

Abstract
Nitric oxide (NO) plays relevant roles in signal transduction in physiopathology and its effects are dependent on several environmental factors. NO has both pro-apoptotic and anti-apoptotic functions but the molecular mechanisms responsible for these opposite effects are not fully understood. The action of NO occurs mainly through redox changes in target proteins, particularly by S-nitrosylation of reactive cysteine residues. Thioredoxin (Trx) and glutaredoxin (Grx) systems are the main cellular controllers of the thiolic redox state of proteins exerting controversial effects on apoptosis with consequences for the resistance to or the development of cancer.
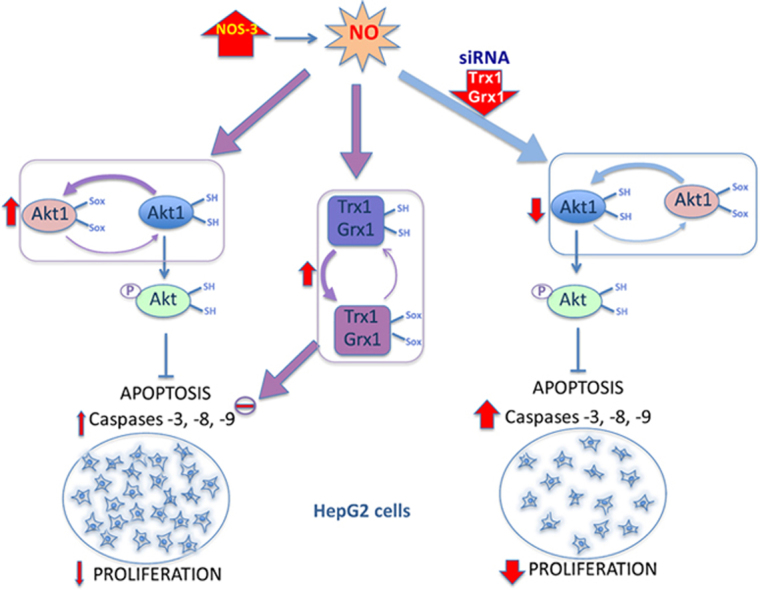
The aim of this study was to ascertain whether Trx and/or Grx systems mediate the antiproliferative effect of NO on hepatoblastoma cells by modulating the redox-state of key proteins.
Proliferation decreased and apoptosis increased in HepG2 cells overexpressing Nitric Oxide Synthase-3 (NOS-3) as a result of multilevel cellular responses to the oxidative environment generated by NO. Enzyme levels and cysteine redox state at several metabolic checkpoints were consistent with prominence of the pentose phosphate pathway to direct the metabolic flux toward NADPH for antioxidant defense and lowering of nucleotide biosynthesis and hence proliferation. Proteins involved in cell survival pathways, proteins of the redoxin systems and phosphorylation of MAPK were all significantly increased accompanied by a shift of the thiolic redox state of Akt1, Trx1 and Grx1 to more oxidized.
Silencing of Trx1 and Grx1 neutralized the increases in CD95, Akt1 and pAkt levels induced by NO and produced a marked increase in caspase-3 and -8 activities in both control and NOS-3 overexpressing cells concomitant with a decrease in the number of cells.
These results demonstrate that the antiproliferative effect of NO is actually hampered by Trx1 and Grx1 and support the strategy of weakening the thiolic antioxidant defenses when designing new antitumoral therapies.
Abbreviations: ACO, aconitase; ASK1, apoptosis signal-regulating kinase 1; Bcl-2, B-cell lymphoma 2; CaM, calmodulin; CD95, cluster of differentiation 95; DTNB, 5,5-dithio-bis-2-nitrobenzoic acid; ELISA, enzyme-linked immunosorbent assay; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; Grx, glutaredoxin; HED, 2-hydroxyethyl disulfide; JAKs, Janus protein tyrosine kinases; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; MATII, methionine adenosyltransferase II; MM(PEG)24, Methyl-PEG24-Maleimide; mTOR, mammalian target of rapamycin; NEM, N-ethylmaleimide; NO, nitric oxide; NOS, nitric oxide synthase; PBS, phosphate buffered saline; PDK, phosphoinositide-dependent kinase; PKB, protein kinase B; PKM2, pyruvate kinase isozyme M2; PMSF, phenylmethylsulfonyl fluoride; PP2A, protein phosphatase 2A; PrSSG, mixed disulfide between protein and glutathione or glutathionylated protein; PrSSPr, inter- or intra-molecular protein disulfide; RNS, reactive nitrogen species; ROS, reactive oxygen species; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; STAT3, signal transducer and activator of transcription 3; TCEP, tris(2-carboxyethyl)phosphine; TKT, transketolase; Trx, thioredoxin; TrxR, thioredoxin reductase; TUNEL, terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling; TXNIP, thioredoxin-interacting protein; ROD, uroporphyrin decarboxylase
Keywords: Thioredoxin, Glutaredoxin, Oxidative/nitrosative stress, Apoptosis, Cancer
Graphical abstract
Highlights
-
•
Endogenous NO induces NADPH and reduces nucleotide biosynthesis in HepG2 cells.
-
•
Trx1 and Grx1 have a pro-oxidant action on key proteins under nitrosative conditions.
-
•
Trx1 and Grx1 hamper the antiproliferative action of NO in tumoral cells.
-
•
Weakening of thiolic antioxidant defenses could help in the design of anti-tumoral therapies.
1. Introduction
Nitric oxide (NO) is a very small, lipophilic, readily diffusible, chemically unstable molecule with a very short half-life (seconds) that plays a relevant role in signal transduction in physiopathology such as vasodilation, respiration, cell migration, immune response and apoptosis [1]. NO is known to be synthesized in a large number of different tissues by the NO synthases (NOS) using l-arginine as substrate. Three different isoforms of NOS have been identified, products of different genes, with different localization, regulation, catalytic properties and inhibitor sensitivity [2]. Their expression and activity are cellular and tissue specific with differential regulation at transcriptional, translational and posttranslational levels [3,4]. These isoforms in mammals are: neuronal NOS (nNOS or NOS-1), inducible NOS (iNOS or NOS-2) and endothelial NOS (eNOS or NOS-3) [5–8]. The NOS-1 and NOS-3 isoforms are constitutively expressed and can be activated as a result of calmodulin (CaM) binding following a rise in intracellular calcium, and also by phosphorylation/dephosphorylation modifications. The expression of NOS-2 isoform is induced by inflammatory stimuli and is maximally activated by Ca2+/CaM even at basal levels of intracellular Ca2+ [1,2,9]. The intracellular localization is relevant for the activity of NOS and accumulating evidence indicates that NOSs are subject to specific targeting to subcellular compartments (plasma membrane, Golgi, cytosol, nucleus and mitochondria) and that this trafficking is crucial for NO production and specific posttranslational modifications of target proteins [10–12].
NO can have opposite biological effects, depending on their local concentration, the target cell type involved and the levels of reactive oxygen species (ROS). The role of NO as a bioregulator of apoptosis is well established, having both antiapoptotic and proapoptotic functions [13]. The molecular mechanisms responsible for its opposite effect are not fully understood but there is strong evidence indicating the involvement of redox changes in key proteins [14].
The cellular redox state plays a critical role in regulating many signaling pathways including activation, differentiation, proliferation, and apoptosis [15–17]. ROS and reactive nitrogen species (RNS), cause irreversible damage when their amounts exceed the cellular antioxidant defense capacity, and are harmful to biomolecules, including genomic and mitochondrial DNA, membrane lipids and proteins. But they can also lead to reversible oxidations that play regulatory roles of protein function. Within proteins, the thiol group (–SH) of cysteine (Cys) can be oxidized in several ways: two thiols can form a disulfide bond as in some proteins (PSSP), or mixed disulfide in glutathionylated proteins (PSSG). Additionally, cysteine can be reversibly oxidized by ROS or RNS to sulfenic acid (–SOH) and nitrosothiol (–SNO). The nitrosylation of reactive cysteine residues in proteins takes part in NO signaling processes and can affect a multitude of intracellular events, beneficial or harmful, depending upon biological context [18–22].
The redox states of Cys residues are controlled by two major cellular systems, the Trx/thioredoxin reductase system and the glutathione (GSH)/Grx system [15,23,24]. The Trx system consists of redox active Trx, thioredoxin reductase (TrxR) and NADPH, which are critical for maintaining DNA synthesis and the cellular redox balance. Human Trx1 and TrxR1 are located in cell cytosol/nucleus. The Grx system consists of Grx, GSH and NADPH-dependent glutathione reductase. Human Grx1 is located in the cytosol [25] and is involved in redox-regulation through the reduction of protein disulfides and mixed disulfides, e.g. deglutathionylation of proteins [24,26].
A relationship exists between redoxins levels and apoptosis with consequences for the resistance or the development of cancer. As an antioxidant system Trx/TrxR catalyzes the denitrosylation of SNO-caspase-3 [27] and some experimental data suggested that it also participates in the denitrosylation of SNO-caspase-9 and the reductive reactivation of caspase-8 [28]. But as a pro-oxidant Trx has been described trans-nitrosylating and inactivating caspase-3 thus showing an anti-apoptotic action [29].
It has been shown that Trx1 and TrxR1 are often overexpressed in tumor cells and that high Trx could be linked to drug resistance during cancer treatment [30]. Other studies suggest that high Trx and TrxR may induce apoptosis and reduce the mitotic index of certain tumors linked to p53 dependent cell death [31]. Reduced Trx is a negative regulator of ASK1 (apoptotic-inducing kinase), which relates the Trx system to evasion of apoptosis [32]. Another apoptosis-regulatory enzyme whose nitrosylation status is reversibly regulated by Trx1 is glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [33]. Because reduced Trx1 plays a critical role in cellular proliferation and viability, excessive oxidation of Trx will lead to cell death [30,34].
On the other hand, Grx1 plays an important role in protecting cells from apoptosis by regulating the redox state of Akt1, also called protein kinase B (PKB), that has consequences for cell survival and also affect the multiple roles played by Akt1, as in the Akt-mTOR signaling cascade [35]. Mitochondrial Grx2 also exerts a protective effect on mitochondrial mediated apoptosis, preventing cardiolipin oxidation and cytochrome c release [36].
The intracellular mechanism regulating cell death and cell proliferation are intimately connected and different studies have shown that NO production has an important role in the regulation of the carcinogenic process. For instance, S-nitrosylation of some proteins, such as GAPDH and CD95, stimulates apoptosis whereas S-nitrosylation of other proteins, such as caspases and Bcl-2, inhibits apoptosis [33]. NO exerts an antineoplastic effect in tumoral cells by increasing cell death [37] and a specific pattern of S-nitrosylation has been observed during induction of apoptosis in hepatocytes [38].
The role of antioxidants in cancer has been controversial for decades. On one hand, ROS could mediate the activation of multiple signaling cascades that promote cell proliferation and on the other hand, the consequent increase in oxidative stress could cause senescence or apoptosis and became a tumor suppressor. Recent evidence indicates that antioxidants such as GSH and Trx can actually contribute to tumorigenesis by preventing ROS accumulation in cancer cells. The cellular response will depend on the levels of ROS and antioxidant status in the cell [31,39,40].
The main objective of this study was to ascertain whether Trx and/or Grx systems mediate the antiproliferative effect of NO on hepatoblastoma cells by modulating the redox-state of key proteins. We demonstrate that Trx1 and Grx1 behave differentially depending on the intracellular oxidative/nitrosative stress in HepG2 cells. They are required for proliferation but they also contribute to the antiproliferative effect of NO, associated with Akt1 redox changes.
2. Material and methods
2.1. Materials
All reagents were of analytical grade and were purchased from Sigma, unless otherwise specified.
HepG2 cell line used in this work was obtained from ATCC LGC Standards Company (Teddington, UK). Cell culture dish and flasks were from TPP (Switzerland). Anti-Trx1 and anti-Grx1 were obtained from rabbit in our laboratory. Antibodies against STAT3, MAPK, Thr202/Tyr204p-MAPK (p-MAPK) and Ser473p-Akt (p-Akt) were from Cell Signaling Technology. Antibodies against ACO1 and UROD were from Aviva Systems Biology (San Diego, CA, USA). Antibodies against ACO2, TKT, TXNIP, Akt1, MATII, Bcl2, PKM2, caspase-3, CD95, NOS-3 and β-actin were from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Anti-TrxR1 was from Abcam, Inc. Secondary antibodies were from Sigma. ECL was from GE Healthcare (Wauwatosa, Wisconsin, USA). Caspase substrates Ac-DEVD-AFC, Ac-LETD-AFC and Ac-LEHD-AFC were from Alexis Biochemicals (Enzo Life Sciences, Farmingdale, NY, USA). DNAse I was from Ambion Life Technologies, Inc. (Foster City, California). siRNA for Grx1 and Trx1, and DharmaFECT 1 were from GE Healthcare Dharmacon, Inc. (Wauwatosa, Wisconsin, USA).
2.2. Cell growth conditions
HepG2 cells were transfected with the pcDNA/4TO (5100 bp; Invitrogen, Molecular Probes, Inc.) expression vector containing NOS-3 cDNA sequence (3462 bp; NCBI, ImaGenes, full length cDNA clone sequence BC063294) under the control of the cytomegalovirus promoter (4TO-NOS). Cell lineages 4TO and 4TO-NOS were selected with zeocin (15 mg/L; Invitrogen) as described by González et al. [37]. Cells were maintained in EMEM Medium (Minimum Essential Medium Eagle), pH 7.4, supplemented with 10% fetal bovine serum, 2.2 g/L HCO3Na, 1 mM sodium pyruvate, 100 U/L penicillin, 100 μg/mL streptomycin, 0.25 μg/mL amphotericin, and the corresponding selective zeocin antibiotic in 5% CO2 atmosphere at 37 °C. The experiments were routinely carried out at 100,000 cells/cm2. Cell extracts were obtained using lysis solution containing 50 mM HEPES (pH 7.5), 2 mM EDTA, 100 mM NaCl, 0.6% Nonidet NP-40, 1 mM phenylmethylsulfonyl fluoride (PMSF), 5 μg/mL aprotinin, 10 μg/mL leupeptin. Afterward, samples were homogenized and centrifuged at 15,000 g for 5 min at 4 °C and the supernatant was stored at −80 °C until use.
2.3. Assay of enzymatic activities
Grx activity was determined spectrophotometrically by measuring the reduction of 0.5 mM 2-hydroxyethyl disulfide (HED) by 0.5 mM GSH in the presence of NADPH and 0.5 units of yeast glutathione reductase, at 25 °C. The disappearance of NADPH was monitored at 340 nm [41]. Trx activity was determined spectrophotometrically by measuring its ability to reduce insulin disulfides in the presence of NADPH and rat thioredoxin reductase [42]. An assay mixture was prepared by mixing 200 μL 1 M HEPES pH 7.6, 40 μL 0.2 M EDTA, 40 μL 40 mM NADPH and 500 μL insulin (10 mg/mL). 40 μL of this mixture was added to test tubes containing 20 μL mammalian TrxR (1 μg) and the sample of protein and the assay volume was completed up to 120 μL with water. The tubes were incubated at 37 °C for 20 min and the reaction was stopped by the addition of 500 μL of a solution containing 0.4 mg/mL DTNB in 6 M guanidine–HCl, 50 mM Tris–HCl, pH 7.5. The absorbance at 412 nm was determined.
2.4. SDS-PAGE and Western blotting
The protein expression of Trx1, TrxR1, TXNIP, Grx1, Bcl-2, STAT3, Akt1, p-Akt, MAPK, p-MAPK, MATII, UROD, ACO, PKM2, TKL, caspase-3, CD95, NOS-3 and TKT were determined by SDS-PAGE coupled to Western blotting analysis. SDS-PAGE was performed with homogeneous 6% (NOS-3) 8% (STAT3, MAPK, ACO1, and TKT), 10% (Trx1, TXNIP, Akt, MATII, UROD, and ACO2), 12% (CD95) and 14% (Trx1, Grx1, Bcl-2, PKM2, and caspase-3) acrylamide gels. After electrophoresis, proteins were transferred to nitrocellulose membrane with a semi-dry electrophoretic transfer system (Bio-Rad). The membranes were incubated overnight at 4 °C with the corresponding primary antibodies against Thr202/Tyr204p-MAPK, 1:2000 dilution; or against ACO1 and UROD, 1:500 dilution; or against Trx1, Grx1, STAT3, MAPK, ACO2, TKT, TXNIP, TrxR1, Akt1, Ser473p-Akt, MATII, Bcl-2, PKM2, caspase-3, CD95 and NOS-3, 1:1000 dilution. Then washed and incubated with the corresponding secondary antibodies conjugated to peroxidase (anti-rabbit, anti-goat or anti-mouse) used at 1:8000 dilution and the chemiluminescent signal was induced by ECL reagent. β-actin detected with the corresponding antibody at 1:5000 dilution, was used as cell protein-loading control.
2.5. Measurement of cell death
Caspase-8, caspase-9 and caspase-3-associated activities were determined using the corresponding peptide-based substrates (100 μM) in the reaction mixture (50 mM HEPES, pH 7.5, 100 mM NaCl, 10% sucrose, 0.1% Chaps, 1 mM EDTA and 5 mM DTT). The substrates used were Ac-DEVD-AFC, Ac-LETD-AFC and Ac-LEHD-AFC for caspase-3, -8, and -9, respectively. The fluorescence due to the reaction product was recorded with a GENIos Microplate Reader (TECAN) set at 400 nm excitation and 505 nm emission [37].
The level of protein caspase-3-active fragment (p17) and cell death receptor, CD95, were also determined by SDS-PAGE coupled to Western blotting analysis as described in the previous section.
Apoptosis in HepG2 cells was also analyzed through DNA fragmentation detected by terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) (Biotool). HepG2 cells 72 h after siRNA interference were fixed in 4% paraformaldehyde solution in PBS (pH 7.4) and later these cells were permeabilized with 0.2% Triton X-100 solution in PBS. Processing the cells was performed according to the recommendations of the manufacturer (Biotool) using DAPI (Molecular Probes) to stain the cell nuclei. Fluorescence provided by the cells was analyzed with fluorescence microscope (Olympus BX43) using standard fluorescein filter set to view the Apo-green fluorescence at 520+20 nm and view blue DAPI at 460 nm.
2.6. Cell viability and total number of cells
Total number of cells and cell viability in a HepG2 cell suspension were determined using the trypan blue dye exclusion method. The cells attached to the plate were washed with PBS and trypsinized for 7 min at 37 °C followed by incubation with complemented culture medium to halt the effect of trypsin. The cell suspension was centrifuged at 238g for 5 min at room temperature and the pellet was resuspended in 0.5 mL complemented culture medium. Cell viability and total number of cells were measured by mixing equal volumes of cell suspension and trypan blue. A viable cell will have a clear cytoplasm whereas a non-viable cell will have a blue cytoplasm. Cell viability was obtained by referring the number of living cells to the total number of cells in the initial cell suspension.
2.7. Cell proliferation
This parameter was analyzed using a cell proliferation colorimetric ELISA (Roche Applied Science) based on the measurement of BrdU incorporation during DNA synthesis in proliferating cells. Cells were cultured (20,000 cells/cm2) in flat-bottomed 96-well multiplates at 37 °C and 5% CO2. 72 h after siRNA Trx1 and Grx1 downregulation HepG2 cells were incubated with 10 μM BrdU labeling solution for 6 h at 37 °C. Later on were followed the steps according to the recommendations of the manufacturer (Roche) and detected the absorbance of the samples at 370 nm (reference wavelength 492 nm).
2.8. Determination of nitrotyrosine
The levels of nitrotyrosine were determined by Western blotting after 12% non-reducing SDS-PAGE as described before. The primary antibody was anti-3-nitrotyrosine (1:1000) from Sigma.
2.9. Silencing of Grx1 and Trx1
Human Grx1 and Trx1 were knocked-down in wild type, 4TO and 4TO-NOS cells using specific siRNA in 6-well plate (20,000 cells/cm2) according to the manufacturer's recommendations (Dharmacon, GE Healthcare Life Sciences). Grx and Trx siRNA (25 nmol) were mixed with the transfection reagent DharmaFECT 1, previously pre-incubated with culture medium (antibiotic/antimycotic and serum free), and incubated for 20 min at room temperature. These interference solutions were added to cells in 2% of culture medium without antibiotic/antimycotic solution and kept for 72 h [43].
2.10. Redox mobility shift assay
Cells were treated with lysis buffer consisting of 50 mM HEPES pH 7.5, 2 mM EDTA, 100 mM NaCl, 1% Nonidet NP-40, 8 M urea, 1 mM PMSF and containing 5 mM N-ethylmaleimide (NEM) to block all free cysteine thiols. The lysates were centrifuged at 15,000 g for 5 min at 4 °C and the supernatant was desalted with Zeba spin desalting columns (Thermo Scientific Pierce) to eliminate the remaining NEM reagent, and then treated with 5 mM Tris(2-carboxyethyl)phosphine (TCEP) at room temperature for 30 min to reduce all cysteine disulfides. Recovered cysteine thiols were reacted at room temperature for 4 h with 6.25 mM Methyl-PEG24-Maleimide (MM(PEG)24, Thermo Scientific Pierce), which adds a 1239.44 Da label to any free thiol present, and desalted again on Zeba spin desalting columns. Labeled proteins in the samples processed this way would show an increase of 1.2 kDa in their apparent Mw per each reversibly oxidized cysteine, compared to the same protein with their cysteines originally in the free thiol form, which had been initially blocked with the non-PEG maleimide reagent present in the lysis buffer. Protein concentration was determined and the samples were processed for SDS-PAGE and Western blotting as described above.
2.11. Statistical analysis
Results are expressed as mean±SEM of three independent experiments. Data were compared using ANOVA with the least significant difference test a post hoc multiple comparison analysis. The threshold for statistically significant differences was set at p≤0.05. The values labeled with “a” were significantly different versus the corresponding internal control in the same cell lineage. The values labeled with “b” were significantly different versus the corresponding control cell lineage.
3. Results
3.1. Characterization of NOS-3 overexpressing HepG2 cells
NOS-3 overexpression was performed as described before and was confirmed by measurement of mRNA and protein levels by Western blotting (supplementary Fig. S1A and B) CD95 protein levels and caspase-3, -8 and -9 activities were also determined and confirmed that their levels increased significantly in NOS-3 overexpressing HepG2 cells (supplementary Fig. S1C–F). The concentrations of NO and ROS were ≈40% higher and the levels of nitrite and nitrate in the culture medium also increased significantly in NOS-3 overexpressing cells compared to the cells carrying the empty plasmid, as described before [37]. These results indicate that NOS3 overexpressing cells are subject to oxidative stress and that the apoptotic pathways are activated.
3.2. Effect of high levels of NO produced by overexpression of NOS-3 on different metabolic pathways
Metabolic impairment is a characteristic feature of tumor cells due to the demand of biomass for cellular division, so we analyzed several checkpoints of biosynthetic, energetic and iron metabolic pathways. ACO1, a marker of cytosolic iron homeostasis and MATII involved in one carbon metabolism were diminished in NOS-3 overexpressing cells (Fig. 1A and B). By contrast, mitochondrial ACO2 from energy metabolism and cytosolic UROD (heme biosynthesis), PKM2 (glycolysis) and TKT (pentose phosphate pathway) increased significantly under NOS-3 overexpression conditions (Fig. 1C–F). These results confirm that NOS3 overexpression induces a marked metabolic rearrangement in HepG2 cells.
Fig. 1.

Effect of high levels of NO on different metabolic pathways. Western blot analysis of the expression of different metabolism proteins and their relative quantitation in the HepG2 wild type cells (WT) and control cells (4TO), as well as in NOS-3 overexpressing cells (4TO-NOS). The levels of two key cytosolic proteins involved in iron metabolism, ACO1 (A), and one carbon metabolism, MATII (B), were significantly lower in NOS-3 overexpressing cells compared to control cells. However, mitochondrial ACO2 from energy metabolism and cytosolic UROD (heme biosynthesis), PKM2 (glycolysis) and TKT (pentose phosphate pathway) were up-regulated in NOS-3 overexpressing cells (C–F). Data are presented as mean±SEM (n=3 independent experiments). The values labeled with “a” were significantly different (p≤0.05) versus the corresponding control cell lineage. The images are representative of three different experiments.
3.3. Effect of high levels of NO produced by overexpression of NOS-3 on different signaling pathways
Growing evidence shows that some transcription factors and proteins involved in signaling pathways could be modulated by both oxidation/reduction and phosphorylation [44]. We choose several well-established representative proteins involved in cell survival pathways to study their response to the oxidative conditions prevailing in NOS-3 overexpressing cells. MAPK, p-MAPK, Bcl-2, STAT3, Akt1 and p-Akt, were all significantly increased in NOS-3 overexpressing cells compared to the control cells (Fig. 2A–F). The ratio of p-MAPK/MAPK increased markedly as a consequence of empty vector introduction but was further increased in NOS3 overexpressing cells (Fig. 2G). However, the ratio p-Akt/Akt1 did not change significantly despite the increase in both forms (Fig. 2H). These results demonstrate that the cell signaling pathways are thoroughly affected by overexpression of NOS3.
Fig. 2.

Effect of NOS-3 overexpression on different signaling pathways. Western blot analysis of the expression of different signaling proteins and their relative quantitation in the HepG2 wild type cells (WT) and control cells (4TO), as well as in NOS-3 overexpressing cells (4TO-NOS). The levels of MAPK (A), p-MAPK (B), Bcl-2 (C), STAT3 (D), Akt1 (E) and p-Akt (F) were all significantly increased in NOS-3 overexpressing cells compared to cells transfected with the empty vector. The proportion of p-MAPK/MAPK ratio increased markedly (G) whereas the p-Akt relative to total Akt1 did not change (H). Data are presented as mean±SEM (n=3 independent experiments). The values labeled with “a” were significantly different versus the corresponding control cell lineage. The images are representative of three different experiments.
3.4. Effect of overexpression of NOS-3 on the redoxin levels
Proteins related to the thioredoxin and glutaredoxin systems are prominent players in thiol redox homeostasis in cells and were studied. Protein levels and activity of Trx1, Grx1 and levels of TrxR1 and TXNIP, increased significantly in NOS-3 overexpressing HepG2 cells (Fig. 3A–F). The simultaneous increase of both Trx1 and its opposing protein TXNIP is a conflicting situation likely a reflection of the cellular response to redox changes under the prevailing nitrosative conditions where Trx1 and TXNIP may not be interacting (see below).
Fig. 3.

High levels of nitric oxide induced activity and expression of redox proteins. Trx activity (A) and Trx1 protein expression (B), Grx activity (C) and Grx1 protein expression (D), TrxR (E) and TXNIP (F) protein expression increased in NOS-3 overexpressing cells compared to cells transfected with the empty vector. Data are presented as mean±SEM (n=3 independent experiments) and the groups labeled with “a” were significantly statistical (p≤0.05) versus the corresponding control group. Representative Western blotting images are shown.
3.5. Effect of overexpression of NOS-3 on thiolic redox state of several proteins
Thiol redox changes may be part of regulatory mechanisms of proteins, so we analyzed the thiolic redox state of several proteins to check whether this type of mechanism could be potentially operative in NOS3 overexpressing cells. We have detected a redox shift of ACO1 and UROD towards more oxidized states (Fig. 4A and B), which could result in iron metabolism perturbation. Signaling pathways could also be affected by redox changes since Akt1 shifted to more oxidized state under nitrosative pressure (Fig. 4C). Changes in the redox state of MAPK, STAT3 and Bcl-2 could not be detected under our experimental conditions and with our redox electrophoretic mobility shift assay. However, the thiolic redox state of Trx1 and Grx1 shifted to more oxidized in the NOS-3 overexpressing cells (Fig. 4D and E) a likely consequence of the nitrosative and oxidative stress [37].
Fig. 4.

Effect of overexpression of NOS-3 on thiolic redox state of different proteins. Electrophoretic redox mobility shift to detect the oxidized and reduced forms (arrows) are showed. The high levels of NO known to prevail in these cells caused a redox shift to more oxidized in ACO1 (A), UROD (B), Akt1 (C) and in both studied redoxins, Trx (D) and Grx (E).
3.6. Specific silencing of Trx1 and Grx1
As shown above, Trx1 and Grx1 levels increased upon NOS3 overexpression in HepG2 cells. To go deep into the role of these proteins in the adaptation to high NO levels, we studied the effect of down-regulation of both proteins. Treatment of WT, 4TO and NOS-3 overexpressing HepG2 cells with vectors harboring siRNA specifically directed to Trx1 and Grx1 mRNA produced a ≈75% decrease in Trx1 and Grx1 protein levels in the three cell lineages (Fig. 5A and C). These changes in protein levels were accompanied by equivalent functional fall off, as indicated by their canonical enzymatic activities (Fig. 5B and D).
Fig. 5.

Down regulation of Trx1 and Grx1 in HepG2 cells by treatment with specific siRNAs. Protein levels and activity of Trx1 (A, B) and Grx1 (C, D) were determined in HepG2 cells with and without NOS-3 overexpression and treated or not with siRNATrx and siRNAGrx. Representative images of Western blot membranes used to quantify protein levels are shown in A and C. The protein levels and activities of both redoxins were significantly higher in NOS-3 overexpressing cells (4TO-NOS, black bars). Treatment with the specific siRNA produced a ≈75% decrease in protein levels and activities of both redoxins in control and NOS-3 overexpressing cells, confirming the efficacy of the treatments. Data are presented as mean±SEM (n=3 independent experiments). The values labeled with “a” were significantly different versus the corresponding internal control in the same cell lineage. The values labeled with “b” were significantly different versus the corresponding control cell lineage. The images are representative of three different experiments.
3.7. Effect of Trx1 and Grx1 silencing on the levels of tyrosine nitration in normal and NOS-3 overexpressing HepG2 cells
Tyrosine nitration is mediated by reactive nitrogen species such as peroxynitrite (ONOO−) anion and nitrogen dioxide, formed as secondary products of NO metabolism in the presence of oxidants including superoxide radicals (O2·−) and hydrogen peroxide (H2O2) [45]. For this reason, the level of Tyr nitration is indicative of the degree of nitrosative and oxidative stress and we measured it to check the effect of Trx1 and Grx1. Overexpression of NOS-3 in HepG2 cells induced protein Tyr nitration (Fig. 6, black bars). Silencing of Trx1 and Grx1 also increased protein tyrosine nitration in wild type and 4TO cells, but, interestingly, had the opposite effect on NOS-3 overexpressing cells (Fig. 6). These results uncover an apparent conflict, e.g., a decrease in antioxidant defenses, Trx1 and Grx1, results in alleviation of oxidative stress.
Fig. 6.

Detection of nitrotyrosine levels in NOS-3 overexpressing HepG2 cells with Grx and Trx down regulation. The detection of this postranslational modification was performed by Western blot with the analysis focused on a representative 55 kDa band (arrow). The image is representative of three different experiments and densitometric data of n=3 independent experiments are presented in the histogram below as mean±SEM. The numbers 1, 2 and 3 indicate the cell treatments control, siRNAGrx and siRNATrx respectively. NOS-3 overexpression induced Tyr nitration of proteins in HepG2 cells (black bars). This effect was offset by treatment with siRNAGrx and siRNATrx as shown in the 4TO-NOS group. However, in WT and 4TO HepG2 cells siRNAGrx and siRNATrx treatment had the opposite effect, inducing Tyr nitration of proteins. Statistical significance was assessed by (p≤0.05). The values labeled with “a” were significantly different versus the corresponding internal control in the same cell lineage. The values labeled with “b” were significantly different versus the corresponding control cell lineage.
3.8. Cellular proliferation and apoptosis in NOS-3 overexpressing cells: effect of Trx1 and Grx1
Nitrosative stress in HepG2 cells as a consequence of NOS3 overexpression results in reduction of cell number and proliferation (Fig. 7A and C) in agreement with a previous report [37]. In the current study we show that silencing Trx1 or Grx1 further decreased the number of cells and proliferation in all cell lineages and siRNATrx further reduced cell viability (Fig. 7A–C). siRNAGrx or siRNATrx treatments also provoked a striking increase in caspase-3 and caspase-8 activities in all cells (Fig. 7D and E), but lowered the levels of CD95 in NOS-3 overexpressing cells (Fig. 7F). Increase in caspases activities was actually accompanied by signs of increased apoptosis like DNA fragmentation as determined by TUNEL assay (Fig. 8). These results are worthy of consideration as they point to a moonlighting role of the redoxins depending on the prevailing cellular redox conditions as will be discussed in the next section (Fig. 9).
Fig. 7.

Effect of down-regulation Grx and Trx on cell proliferation and cell death induced by NOS-3. Number of total cells (A), cell viability (B) and incorporation of BrdU (C) in WT, 4TO and 4TO-NOS cells. Treatment with siRNAGrx and siRNATrx significantly diminished the number of cells (gray and white bars) and siRNATrx further reduced cell viability in all cell lineages (white bars). Caspase-3 (D) and caspase-8 (E) activities, known markers of apoptosis, were also markedly increased in all cell types by treatment with either siRNAGrx or siRNATrx, whereas the same treatments offset the increase in CD95 receptor levels induced by NOS-3 overexpression (F). The numbers 1, 2 and 3 refer to control, siRNAGrx and siRNATrx treatments, respectively. Data are presented as mean±SEM (n=3 independent experiments). The values labeled with “a” were significantly different versus the corresponding internal control in the same cell lineage. The values labeled with “b” were significantly different versus the corresponding control cell lineage. The images are representative of three different experiments.
Fig. 8.

Effect of down-regulation of Grx and Trx on apoptosis. DNA fragmentation in cells detected by terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL). We have selected representing merged images of FITC and DAPI fluorescence of these treatments in the cell lineages studied (A). Apoptotic nuclei vs. total nuclei in percentage detected in different fluorescence images of control, siRNAGrx and siRNATrx treated cells in 3 independent experiments (B). The values labeled with “a” were significantly different (p≤0.05) versus the corresponding internal control in the same cell lineage. The values labeled with “b” were significantly different versus the corresponding control cell lineage.
Fig. 9.

Effect of down-regulation of Grx and Trx on Akt phosphorylation and redox state. Western blot analysis of the expression of Akt1 (A) and p-Akt (B) proteins and their relative quantitation in the HepG2 wild type cells (WT) and control cells (4TO), as well as in NOS-3 overexpressing cells (4TO-NOS). The numbers 1, 2 and 3 refer to control, siRNAGrx and siRNATrx treatments, respectively. The enhancing effect of NOS-3 overexpression on Akt1 (A) and p-Akt (B) protein levels was offset by siRNA Grx (gray bars, 2) and even more by siRNA Trx (white bars, 3). None of these siRNA treatments had effect on the p-Akt/Akt1 ratio (C). However, silencing Grx1 and more prominently Trx1, shifted the redox state of Akt1 to the reduced form (arrows) in NOS-3 overexpressing cells (D). Data are presented as mean±SEM (n=3 independent experiments). The values labeled with “a” were significantly different (p≤0.05) versus the corresponding internal control in the same cell lineage. The values labeled with “b” were significantly different versus the corresponding control cell lineage. The images are representative of three different experiments.
3.9. Effect of Trx1 and Grx1 silencing on the levels and redox state of Akt1 in normal and NOS-3 overexpressing HepG2 cells
Akt1 is known to be affected by thiol redox changes and is a major agent in the control of apoptosis, so it was pertinent to study its response to redoxins dow-regualtion. Trx1 or Grx1 specific siRNA counteracted the effect of the NOS-3 overexpression, regarding both, total levels (Fig. 8A–C) and redox state (Fig. 8D) of Akt. Grx1 and Trx1 down-regulation shifted Akt towards a more reduced state but at the same time, there was a parallel marked decrease in total Akt protein.
4. Discussion
NOS3 overexpression in HepG2 cells induces oxidative stress and slight but significant activation of apoptosis. In these conditions the cells undergo a pronounced metabolic remodeling as deduced from the changes observed in the levels of checkpoint enzymes from key metabolic pathways.
In mammals, there are two genes encoding pyruvate kinase that produce two isoforms. Whereas PKM1 is a constitutively active enzyme, PKM2 is expressed in all cancers and cancer cell lines studied to date. PKM2 is highly regulated at both the activity and expression levels allowing this isoform to act as floodgate keeper to change the metabolic flux from lactate production to NADPH and anabolic pathways. This metabolic change is particularly relevant under conditions of oxidative stress [46], thus explaining the induction of PKM2 in NOS3 overexpressing cells and in coherent with the observed concomitant induction of TKT.
UROD has been considered a target for head and neck cancer treatment [47]. It plays a key role in the biosynthesis of heme group and its down-regulation has been shown to increase oxidative stress and affect the cellular iron homeostasis. It's up-regulation in NOS3 overexpressing cells (Fig. 1D) could be counterbalanced by inactivation through thiol oxidation [48] as shown in Fig. 4B. These results, together with the observed down-regulation of ACO1, indicate that cytosolic iron metabolism is perturbed.
MATs (methionine adenosyltransferases) are regulated by oxidative stress and are associated to different pathologies such as hepatocellular carcinoma and diabetes [49]. The decrease in MATII levels we have observed in NOS-3 overexpressing cells mean that AdoMet levels were reduced in those cells, with negative consequences for nucleotide biosynthesis and hence for proliferation, which would agree with the results on cell proliferation shown in Fig.7. This situation would make sense under the oxidative conditions prevailing in those cells, since the metabolic flux would be compensated by diversion towards pentose phosphate pathway for NADPH production and antioxidant defense.
The serine/threonine kinase Akt is a critical component of an intracellular signaling pathway that exerts effects on cell survival and apoptosis. Akt is activated by insulin and various growth and survival factors. The unphosphorylated form of Akt is inactive and it is activated by phosphorylation at Thr308 by PDK1 and at Ser473 by mTOR2. It can also be inactivated by protein phosphatase 2A (PP2A) dephosphorylation when Akt is in its oxidized form [50]. It has been shown that Grx prevented Akt from forming a specific disulfide bond and suppressed its association with PP2A under oxidative stress, resulting in phosphorylation of Akt and inhibition of apoptosis. In Fig. 2E–H, we show that nitrosative stress caused the increase of both Akt forms (Akt1 and p-Akt), accompanied by a shift towards a more oxidized state (Fig. 4C). This would mean that Akt is prone to inactivation by dephosphotylation under NOS-3 overexpression conditions, which indicates that apoptosis is being favored.
The redox state and total protein levels of Akt are both under the influence of Trx1 and Grx1 as will be detailed in another section below.
The mitogen-activated protein kinase (MAPK) cascades are multifunctional signaling pathways involved in cell growth, differentiation, apoptosis and cellular response to stress. Two different MAP kinase cascades that converge to c-Jun N-terminal kinases (JNK) and p38 MAP Kinases are mainly activated by citotoxic stresses, oxidative/nitrosative stress and proinflammatory cytokines such as tumor necrosis factor (TNF)-α [32].
Our results show that MAPK is more phosphorylated in NOS3 overexpressing cells and hence less active. At first sight, this result would conflict with the fact that Trx1 and Grx1 had shifted to more oxidized state in these cells (Fig. 4D and E) because oxidation of both redoxins would suppress their inhibiting interaction with apoptosis signal-regulating kinase 1 (ASK1) [50]. ASK1 would then be free to initiate the MAPK cascade leading to activation of JNK or p38 [51]. The explanation of this apparently contradictory result could be that downstream p38MAPK, which is prone to tyrosine nitration as opposed to phosphorylation, could not be activated under the nitrosative pressure with elevated levels of nitrotyrosine (see Fig. 6).
Human Bcl-2 is an anti-apoptotic, membrane-associated oncoprotein that can be directly affected by oxidative stress induced by NO [44] and could play an important role in NO-induced apoptosis. In Fig. 2C, we show a significant increase in Bcl-2 in HepG2 cells under nitrosative stress. STAT3, the key cell growth regulator signal transducer and activator of transcription, is subject to redox regulation by glutathionylation and ROS trigger tyrosine phosphorylation and nuclear translocation of STAT3. Glutathionylated STAT3 was a poor Janus protein tyrosine kinase 2 substrate, and it exhibited low DNA-binding activity [52]. Furthermore, the effect of inactivation of STAT3 has resulted in inhibition of growth and metastasis of human hepatocellular carcinoma cells, and increase in their chemo-sensitivity.
The observed increases in Bcl-2 and STAT3 in NOS3 overexpressing cells may be part of a counteracting cellular response to induction of cell death, although the activation state of Bcl-2 and the glutathionylation state of STAT3 should have to be determined to support this reasoning. So far, we could not detect redox changes in both proteins by the electrophoretic redox mobility shift assay, but ongoing redox proteomic studies could throw light to this question.
Trx1 was detected in a more oxidized state in NOS3 overexpressing cells than in normal HepG2 cells (Fig. 4D). Trx1 is mostly found in the cytoplasm; however, it moves to the nucleus as a response to nitrosative/oxidative stress and this Trx1 nuclear migration has been described as part of a survival signaling pathway, associated with intracellular compartmentalization and activation of ERK1/2 MAP Kinases. TXNIP prevents Trx1 nuclear migration and its expression was down-regulated in different human carcinomas [53], however, it binds to reduced Trx1 but not to oxidized Trx1. Hence, in the oxidative environment of NOS-3 overexpressing cells, inhibition of Trx1 by TXNIP might not be fully operative despite the increase in TXNIP levels.
The increase in protein Tyr nitration occurring in WT cells upon Trx1 and Grx1 down-regulation was an indication of oxidative stress [54], the expected consequence of a drop in thiol antioxidant defenses. However, in NOS-3 overexpressing cells, whose redoxins levels are markedly higher (Fig. 3), silencing of Trx1 and Grx1 provokes a decrease in nitro-Tyr levels (Fig. 6). It has been noted that nitration processes are promoted over other oxidative modifications in specific protein targets [55] and it has also been suggested that nitration is a reversible process [56]. Whether the observed parallel drop in protein nitration and Trx1 and Grx1 levels in NOS-3 overexpressing cells is an indication of specificity of the nitration process, its reversibility or the degradation of nitrated proteins, is an interesting matter for further research and adds up to the debate [57].
The observed neutralization of CD95 induction in NOS-3 overexpressing cells by Trx1 or Grx1 down regulation (Fig. 7F) suggest that the high levels of both thiol-disulfide oxidoreductases prevailing in these cells (Fig. 3) are required to induce the expression of CD95. Activation of the extrinsic apoptotic pathway, indicated by the increase in caspase-8 activity (Fig. 7E), despite the drop in dead receptor CD95 levels, points to the operation of other pro-apoptotic mechanism involving thiol redox changes.
Many reports have documented the influence of redox changes on the apoptotic signaling pathways in a variety of cell types through glutathionylation and S-nitrosylation of cysteines in various proteins along the signaling cascade, from receptors to caspases, with the involvement of Grx and Trx in most cases [58]. Trx1 acting as denitrosylating enzyme counteracted inactivation of caspase-3 and -8 by S-nitrosylation in HepG2 cells [28]. But on the other hand, Trx1 can be S-nitrosylated itself [59] and this oxidized Trx1 can impede apoptosis by S-transnitrosylation of caspase-3 [29,60]. Thus, depending on the redox state of the cell, Trx1 can catalyze either trans-S-nitrosylation or S-denitrosylation reactions [29,61]. Total levels of Trx1 increase in NOS-3 overexpressing cells but a measurable proportion of the protein is in the oxidized state (Fig. 4D) and could be acting on caspase-3 to inhibit it. This inhibitory effect would be alleviated in siRNATrx treated cells in which the total amount of Trx1 drops down to ≈25%.
Most of the effects of siRNATrx treatment were also attained by treatment with siRNAGrx, reflecting a degree of similarity of both redoxins in the control of redox homeostasis, although differences also exist. Both proteins have been related to apoptosis regulation through redox modulation of stress activated and ASK1 [32] although the role of Grx1 has been documented to more extent [50].
This increase of thiolic form of Akt when the levels of thiol antioxidant redoxins decrease would favor activation by phosphorylation and apoptosis inhibition, whereas the parallel decrease in total Akt protein would attenuate the antiapoptotic potential of Akt. This increase of thiolic form of Akt when the levels of thiol antioxidant redoxins decrease may sound contradictory, but could be caused by the proteasome differentially degrading the oxidized form, thus unbalancing the redox equilibrium towards the reduced form. Inactivation and proteasomal degradation of Akt upon Grx1 down-regulation had been observed before [35]. The effect of down-regulation of Grx1 and Trx1 on Akt redox state followed the same trend but the changes were more pronounced when Trx1 was silenced, which could be correlated with fading of its denitrosylase activity.
5. Conclusions
Overexpression of NOS-3 has antiproliferative effect on HepG2 cells. Activation of apoptosis, as demonstrated by increases in caspases activity, is the final outcome resulting from the balance of multilevel and conflicting cellular responses to the oxidative stress generated by the elevated level of NO. Among these, metabolic remodeling potentiates the pentose phosphate pathway to increase the metabolic flux towards NADPH production for antioxidant defense at the expenses of lowering nucleotide biosynthesis and therefore proliferation. Proteins of the antioxidant redoxin systems and proteins involved in cell survival pathways and phosphorylation of MAPK increase significantly. At the same time, the thiolic redox state of Akt1, Trx1 and Grx1 shifts to more oxidized.
The important finding of our study is that Trx1 and Grx1, principal actors of the antioxidant response induced by increased NO levels, are actually a hindrance to the antiproliferative action of NO. Down regulation of either redoxin by specific siRNAs activates caspases dramatically and reduces cell proliferation. These results support the contention that weakening the thiolic antioxidant defenses enhances the antiproliferative effects on tumoral cells.
Acknowledgments
This research was supported by grants from the Spanish Ministry of Economy and Competitiveness (BFU2012-32056) and from the Andalusian Government (BIO-0216 and CTS-6264). We thank Biomedical Research Network Center for Liver and Digestive Diseases (CIBERehd) founded by “Instituto de Salud Carlos III”.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2015.07.007.
Supplementary material
Supplementary material NOS-3 overexpression and induction of cell death in 4TO-NOS cells. (A) and (B) NOS-3 mRNA and protein expression in 4TO-NOS cells confirm the phenotype of transformed cells. (C– F) CD95 protein expression and caspase-3-, caspase-8- and caspase-9-associated activities were increased in NOS-3 overexpressing cells. Cells were collected 48 h after stabilization. β-actin was used as internal protein loading control. Data are expressed as mean±SEM (p≤0.05) of three different experiments. The groups with “a” were significantly different versus the corresponding control group. Representative Western blotting for NOS-3 and CD95 protein level analysis are shown.
References
- 1.Muntané J., la Mata M. Nitric oxide and cancer. World J. Hepatol. 2010;2:337–344. doi: 10.4254/wjh.v2.i9.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alderton W., Cooper C., Knowles R. Nitric oxide synthases: structure, function and inhibition. Biochem. J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geller D.A., Billiar T.R. Molecular biology of nitric oxide synthases. Cancer Metastasis Rev. 1998;17:7–23. doi: 10.1023/a:1005940202801. [DOI] [PubMed] [Google Scholar]
- 4.Forstermann U., Boissel J.P., Kleinert H. Expressional control of the ‘constitutive’ isoforms of nitric oxide synthase (NOS I and NOS III) FASEB J. 1998;12:773–790. [PubMed] [Google Scholar]
- 5.Bredt D., Hwang P., CE. G., Lowenstein C., Reed R., Snyder S. Cloned and expressed nitric oxide synthase structurally resembles cytochrome P-450 reductase. Nature. 1991;351:714–718. doi: 10.1038/351714a0. [DOI] [PubMed] [Google Scholar]
- 6.Lamas S., Marsden P., Li G., Tempst P., Michel T. Endothelial nitric oxide synthase: molecular cloning and characterization of a distinct constitutive enzyme isoform. Proc. Natl. Acad. Sci. USA. 1992;89:6348–6352. doi: 10.1073/pnas.89.14.6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lyons C., Orloff G., Cunningham J. Molecular cloning and functional expression of an inducible nitric oxide synthase from a murine macrophage cell line. J. Biol. Chem. 1992;267:6370–6374. [PubMed] [Google Scholar]
- 8.Xie Q., Cho H., Calaycay J., Mumford R., Swiderek K., Lee T., Ding A., Troso T., Nathan C. Cloning and characterization of inducible nitric oxide synthase from mouse macrophages. Science. 1992;256:225–228. doi: 10.1126/science.1373522. [DOI] [PubMed] [Google Scholar]
- 9.Xu W., Liu L., Loizidou M., Ahmed M., Charles I. The role of nitric oxide in cancer. Cell Res. 2002;12:311–320. doi: 10.1038/sj.cr.7290133. [DOI] [PubMed] [Google Scholar]
- 10.Boo Y., Kim H., Song H., Fulton D., Sessa W., Jo H. Coordinated regulation of endothelial nitric oxide synthase activity by phosphorylation and subcellular localization. Free Radic. Biol. Med. 2006;41:144–153. doi: 10.1016/j.freeradbiomed.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 11.Oess S., Icking A., Fulton D., Govers R., Müller-Esterl W. Subcellular targeting and trafficking of nitric oxide synthases. Biochem. J. 2006;396:401–409. doi: 10.1042/BJ20060321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iwakiri Y., Satoh A., Chatterjee S., Toomre D., Chalouni C., Fulton D., Groszmann R., Shah V., Sessa W. Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc. Natl. Acad. Sci. USA. 2006;103:19777–19782. doi: 10.1073/pnas.0605907103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li C., Woga G. Nitric oxide as a modulator of apoptosis. Cancer Lett. 2005;226:1–15. doi: 10.1016/j.canlet.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 14.Li H., Wan A. Apoptosis of rheumatoid arthritis fibroblast-like synoviocytes: possible roles of nitric oxide and the thioredoxin 1. Mediat. Inflamm. 2013;2013:1–8. doi: 10.1155/2013/953462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buchanan B., Balmer Y. Redox regulation: a broadening horizon. Annu. Rev. Plant Biol. 2005;56:187–220. doi: 10.1146/annurev.arplant.56.032604.144246. [DOI] [PubMed] [Google Scholar]
- 16.Burgoyne J., Madhani M., Cuello F., Charles R., Brennan J., Schröder E., Browning D., Eaton P. Cysteine redox sensor in PKGI(alfa) enables oxidant-induced activation. Science. 2007;317:1393–1397. doi: 10.1126/science.1144318. [DOI] [PubMed] [Google Scholar]
- 17.Ghezzi P., Bonetto V., Fratelli M. Thiol-disulfide balance: from the concept of oxidative stress to that of redox regulation. Antioxid. Redox Signal. 2005;7:964–972. doi: 10.1089/ars.2005.7.964. [DOI] [PubMed] [Google Scholar]
- 18.Martinez-Ruiz A., Cadenas S., Lamas S. Nitric oxide signaling: classical, less classical, and nonclassical mechanisms. Free Radic. Biol. Med. 2011;51:17–29. doi: 10.1016/j.freeradbiomed.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 19.Hess D., Matsumoto A., Kim S., Marshall H., Stamler J. Protein S-nitrosylation: purview and parameter. Nat. Rev. Mol. Cell. Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 20.Derakhshan B., Hao G., Gross S. Balancing reactivity against selectivity: the evolution of protein S-nitrosylation as an effector of cell signaling by nitric oxide. Cardiovasc. Res. 2007;75:210–219. doi: 10.1016/j.cardiores.2007.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Foster M., McMahon T., Stamler J. S-nitrosylation in health and disease. Trends Mol. Med. 2003;9:160–168. doi: 10.1016/s1471-4914(03)00028-5. [DOI] [PubMed] [Google Scholar]
- 22.Nakamura H., Nakamura K., Yodoi J. Redox regulation of cellular activation. Annu. Rev. Immunol. 1997;15:351–369. doi: 10.1146/annurev.immunol.15.1.351. [DOI] [PubMed] [Google Scholar]
- 23.Holmgren A. Thioredoxin and glutaredoxin systems. J. Biol. Chem. 1989;264:13963–13966. [PubMed] [Google Scholar]
- 24.Holmgren A., Johansson C., Berndt C., Lönn M., Lillig C. Thiol redox control via thioredoxin and glutaredoxin systems. Biochem. Soc. Trans. 2005;33:1375–1377. doi: 10.1042/BST0331375. [DOI] [PubMed] [Google Scholar]
- 25.Padilla C., Martínez-Galisteo E., Bárcena J., Spyrou G., Holmgren A. Purification from placenta, amino acid sequence, structure comparisons and cDNA cloning of human glutaredoxin. Eur. J. Biochem. 1995;227:27–34. doi: 10.1111/j.1432-1033.1995.tb20356.x. [DOI] [PubMed] [Google Scholar]
- 26.Gallogly M., Mieyal J. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 2007;7:381–391. doi: 10.1016/j.coph.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 27.Benhar M., Forrester M., Stamler J. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science. 2008;320:1050–1054. doi: 10.1126/science.1158265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sengupta R., Billiar T., Kagan V., Stoyanovsky D. Nitric oxide and thioredoxin type 1 modulate the activity of caspase 8 in HepG2 cells. Biochem. Biophys. Res. Commun. 2010;391:1127–1130. doi: 10.1016/j.bbrc.2009.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitchell D., Marletta M. Thioredoxin catalyzes the S-nitrosation of the caspase-3 active site cysteine. Nat. Chem. Biol. 2005;1:154–158. doi: 10.1038/nchembio720. [DOI] [PubMed] [Google Scholar]
- 30.Du Y., Zhang H., Lu J., Holmgren A. Glutathione and glutaredoxin act as a backup of human thioredoxin reductase 1 to reduce thioredoxin 1 preventing cell death by aurothioglucose. J. Biol. Chem. 2012;287:38210–38219. doi: 10.1074/jbc.M112.392225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arner E., Holmgren A. The thioredoxin system in cancer. Semin. Cancer Biol. 2006;16:420–426. doi: 10.1016/j.semcancer.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 32.Saitoh M., Nishitoh H., Fujii M., Takeda K., Tobiume K., Sawada Y., Kawabata M., Miyazono K., Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H., Wan A., Xu G., Ye D. Small changes huge impact: the role of thioredoxin 1 in the regulation of apoptosis by S-nitrosylation. Acta Biochim. Biophys. Sin. 2013;45:153–161. doi: 10.1093/abbs/gms103. [DOI] [PubMed] [Google Scholar]
- 34.Greetham D., Kritsiligkou P., Watkins R., Carter Z., Parkin J., Grant C. Oxidation of the yeast mitochondrial thioredoxin promotes cell death. Antioxid. Redox Signal. 2013;18:376–385. doi: 10.1089/ars.2012.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ahmad F., Nidadavolu P., Durgadoss L., Ravindranath V. Critical cysteines in Akt1 regulate its activity and proteasomal degradation: implications for neurodegenerative diseases. Free Radic. Biol. Med. 2014;74:118–128. doi: 10.1016/j.freeradbiomed.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 36.Enoksson M., Fernandes A., Prast S., Lillig C., Holmgren A., Orrenius S. Overexpression of glutaredoxin 2 attenuates apoptosis by preventing cytochrome c release. Biochem. Biophys. Res. Commun. 2005;327:774–779. doi: 10.1016/j.bbrc.2004.12.067. [DOI] [PubMed] [Google Scholar]
- 37.González R., Ferrín G., Aguilar-Melero P., Ranchal I., Linares C., Bello R., De la Mata M., Gogvadze V., Bárcena J., Álamo J., Orrenius S., Padillo F., Zhivotovsky B., Muntané J. Targeting hepatoma using nitric oxide donor strategies. Antioxid. Redox Signal. 2013;18:491–506. doi: 10.1089/ars.2011.4476. [DOI] [PubMed] [Google Scholar]
- 38.López-Sánchez L., Corrales F., De la Mata M., Muntané J., Rodríguez-Ariza A. Detection and proteomic identification of S-nitrosated proteins in human hepatocytes. Methods Enzymol. 2008;440:273–281. doi: 10.1016/S0076-6879(07)00817-8. [DOI] [PubMed] [Google Scholar]
- 39.Harris I., Treloar A., Inoue S., Sasaki M., Gorrini C., Lee K., Yung K., Brenner D., Knobbe-Thomsen C., Cox M., Elia A., Berger T., Cescon D., Adeoye A., Brüstle A., Molyneux S., Mason J., Li W., Yamamoto K., Wakeham A., Berman H., Khokha R., Done S., Kavanagh T., Lam C., Mak T. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell. 2015;27:211–222. doi: 10.1016/j.ccell.2014.11.019. [DOI] [PubMed] [Google Scholar]
- 40.Watson J. Oxidants, antioxidants and the current incurability of metastatic cancers. Open Biol. 2013;3:1–9. doi: 10.1098/rsob.120144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luthman M., Holmgren A. Glutaredoxin from calf thymus. Purification to homogeneity. J. Biol. Chem. 1982;257:6686–6690. [PubMed] [Google Scholar]
- 42.Holmgren A. Thioredoxin catalyzes the reduction of insulin disulfides by dithiothreitol and dihydrolipoamide. J. Biol. Chem. 1979;254:9627–9632. [PubMed] [Google Scholar]
- 43.Yang L., Wu D., Wang X., Cederbaum A. Depletion of cytosolic or mitochondrial thioredoxin increases CYP2E1-induced oxidative stress via an ASK-1–JNK1 pathway in HepG2 cells. Free Radic. Biol. Med. 2011;51:185–196. doi: 10.1016/j.freeradbiomed.2011.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trachootham D., Lu W., Ogasawara M., Rivera-Del Valle N., Huang P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008;10:1343–1374. doi: 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Radi R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. USA. 2004;101:4003–4008. doi: 10.1073/pnas.0307446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anastasiou D., Poulogiannis G., Asara J., Boxer M., Jiang J., Shen M., Bellinger G., Sasaki A., Locasale J., Auld D., Thomas C., Vander Heiden M., Cantley L. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334:1278–1283. doi: 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ito E., Yue S., Moriyama E., Hui A., Kim I., Shi W., Alajez N., Bhogal N., Li G., Datti A., Schimmer A., Wilson B., Liu P., Durocher D., Neel B., O’Sullivan B., Cummings B., Bristow R., Wrana J., Liu F.-F. Uroporphyrinogen decarboxylase is a radiosensitizing target for head and neck cancer. Sci. Transl. Med. 2011;3:1–7. doi: 10.1126/scitranslmed.3001922. [DOI] [PubMed] [Google Scholar]
- 48.McDonagh B., Pedrajas J., Padilla C., Barcena J. Thiol redox sensitivity of two key enzymes of heme biosynthesis and pentose phosphate pathways: uroporphyrinogen decarboxylase and transketolase. Oxid. Med. Cell. Longev. 2013:1–13. doi: 10.1155/2013/932472. (ID 932472) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pajares M., Markham G. Methionine adenosyltransferase (s-adenosylmethionine synthetase) Adv. Enzymol. Relat. Areas Mol. Biol. 2011;78:449–521. doi: 10.1002/9781118105771.ch11. [DOI] [PubMed] [Google Scholar]
- 50.Murata H., Ihara Y., Nakamura H., Yodoi J., Sumikawa K., Kondo T. Glutaredoxin exerts an antiapoptotic effect by regulating the redox state of Akt. J. Biol. Chem. 2003;278:50226–50233. doi: 10.1074/jbc.M310171200. [DOI] [PubMed] [Google Scholar]
- 51.Song J., Lee Y. Differential role of glutaredoxin and thioredoxin in metabolic oxidative stress-induced activation of apoptosis signal-regulating kinase 1. Biochem. J. 2003;373:845–853. doi: 10.1042/BJ20030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xie Y., Kole s, Precht P., Pazin M., Bernier M. S-glutathionylation impairs signal transducer and activator of transcription 3 activation and signaling. Endocrinology. 2009;150:1122–1131. doi: 10.1210/en.2008-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ogata F., Batista W., Sartori A., Gesteira T., Masutani H., Arai R., Yodoi J., Stern A., Monteiro H. Nitrosative/oxidative stress conditions regulate thioredoxin-interacting protein (TXNIP) expression and thioredoxin-1 (TRX-1) nuclear localization. PLoS One. 2013;8:e84588. doi: 10.1371/journal.pone.0084588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ischiropoulos H. Biological selectivity and functional aspects of protein tyrosine nitration. Biochem. Biophys. Res. Commun. 2003;305:776–783. doi: 10.1016/s0006-291x(03)00814-3. [DOI] [PubMed] [Google Scholar]
- 55.Haqqani A., Kelly J., Birnboim H. Selective nitration of histone tyrosine residues in vivo in mutatect tumors. J. Biol. Chem. 2002;277:3614–3621. doi: 10.1074/jbc.M105730200. [DOI] [PubMed] [Google Scholar]
- 56.Kamisaki Y., Wada K., Bian K., Balabanli B., Davis K., Martin E., Behbod F., Lee Y.-C., Murad F. An activity in rat tissues that modifies nitrotyrosine-containing proteins. Proc. Natl. Acad. Sci. USA. 1998;95:11584–11589. doi: 10.1073/pnas.95.20.11584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Radi R. Protein tyrosine nitration: biochemical mechanisms and structural basis of its functional effects. Acc. Chem. Res. 2013;46:550–559. doi: 10.1021/ar300234c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sengupta R., Holmgren A. The role of thioredoxin in the regulation of cellular processes by S-nitrosylation. Biochim. Biophys. Acta. 2012;1820:689–700. doi: 10.1016/j.bbagen.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 59.Hashemy S., Holmgren A. Regulation of the catalytic activity and structure of human thioredoxin 1 via oxidation and S-nitrosylation of cysteine residues. J. Biol. Chem. 2008;283:21890–21898. doi: 10.1074/jbc.M801047200. [DOI] [PubMed] [Google Scholar]
- 60.Haendeler J., Hoffmann J., Tischler V., Berk B., Zeiher A., Dimmeler S. Redox regulatory and anti-apoptotic functions of thioredoxin depend on S-nitrosylation at cysteine 69. Nat. Cell Biol. 2002;4:743–749. doi: 10.1038/ncb851. [DOI] [PubMed] [Google Scholar]
- 61.Barglowa K., Knutsonb C., Wishnokb J., Tannenbaumb S., Marletta M. Site-specific and redox-controlled S-nitrosation of thioredoxin. Proc. Natl. Acad. Sci. USA. 2011;108:600–606. doi: 10.1073/pnas.1110736108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material NOS-3 overexpression and induction of cell death in 4TO-NOS cells. (A) and (B) NOS-3 mRNA and protein expression in 4TO-NOS cells confirm the phenotype of transformed cells. (C– F) CD95 protein expression and caspase-3-, caspase-8- and caspase-9-associated activities were increased in NOS-3 overexpressing cells. Cells were collected 48 h after stabilization. β-actin was used as internal protein loading control. Data are expressed as mean±SEM (p≤0.05) of three different experiments. The groups with “a” were significantly different versus the corresponding control group. Representative Western blotting for NOS-3 and CD95 protein level analysis are shown.