (up to 32 weeks after cessation of PhIP consumption) demonstrated that mast cells were still significantly elevated in the ventral prostate lobe compare to animals on control diet (Fig. 3B). Mast cells were also elevated in E. coli and PhIP+E. coli treated animals at levels significantly higher than both control and PhIP-alone treated animals (Fig. 3B). IHC for CD68 in the rat ventral prostate tissues also indicated an increased number of prostate-infiltrating macrophages, particularly in inflamed areas in E. coli and PhIP+E. coli-treated animals (Fig. 3C).
PhIP+E. coli treatment leads to an increase in invasive cancers in multiple bodily locations
As previously mentioned, PhIP-treated male F344 rats are known to develop tumors at sites in addition to the prostate including the skin, small intestines and colon (26). In regards to skin tumors, 42.9% of PhIP-alone treated animals developed lesions and 30% of PhIP+E. coli animals developed lesions. These skin tumors consisted of non-invasive and invasive basaloid, sebaceous, and squamous cell carcinomas (Fig. 4A). In regards to small intestinal and colon tumors, 35.7% of PhIP treated animals and 50% of PhIP+E. coli animals developed small intestinal adenocarcinoma and/or colon adenomas (Fig. 4B). PhIP+E. coli animals also developed Zymbal’s gland tumors, which are similar to squamous cell carcinoma but developed in the Zymbal’s gland, (20% of animals, Fig. 4C) and a sarcoma in a single animal (Fig. 4D). Overall, the PhIP+E. coli treated animals exhibited a 2-fold increase in the development of invasive cancers on average per rat, although this did not reach statistical significance (Fig. 4E, p=0.159).
Figure 4.
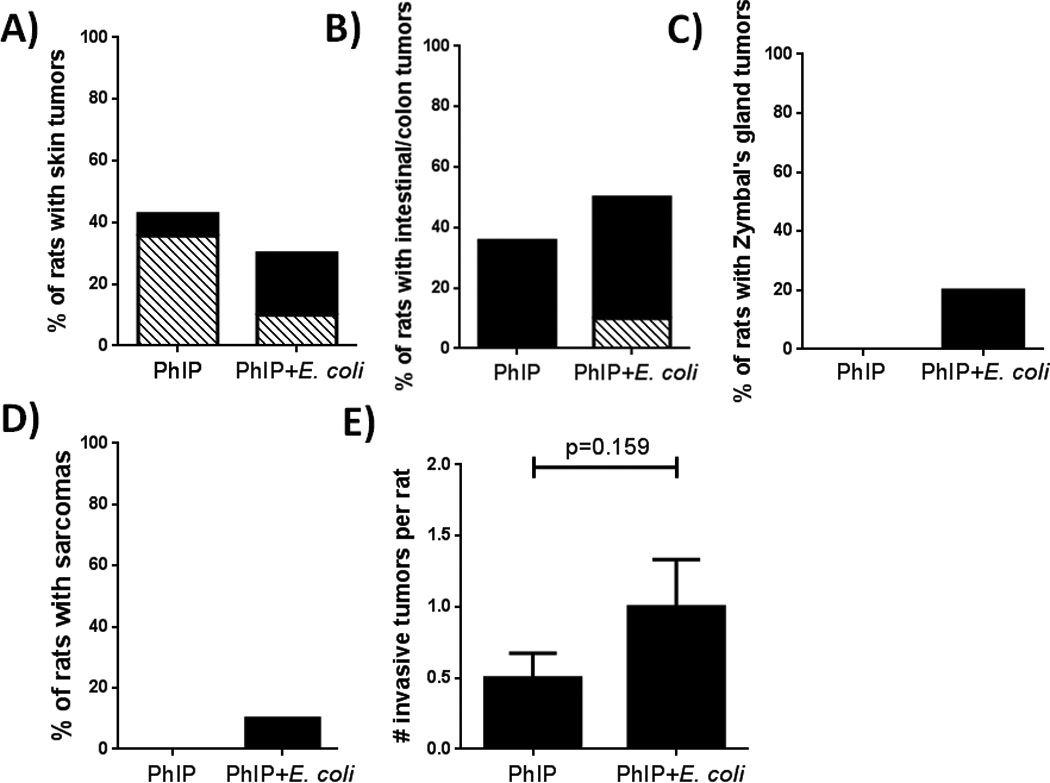
Development of tumors at non-prostate sites. Incidence of skin tumors (A), small intestinal carcinoma/adenomas and colon adenomas (B), Zymbal’s gland tumors (C), sarcoma (D), and total invasive cancers (E). Black bars = invasive cancer, striped bars = non-invasive tumors. None of the differences were found to be statistically significant either by Fisher’s exact test (A-D) or by unpaired two-tailed t tests (E).
Serum cytokine analyses reveal up-regulation of systemic pro-inflammatory cytokines in PhIP+E. coli treated animals
We hypothesized that the increase in the severity and number of invasive carcinomas in PhIP+E. coli-treated animals may have been mediated by an elevation in systemic cytokine levels due to the chronic, persistent prostatitis in UPEC treated animals. We therefore tested a panel of serum cytokines that have been previously linked to cancer development or severity using a multiplexed Luminex assay (GM-CSF, IFNγ, IL-1a, IL-1b, IL-4, IL-6, IL-10, IL-17, IL-12 (p70), Leptin, and TNFα). As shown in Fig. 5A, serum IL-6 levels were elevated in a subset of PhIP-alone treated animals and significantly elevated in PhIP+E. coli treated animals (p=0.001) in comparison to control animals. Serum IL-12 levels were also significantly elevated in PhIP+E. coli treated animals in comparison to PhIP-alone treated animals (p=0.017). Interestingly, serum IL-6 levels were significantly up-regulated in animals that developed cribriform PIN/CIS lesions in the prostate compared to animals that did not develop these prostatic lesions (p=0.012, Fig. 5B). This association was not observed for systemic IL-12 levels (Fig. 5B), or for any of the other cytokines analyzed. With CISH analysis, the only situation where IL-6 was observed to be expressed in the ventral prostate was in inflammatory cells within acutely inflamed areas in E. coli-alone treated animals (Fig. 6A). IL-6 was not found to be overexpressed locally in cribriform PIN/CIS lesions in PhIP or PhIP+E. coli treated animals via CISH analysis (Fig. 6B,C) and was undetectable by reverse transcription PCR on RNA extracted from ventral prostate tissue (data not shown). There were no significant differences in any of the other cytokines analyzed in any of the treatment groups or in association with any other tumor type aside from a significant decrease in leptin levels in PhIP+E. coli treated animals compared to PhIP-alone or E. coli-alone treated animals (Fig. 5A). In this regard, many of the PhIP+E. coli animals exhibited marked weight loss (presumably due to tumor-related sequelae) near time of death/euthanasia.
Figure 5.
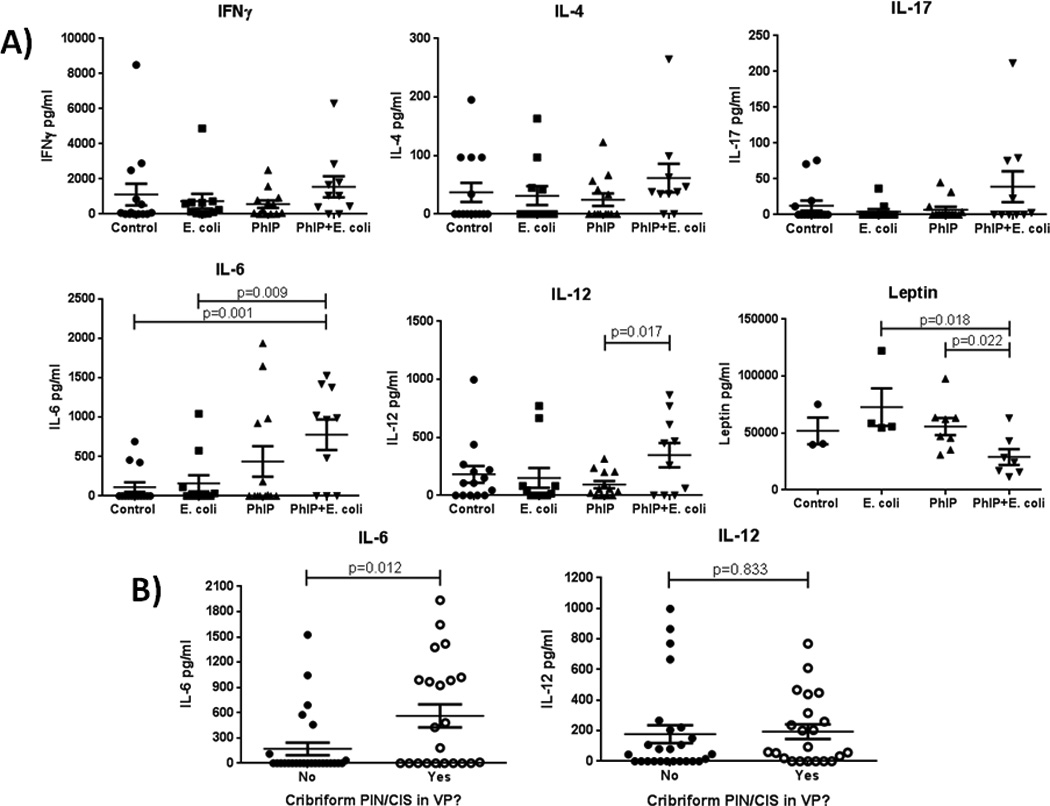
Serum cytokine analyses reveal increased systemic IL-6 and IL-12(p70) levels in PhIP+E. coli treated animals and a positive correlation between systemic IL-6 levels and the development of prostate cribriform PIN/CIS lesions. Serum levels of IFNγ, IL-4, IL-17, IL-6, IL-12(p70), and leptin as determined by Luminex assay (A). Significant differences are indicated, otherwise p > 0.05. Serum IL-6 and IL-12(p70) levels versus the presence or absence of cribriform PIN/CIS lesions in the ventral prostate (B). Only a subset of study animals were evaluated for leptin levels. Detectable levels of GM-CSF, IL-1a, IL-1b, IL-10, and TNFα were not observed in more than 1 study animal in any of the treatment groups, therefore data are not shown.
Figure 6.
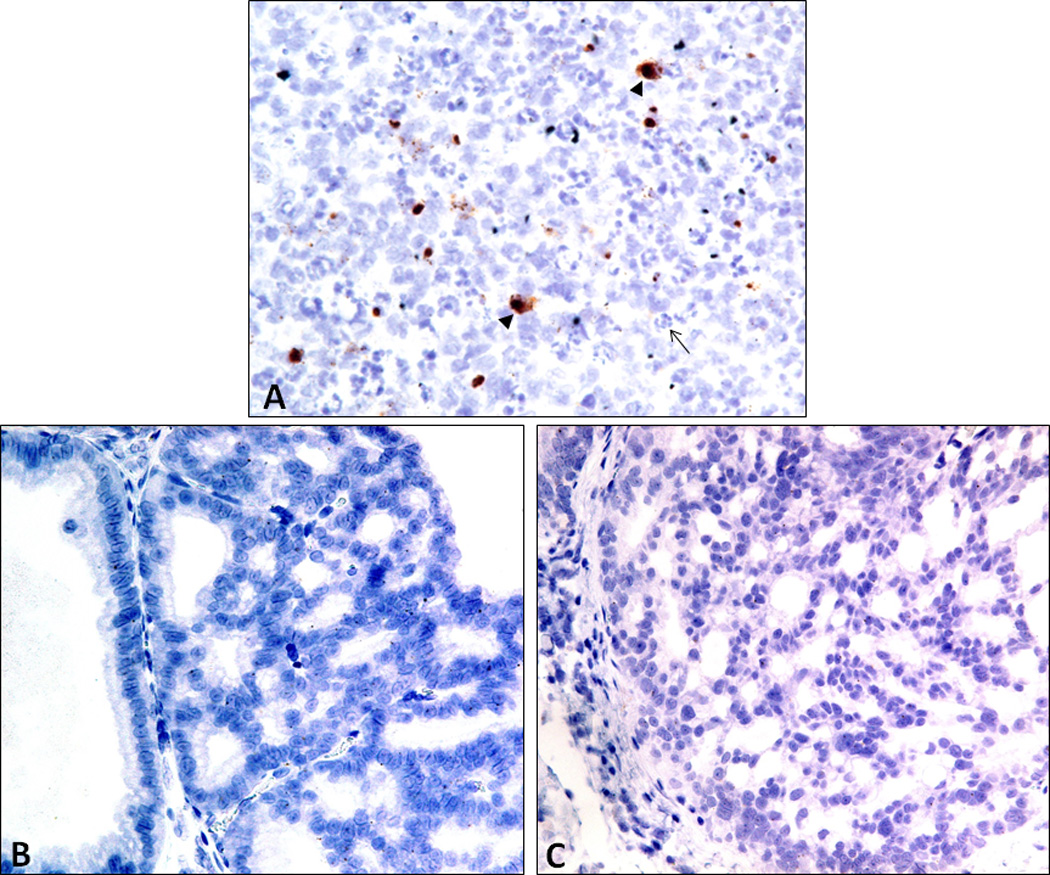
CISH analysis indicates that IL-6 is not expressed locally in cribriform PIN/CIS lesions. IL-6 expression in the rat ventral prostate was only observed in infiltrating immune cells (arrowheads) in areas of acute inflammation (as evidenced by the dense infiltration of neutrophils, arrow) in E. coli-alone treated animals (A). IL-6 was not observed by CISH in cribriform PIN/CIS lesions in either PhIP (B) or PhIP+E. coli (C) treated rats. Single spots in the nucleus likely represent the genomic copy of IL-6.
Discussion
In the present study, we describe a rat model for E. coli-induced prostatitis whereby a single infection generates a chronic inflammatory response that persists for up to 42 weeks post-infection. Whereas all lobes of the prostate are subject to marked inflammation immediately following infection in this model, chronic inflammation was observed predominantly in the ventral lobe of the prostate at time of death. When rats were fed the dietary carcinogen PhIP concurrent to prostate infection, they exhibited a marked decrease in survival compared to PhIP-alone treated animals as a result of an increase in the number of invasive cancers that developed at multiple distant non-prostatic sites including the skin, small intestine, and Zymbal’s gland. The model used in the present study for analyzing the effect of PhIP consumption on tumor development in rats has been well established. Shirai et al. (30) previously reported that a dose of 400 ppm (but not 100 or 25 ppm) significantly induced cancerous lesions in the rat prostate compared to non-treated controls, but also suppressed animal growth. Borowsky et al. (24) planned a 20 week study of PhIP given at 400 ppm, but reported that due to decreased food intakes, body weights, and health of rats consuming 400 ppm PhIP, the dose was decreased to 200 ppm in week 13 of the study. Thus we chose to use 200 ppm for 20 weeks in our study. Importantly, it should be noted that the dose of 200 ppm PhIP utilized in this study is not a physiologically relevant dose compared to typical human PhIP consumption levels. The high dose given over 20 weeks is meant to induce a very high incidence of rodent cancers in a relatively short period of time. Thus, the high incidence of cancers observed in this study would not be expected to occur in humans due to the lower levels of PhIP consumed by humans routinely. However, humans may consume PhIP over a much longer time period and it is difficult to determine how high dose short term exposure is related to much longer term exposure of lower doses. Thus, additional studies need to be carried out to explore the hypothesis that chronic prostatic infections could increase the risk of carcinogen-induced cancer at distant sites in humans.
At the onset of our study, our aim was to determine whether the induction of chronic inflammation via bacterial infection in the rat prostate would alter the biology of PhIP-induced prostate precancerous lesions. Yet, as a result of the increased tumorigenesis in other organs, we were not able to specifically address age-matched prostate neoplastic lesion formation in the PhIP vs. the PhIP + E. coli animals. Of interest, our study demonstrates that UPEC-induced long-term chronic inflammation in the prostate does not in itself induce carcinogenesis (at least not by the 52 week time point in the E. coli-alone arm of our study). Further, the lateral prostatic lobes, which exhibited marked acute inflammation at all time points, do not develop neoplastic lesions in response to PhIP. The finding of ubiquitous lateral lobe inflammation in all of the study animals is possibly due to the absence of soy/phytoestrogens in the AIN93G diet, which has been previously described in Sprague-Dawley rats (31). The mechanism whereby dietary soy reduces prostatitis in rats is unknown.
What we unexpectedly observed was that the establishment of prostatic infection and chronic inflammation via UPEC infection in rats who were also fed PhIP appeared to have a systemic effect, leading to an increase in the development of carcinoma at multiple sites distant to the prostate. To our knowledge, these findings represent the first documented occurrence of an infection limited to the prostate that results in an increase in carcinogen-induced tumorigenesis elsewhere in the body. This phenomenon of bacterial infections in one anatomic site leading to tumor promotion in an anatomically distinct site via systemic inflammatory effects has in fact been previously described in the literature in models of the mouse intestinal pathogen Helicobacter hepaticus (H. hepaticus). In one study, intestinal colonization by H. hepaticus in mice was shown to be sufficient to promote aflatoxin-induced hepatocellular carcinoma (32). This promotion of liver carcinogenesis by H. hepaticus was reported to have occurred without H. hepaticus leaving the lower bowel and may have been due to H. hepaticus-induced TH1-type responses and secreted factors from the intestine that may have acted upon the liver. Intriguingly, serum cytokine analysis in male mice identified elevated levels of IL-12 p40 that correlated to the development of liver lesions (32). In the present study, IL-12 (p70) was found to be significantly elevated in the serum of PhIP+E. coli versus PhIP-alone treated animals. Another study of intestinal H. hepaticus infection in Rag2-deficient C57BL/6 ApcMin/+ mice demonstrated an increase in mammary carcinoma in female mice (33). Mammary tumor promotion in this model was hypothesized to be mediated by systemic increases in pro-inflammatory cytokines and/or trafficking of activated immune cells to mammary tissues (33). Collectively, these studies along with our present study highlight a potential mechanism whereby localized bacterial infections (or other mechanisms of chronic/persistent inflammation) may drive systemic cytokine levels that may influence tumor development at sites spatially distant to the site of infection. Furthermore, IL-12 may be a cytokine of particular interest in mediating this process. These findings raise an interesting question of whether chronic infection in one organ can cause an elevated risk of cancer is distant organs in humans. To our knowledge, no specific examples of this exist in humans. However, this is somewhat reminiscent of organ transplant patients who are known to have a higher risk of cancer in multiple locations (34). While this increase is largely the result of infection-related cancers, not all of the risk has been attributed to such infection-related cancers. At present, it is unknown whether men with chronic bacterial or other forms of chronic prostatitis have elevated levels of cancer at sites beyond the prostate, although this could be addressed to some extent in existing longitudinal epidemiological studies.
Intriguingly, we also observed an increase in serum IL-6 levels in rats consuming PhIP and a more pronounced increase in serum IL-6 levels in rats treated with PhIP+E. coli. It is not currently known whether PhIP consumption in humans can lead to increased systemic IL-6 levels. Furthermore, serum IL-6 levels were positively correlated with the presence of precancerous prostate lesions. This positive correlation between systemic IL-6 levels and tumor development was not observed for any of the other PhIP-induced tumor types (skin, small intestinal, etc.). Circulating levels of IL-6 are elevated in patients with metastatic prostate cancer and hormone-refractory prostate cancer (35–37), and systemic levels of IL-6 have been shown to be correlated with measures of prostate cancer morbidity (38). Our findings that increased systemic levels of IL-6 likely cannot be accounted for by IL-6 expression locally from the prostate or from prostate cribriform PIN/CIS lesions in this model are consistent with recent evidence that human prostate cancer cells do not express IL-6 in either primary or metastatic cancer (Yu S., Drake C., De Marzo A.M., Sfanos K., manuscript in process). It is possible that PhIP itself may result in systemic elevation of IL-6, at least in some animals, but the mechanism by which this would occur remains unclear. It should be noted that although we observed significant increases in select systemic cytokine levels in correlation with PhIP and/or PhIP+E. coli treatment, there was marked heterogeneity in the cytokine levels among study animals. Further studies will be required to provide mechanistic insight into how elevation of circulating cytokines may contribute to the carcinogenic process.
In summary, we set out to determine whether the combination of a dietary carcinogen and a common infectious agent in the rat prostate could synergize in causing prostate cancer. Yet, we could not fully answer this question as a result of an unexpected increase in neoplasms that developed at multiple distant sites that resulted in decreased survival of the animals. These findings raise the intriguing and novel hypothesis that chronic prostatic infections could increase the risk of cancer at distant sites in humans and additional studies are required to address this.
Supplementary Material
Acknowledgements
We would like to thank Dr. Charles Drake and Dr. Bruce Trock for helpful discussions.
Financial Support: K.S. Sfanos was funded by a Postdoctoral Fellowship Award from the Prevent Cancer Foundation and is supported as the Chris and Felicia Evensen Prostate Cancer Foundation (PCF) Young Investigator and through the Patrick C. Walsh Prostate Cancer Research Fund as the Beth W. and A. Ross Myers Scholar. K. Canene-Adams was supported by a Ruth L. Kirschstein NRSA Institutional Research Training Grant (T32 CA067751). This work was also supported by NIH grant P30 CA006973 (to W.G. Nelson) and the Maryland Cigarette Restitution Fund at Johns Hopkins (to A.M. De Marzo).
Footnotes
Conflicts of Interest: None
References
- 1.Wild CP. Complementing the genome with an exposome: The outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiology Biomarkers & Prevention. 2005;14:1847–1850. doi: 10.1158/1055-9965.EPI-05-0456. [DOI] [PubMed] [Google Scholar]
- 2.Sugimura T. Nutrition and dietary carcinogens. Carcinogenesis. 2000;21:387–395. doi: 10.1093/carcin/21.3.387. [DOI] [PubMed] [Google Scholar]
- 3.Nelson WG. Prostate Cancer Prevention. The Journal of Nutrition. 2004;134:3211S–3212S. doi: 10.1093/jn/134.11.3211S. [DOI] [PubMed] [Google Scholar]
- 4.Schut HAJ, Snyderwine EG. DNA adducts of heterocyclic amine food mutagens: implications for mutagenesis and carcinogenesis. Carcinogenesis. 1999;20:353–368. doi: 10.1093/carcin/20.3.353. [DOI] [PubMed] [Google Scholar]
- 5.Sander A, Linseisen J, Rohrmann S. Intake of heterocyclic aromatic amines and the risk of prostate cancer in the EPIC-Heidelberg cohort. Cancer Causes and Control. 2011;22:109–114. doi: 10.1007/s10552-010-9680-9. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez C, McCullough ML, Mondul AM, Jacobs EJ, Chao A, Patel AV, et al. Meat Consumption among Black and White Men and Risk of Prostate Cancer in the Cancer Prevention Study II Nutrition Cohort. Cancer Epidemiology Biomarkers & Prevention. 2006;15:211–216. doi: 10.1158/1055-9965.EPI-05-0614. [DOI] [PubMed] [Google Scholar]
- 7.Sinha R, Park Y, Graubard BI, Leitzmann MF, Hollenbeck A, Schatzkin A, et al. Meat and meat-related compounds and risk of prostate cancer in a large prospective cohort study in the United States. American Journal of Epidemiology. 2009;170:1165–1177. doi: 10.1093/aje/kwp280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koutros S, Cross AJ, Sandler DP, Hoppin JA, Ma X, Zheng T, et al. Meat and meat mutagens and risk of prostate cancer in the agricultural health study. Cancer Epidemiology Biomarkers & Prevention. 2008;17:80–87. doi: 10.1158/1055-9965.EPI-07-0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.John EM, Stern MC, Sinha R, Koo J. Meat consumption, cooking practices, meat mutagens, and risk of prostate cancer. Nutrition and cancer. 2011;63:525–537. doi: 10.1080/01635581.2011.539311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shirai T, Sano M, Tamano S, Takahashi S, Hirose M, Futakuchi M, et al. The prostate: a target for carcinogenicity of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) derived from cooked foods. Cancer Res. 1997;57:195–198. [PubMed] [Google Scholar]
- 11.Grivennikov SI, Greten FR, Karin M. Immunity, Inflammation, and Cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanahan D, Weinberg Robert A. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 13.de Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, et al. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. The Lancet Oncology. 2012;13:607–615. doi: 10.1016/S1470-2045(12)70137-7. [DOI] [PubMed] [Google Scholar]
- 14.Sfanos KS, Isaacs WB, De Marzo AM. Infections and inflammation in prostate cancer. Am J Clin Exp Urol. 2013;1:3–11. [PMC free article] [PubMed] [Google Scholar]
- 15.Sfanos KS, De Marzo AM. Prostate cancer and inflammation: the evidence. Histopathology. 2012;60:199–215. doi: 10.1111/j.1365-2559.2011.04033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sfanos KS, Sauvageot J, Fedor HL, Dick JD, De Marzo AM, Isaacs WB. A molecular analysis of prokaryotic and viral DNA sequences in prostate tissue from patients with prostate cancer indicates the presence of multiple and diverse microorganisms. The Prostate. 2008;68:306–320. doi: 10.1002/pros.20680. [DOI] [PubMed] [Google Scholar]
- 17.Bergh J, Marklund I, Thellenberg-Karls C, Gronberg H, Elgh F, Alexeyev OA. Detection of Escherichia coli 16S RNA and cytotoxic necrotizing factor 1 gene in benign prostate hyperplasia. Eur Urol. 2007;51:457–462. doi: 10.1016/j.eururo.2006.06.008. discussion 62-3. [DOI] [PubMed] [Google Scholar]
- 18.Yanamandra K, Alexeyev O, Zamotin V, Srivastava V, Shchukarev A, Brorsson A-C, et al. Amyloid formation by the pro-inflammatory S100A8/A9 proteins in the ageing prostate. PLoS ONE. 2009;4:e5562. doi: 10.1371/journal.pone.0005562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sfanos KS, Wilson BA, De Marzo AM, Isaacs WB. Acute inflammatory proteins constitute the organic matrix of prostatic corpora amylacea and calculi in men with prostate cancer. Proceedings of the National Academy of Sciences. 2009;106:3443–3448. doi: 10.1073/pnas.0810473106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boehm BJ, Colopy SA, Jerde TJ, Loftus CJ, Bushman W. Acute bacterial inflammation of the mouse prostate. The Prostate. 2012;72:307–317. doi: 10.1002/pros.21433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elkahwaji JE, Hauke RJ, Brawner CM. Chronic bacterial inflammation induces prostatic intraepithelial neoplasia in mouse prostate. Br J Cancer. 2009;101:1740–1748. doi: 10.1038/sj.bjc.6605370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khalili M, Mutton LN, Gurel B, Hicks JL, De Marzo AM, Bieberich CJ. Loss of Nkx3.1 expression in bacterial prostatitis: A potential link between inflammation and neoplasia. The American Journal of Pathology. 2010;176:2259–2268. doi: 10.2353/ajpath.2010.080747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simons BW, Durham NM, Bruno TC, Grosso JF, Schaeffer AJ, Ross AE, et al. A human prostatic bacterial isolate alters the prostatic microenvironment and accelerates prostate cancer progression. The Journal of Pathology. 2014 doi: 10.1002/path.4472. n/a-n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Borowsky AD, Dingley KH, Ubick E, Turteltaub KW, Cardiff RD, Devere-White R. Inflammation and atrophy precede prostatic neoplasia in a PhIP-induced rat model. Neoplasia. 2006;8:708–715. doi: 10.1593/neo.06373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakai Y, Nelson WG, De Marzo AM. The dietary charred meat carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine acts as both a tumor initiator and promoter in the rat ventral prostate. Cancer Res. 2007;67:1378–1384. doi: 10.1158/0008-5472.CAN-06-1336. [DOI] [PubMed] [Google Scholar]
- 26.Canene-Adams K, Sfanos KS, Liang C-T, Yegnasubramanian S, Nelson WG, Brayton C, et al. Dietary chemoprevention of PhIP induced carcinogenesis in male Fischer 344 rats with tomato and broccoli. PLoS ONE. 2013;8:e79842. doi: 10.1371/journal.pone.0079842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rudick CN, Berry RE, Johnson JR, Johnston B, Klumpp DJ, Schaeffer AJ, et al. Uropathogenic Escherichia coli induces chronic pelvic pain. Infect Immun. 2011;79:628–635. doi: 10.1128/IAI.00910-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson JR, Delavari P, Kuskowski M, Stell AL. Phylogenetic distribution of extraintestinal virulence-associated traits in Escherichia coli. Journal of Infectious Diseases. 2001;183:78–88. doi: 10.1086/317656. [DOI] [PubMed] [Google Scholar]
- 29.Johnson JR, Kuskowski MA, Gajewski A, Soto S, Horcajada JP, de Anta MTJ, et al. Extended virulence genotypes and phylogenetic background of Escherichia coli isolates from patients with cystitis, pyelonephritis, or prostatitis. Journal of Infectious Diseases. 2005;191:46–50. doi: 10.1086/426450. [DOI] [PubMed] [Google Scholar]
- 30.Shirai T, Cui L, Takahashi S, Futakuchi M, Asamoto M, Kato K, et al. Carcinogenicity of 2-amino-1-methyl-6-phenylimidazo [4,5-b]pyridine (PhIP) in the rat prostate and induction of invasive carcinomas by subsequent treatment with testosterone propionate. Cancer letters. 1999;143:217–221. doi: 10.1016/s0304-3835(99)00128-7. [DOI] [PubMed] [Google Scholar]
- 31.Sharma OP, Adlercreutz H, Stranberg JD, Zirkin BR, Coffey DS, Ewing LL. Soy of dietary source plays a preventive role against the pathogenesis of prostatitis in rats. The Journal of Steroid Biochemistry and Molecular Biology. 1992;43:557–564. doi: 10.1016/0960-0760(92)90244-d. [DOI] [PubMed] [Google Scholar]
- 32.Fox JG, Feng Y, Theve EJ, Raczynski AR, Fiala JLA, Doernte AL, et al. Gut microbes define liver cancer risk in mice exposed to chemical and viral transgenic hepatocarcinogens. Gut. 2010;59:88–97. doi: 10.1136/gut.2009.183749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rao VP, Poutahidis T, Ge Z, Nambiar PR, Boussahmain C, Wang YY, et al. Innate immune inflammatory response against enteric bacteria Helicobacter hepaticus induces mammary adenocarcinoma in mice. Cancer Research. 2006;66:7395–7400. doi: 10.1158/0008-5472.CAN-06-0558. [DOI] [PubMed] [Google Scholar]
- 34.Engels EA, Pfeiffer RM, Fraumeni JF, et al. Spectrum of cancer risk among us solid organ transplant recipients. JAMA. 2011;306:1891–1901. doi: 10.1001/jama.2011.1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adler HL, McCurdy MA, Kattan MW, Timme TL, Scardino PT, Thompson TC. Elevated levels of circulating interleukin-6 and transforming growth factor-beta1 in patients with metastatic prostatic carcinoma. J Urol. 1999;161:182–187. [PubMed] [Google Scholar]
- 36.Drachenberg DE, Elgamal A-AA, Rowbotham R, Peterson M, Murphy GP. Circulating levels of interleukin-6 in patients with hormone refractory prostate cancer. The Prostate. 1999;41:127–133. doi: 10.1002/(sici)1097-0045(19991001)41:2<127::aid-pros7>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 37.Nakashima J, Tachibana M, Horiguchi Y, Oya M, Ohigashi T, Asakura H, et al. Serum Interleukin 6 as a Prognostic Factor in Patients with Prostate Cancer. Clinical Cancer Research. 2000;6:2702–2706. [PubMed] [Google Scholar]
- 38.Twillie DA, Eisenberger MA, Carducci MA, Hseih W-S, Kim WY, Simons JW. Interleukin-6: A candidate mediator of human prostate cancer morbidity. Urology. 1995;45:542–549. doi: 10.1016/S0090-4295(99)80034-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.