

Background: Adipogenesis can be modulated by various signaling pathways and transcription factors.
Results: AICAR-activated AMPK inactivates GSK3β, which stimulates Wnt/β-catenin signaling and relieves CtBP repression, thus increasing GATA3 and inhibiting adipogenesis.
Conclusion: AMPK controls the Wnt/β-catenin/GATA3 axis via inactivating GSK3β to coordinately modulate adipogenesis.
Significance: These findings provide new insight into the molecular mechanism and signal transduction of adipogenesis.
Keywords: adipogenesis, AMP-activated kinase (AMPK), β-catenin, GATA transcription factor, glycogen synthase kinase 3 (GSK3), Wnt pathway, CtBP, GATA3
Abstract
A better understanding of the mechanism and manipulation of the tightly regulated cellular differentiation process of adipogenesis may contribute to a reduction in obesity and diabetes. Multiple transcription factors and signaling pathways are involved in the regulation of adipogenesis. Here, we report that the AMP-activated protein kinase activator, 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR) can activate AMPK in preadipocytes and thus increase the expression of GATA3, an anti-adipogenic factor. However, AICAR-increased GATA3 is mediated by the stimulation of Wnt/β-catenin signaling in preadipocytes. Mechanistically, AICAR-activated AMPK inhibits GSK3β through a phosphorylation process that stabilizes β-catenin. This stabilized β-catenin then translocates into nucleus where it interacts with T-cell factors (TCF), leading to the increased β-catenin/TCF transcriptional activity that induces GATA3 expression. In addition, AICAR also relieves the repressing effect of the C-terminal-binding protein (CtBP) co-repressor by diverting CtBP away from the β-catenin·TCF complex at the GATA3 promoter. The anti-adipogenic effect of GATA3 and AICAR is consistently attenuated by the disruption of Wnt/β-catenin signaling. Furthermore, GATA3 suppresses key adipogenic regulators by binding to the promoters of these regulators, such as the peroxisome proliferator-activated receptor-γ (PPARγ) gene, and the disruption of Wnt/β-catenin signaling reduces the GATA3 binding at the PPARγ promoter. In differentiated adipocytes, GATA3 expression inhibition is facilitated by the down-regulation of β-catenin levels, the reduction in β-catenin binding, and the increase in CtBP binding at the GATA3 promoter. Our findings shed light on the molecular mechanism of adipogenesis by suggesting that different regulation pathways and adipogenic regulators collectively modulate adipocyte differentiation through cross-talk.
Introduction
Obesity is a global health problem associated with an increased risk of type II diabetes, cancer, cardiovascular diseases, and other metabolism-related health problems. In adipose tissue, the increased fat mass associated with obesity is characterized by both increased adipocyte size and/or increased adipocyte number (1). The number of adipocytes in body fat depots is largely determined by the process of adipogenesis. Adipogenesis, a differentiation process in which preadipocytes are transformed into differentiated adipocyte cells, is tightly regulated. A better understanding of the pathways that control adipogenesis will greatly contribute to the reduction of fat mass and protection from obesity and its associated diseases. To identify new biomarkers and therapeutic targets for the development of anti-obesity drugs, it is essential to first understand the molecular basis of adipogenesis and fat cell development in obesity. The molecular regulation of adipogenesis is controlled by many signaling molecules and transcription factors. Some studies have demonstrated that the GATA transcription factors GATA2 and GATA3 are expressed in white preadipocytes and in cultured preadipocyte cell lines such as 3T3-L1, but their expression is down-regulated with adipocyte differentiation (2). These transcription factors act as preadipocyte markers and molecular gatekeepers that suppress the transition from preadipocytes to adipocytes by inhibiting the expression and activity of PPARγ2 and C/EBPs (2, 3), the key transcription factors in adipogenesis; this contributes to the balance between energy intake and output and prevents the development of obesity. Defective GATA2 and GATA3 expression is associated with obesity, so increased GATA factors may be important for therapeutic intervention. However, how the GATA factor expression is regulated in preadipocytes is still largely unknown.
Wnt factors, one group of extracellular signaling molecules, also inhibit adipogenesis. Wnt signaling inhibits adipogenesis by stabilizing β-catenin, a multifunctional protein involved in cell fate decisions, development, and transcriptional regulation (4–6). In the absence of Wnt stimulation, cytoplasmic β-catenin is phosphorylated by a degradation complex containing GSK3β (7, 8). Phosphorylated β-catenin is then targeted to the ubiquitin/proteosome degradation pathway (8). Upon Wnt stimulation, the degradation complex is dissociated and β-catenin degradation is prevented. The inactivation of GSK3β by phosphorylation can lead to β-catenin accumulation and stabilization (9–11). This stabilized β-catenin then translocates into the nucleus where it interacts with transcription factors, most notably members of the TCF/lymphoid-enhancing factor (LEF) family, to induce Wnt/β-catenin downstream target gene expression (12, 13).
In the absence of Wnt signaling, TCFs are thought to repress Wnt target gene expression, as has been suggested for other transcription factors that mediate signaling (14). β-Catenin and co-repressors bind competitively to TCF as a result of overlapping binding sites, suggesting that β-catenin displaces the co-repressors once it enters the nucleus, thus relieving transcriptional repression (6). In addition to relieving TCF repression, β-catenin is thought to directly activate Wnt target gene expression by recruiting additional proteins to TCF-bound chromatin. Some studies have shown that 3T3-L1 preadipocytes express Wnt10b and that the ectopic expression of Wnt10b will activate Wnt/β-catenin signaling, leading to the subsequent inhibition of two well characterized, key adipogenic transcription factors, C/EBPα and PPARγ, which are necessary and sufficient for adipogenic differentiation (15–17).
Another factor shown to play an important role in modulating the Wnt/β-catenin pathway is CtBP. CtBP is a well known co-repressor in the repression of gene expression, including the Wnt/β-catenin target genes. By forming dimers, which are also regulated by cellular metabolic status, CtBP brings together gene-specific transcriptional factors and epigenetic modifiers such as HDAC1, G9a, and LSD1. This process is essential to the establishment of gene-specific repression (18–23). CtBP also acts as a co-activator in some conditions; its co-activator function may not require dimerization but does depend on interaction with other co-activators such as p300 (24–26). However, this conclusion has been challenged by studies that show CtBP as a co-repressor even when recruited or acetylated by p300 (27–29). As a co-repressor, CtBP has been reported to inhibit Wnt signaling targets by binding directly to TCFs or by preventing the interaction between β-catenin and TCFs (30–33), leading to the inhibition of Wnt target genes. As an energy sensor, the AMP-activated protein kinase (AMPK) signaling pathway also activates the Wnt/β-catenin signaling that regulates cell differentiation and development (34, 35). In addition, 5-amino-imidazolecarboxamide ribonucleoside (AICAR), an activator of AMPK, has been used as an experimental tool to activate AMPK in vitro and in vivo. Some studies report that AICAR inhibits the differentiation of 3T3-L1 adipocytes and adipogenesis (36–38). AICAR blocks the expression of the late adipogenic markers, fatty acid synthase and acetyl-CoA carboxylase, and of the transcription factors, C/EBPα and PPARγ (37). These studies suggest that AMPK has an inhibitory role in adipocyte differentiation. However, despite these studies on the role of various transcription factors and signaling pathways in the anti-adipogenic effect, the detailed cross-talk among these signaling pathways and transcription factors remains incompletely understood.
In this study, we have demonstrated that AMPK activation by AICAR stimulates Wnt/β-catenin signaling, possibly by increasing phosphorylation and inactivating GSK3β to stabilize β-catenin. AICAR-activated AMPK also increases β-catenin/TCF transcriptional activity and relieves CtBP repression on GATA3 expression, collectively inhibiting the expression of key adipogenic regulators and adipogenesis.
Experimental Procedures
Cell Culture
Mouse 3T3-L1 preadipocytes (ATCC) were grown in 5% CO2 in DMEM with 10% FBS and 1% penicillin/streptomycin (Gibco). For preadipocyte differentiation, cells at 100% confluency were stimulated in DMEM supplemented with 0.5 mm 3-isobutyl-1-methylxanthine, 5 μg ml−1 insulin, and 1 μm dexamethasone (Sigma). After 3 days, a maintenance medium (DMEM, 10% FBS, 1% penicillin/streptomycin, and 5 μg ml−1 insulin) was used and was changed every day for another 5–6 days. Oil Red O staining was used to monitor the matured adipocyte differentiation. Briefly, the cells were washed in PBS, fixed with 10% formalin in PBS for 30 min, and then stained with a 4:6 dilution in water of 0.5% Oil Red O (Sigma) in isopropanol for 1–2 h at room temperature. Finally, the stained cells were washed twice with distilled water and dried. To quantify the lipid accumulation, the stained dishes were dried, the Oil Red O was resuspended in isopropanol (1 min), and an OD solution was determined at 510 nm. Undifferentiated 3T3-L1 preadipocytes were treated with AICAR (0.5 mm), lithium chloride (LiCl, 25 mm) or Dickkopf 1 (Dkk1, 1 μg/ml) at the times indicated in Figs. 1–4. Differentiated 3T3-L1 adipocytes were also treated with the above chemicals at the beginning of the differentiation induction or on the indicated days.
FIGURE 1.

AICAR-activated AMPK increases GATA3 expression in preadipocytes. A, expression of GATA2 and GATA3 was quantified using quantitative PCR in 3T3-L1 preadipocytes without or with AICAR treatment (0.5 mm) for 48 h. B, the GATA3 protein level was determined by Western blotting in 3T3-L1 preadipocytes without (Ctrl, control) and with AICAR treatment (0.5 mm) for 48 h. C, ChIP assay of the GATA3 binding at the PPARγ2 promoter (pro) in 3T3-L1 preadipocytes without and with AICAR treatment (0.5 mm) for 48 h. A neighbor region of the PPARγ2 gene without transcripts (Non-pro) was used as a negative binding control region and a nonspecific IgG was used as a negative control for the chromatin pulldown. D, GATA3 protein level was determined by Western blotting to detect the GATA3 knockdown effect. E, the expression of PPARγ and C/EBPα was determined by RT-PCR in 3T3-L1 adipocytes without or with AICAR treatment (0.5 mm) for 9 days together with or without GATA3 knockdown (KD). F, Oil Red O staining and lipid accumulation quantification of 3T3-L1 adipocytes without or with AICAR (0.5 mm) treatment for 9 days together with or without GATA3 knockdown. The cells were induced to differentiate for 9 days using the full differentiation mixture. Scale bars, 50 μm. G and H, GATA3 expression was determined in 3T3-L1 preadipocytes without or with AICAR (0.5 mm) treatment for 48 h together without or with AMPK knockdown by RT-PCR (G) and Western blotting (H). One-way ANOVA was used in E–G, and other statistics were performed using Student's t test; bar graphs are mean ± S.E. In vitro data are the means of three independent experiments. *, p < 0.05; **, p < 0.01.
FIGURE 2.

AICAR increases β-catenin expression and phosphorylates GSK3β to stabilize β-catenin. A and B, β-catenin expression was determined in 3T3-L1 preadipocytes without or with AICAR (0. 5 mm) treatment for 48 h together without or with AMPK knockdown (KD) by RT-PCR and Western blotting. C, Western blot analysis of nuclear β-catenin levels in 3T3-L1 preadipocytes without or with AICAR (0.5 mm) treatment for 48 h. D, immunofluorescence staining analysis of 3T3-L1 preadipocytes with the anti-β-catenin antibody without (Ctrl, control) or with AICAR (0.5 mm) treatment for 24 h. Scale bars, 50 μm. E, the phosphorylation (Ser9) of GSK3β was determined without or with AICAR (0.5 mm) treatment at the indicated times together with or without AMPK knockdown. F, Oil Red O staining of 3T3-L1 adipocytes and quantification of lipid accumulation without or with AICAR (0.5 mm) treatment for 9 days together with or without β-catenin knockdown. Scale bars, 50 μm. G and H, GATA3 mRNA (G) and protein (H) levels were determined in 3T3-L1 preadipocytes without or with AICAR (0.5 mm) treatment for 48 h together without or with DKK1 treatment. I, ChIP assay of GATA3 binding at the PPARγ2 promoter (pro) under the specified conditions for 48 h. A neighbor region of the PPARγ2 gene without transcripts (Non-pro) was used as a negative binding control region, and nonspecific IgG was used as a negative control for chromatin pulldown. One-way ANOVA was used in A, F, G, and I, and other statistics were performed using Student's t test; bar graphs are mean ± S.E. In vitro data are the means of three independent experiments. *, p < 0.05; **, p < 0.01.
FIGURE 3.

β-Catenin directly regulates GATA3 expression to modulate adipocyte differentiation. A and B, β-catenin level was determined to check the β-catenin knockdown effect in 3T3-L1 preadipocytes by RT-PCR (A) and Western blotting (B). C, GATA3 protein level was determined in 3T3-L1 preadipocytes without or with β-catenin knockdown. D, GATA3 protein level was determined in 3T3-L1 preadipocytes without or with LiCl treatment (25 mm) for 48 h. E, GATA3 protein level was determined in 3T3-L1 preadipocytes without or with AICAR (0.5 mm) treatment for 48 h together without or with β-catenin knockdown (KD). F, the overexpression of GATA3 was detected by Western blotting. G, 3T3-L1 preadipocytes overexpressing GATA3 or an empty vector control (Ctrl) were induced to differentiate in the absence or presence of β-catenin knockdown, as indicated. The extent of differentiation at 9 days post-induction was assessed by Oil Red O staining, and lipid accumulation was quantified. Scale bars, 50 μm. H and I, the expression of PPARγ and C/EBPα was determined in the cells with the same treatment as described in G by RT-PCR (H) and Western blotting (I). J, ChIP assay of GATA3 binding at the PPARγ2 promoter (pro) under the specified conditions for 48 h. A neighbor region of the PPARγ2 gene without transcripts (Non-pro) was used as a negative binding control region, and nonspecific IgG was used as a negative control for chromatin pulldown. One-way ANOVA was used in G, and other statistics were performed using Student's t test; bar graphs are mean ± S.E. In vitro data are the means of three independent experiments. *, p < 0.05; **, p < 0.01.
FIGURE 4.

AICAR increases β-catenin/TCF transcriptional activity to regulate GATA3 expression. A, ChIP assay of β-catenin binding at the GATA3 promoter (pro) under the specified conditions for 48 h. A neighbor region of the GATA3 gene without transcripts (Non-pro) was used as a negative binding control region, and nonspecific IgG was used as a negative control for chromatin pulldown. B, ChIP assay of TCF binding at the GATA3 promoter under the specified conditions for 48 h. A neighbor region of GATA3 gene without transcripts was used as a negative binding control region, and nonspecific IgG was used as a negative control for chromatin pulldown. C, TCF/LEF luciferase reporter assays in 3T3-L1 preadipocytes without or with AICAR (0.5 mm) treatment for 24 h together without or with AMPK knockdown (KD) and relative luciferase activities were measured. D, ChIP assay of the CtBP binding at the GATA3 promoter under the specified conditions for 48 h. A neighbor region of the GATA3 gene without transcripts was used as a negative binding control region, and nonspecific IgG was used as a negative control for chromatin pulldown. One-way ANOVA was used in C, and bar graphs are mean ± S.E. In vitro data are the means of three independent experiments. *, p < 0.05.
Antibodies and Chemicals
The antibodies against β-catenin, phospho-GSK3β (Ser9), and GSK3β were purchased from Cell Signaling. The antibody against TCF was purchased from Millipore, and antibodies against CtBP, GATA3, PPARγ, and C/EBPα were purchased from Santa Cruz Biotechnology, Santa Cruz, CA. AICAR was purchased from Sigma. DKK1 was purchased from R&D Systems, and LiCl was purchased from Sigma.
Quantitative Real-time RT-PCR
The total RNA extracted from the cells using TRIzol (Invitrogen) was treated with Turbo DNase (Ambion), and 2 μg was reverse-transcribed in a 40-μl volume system (QuantiTect reverse transcription kit (Qiagen)) for quantitative PCR assays. The electrophoresis method was used to ensure the integrity of the RNA by checking multiple major rRNA bands and other high-copy RNAs. Quantitative real-time PCR analyses were carried out using gene-specific primers (working concentration at 250 nm) in a 7900 Sequence Detector (Applied Biosystems, Foster City, CA). The PCR running procedure was as follows: 4 min denaturing at 95 °C followed by 40 cycles of 30 s of denaturing at 95 °C and then annealing and extension at 60 °C for 1 min. The fluorescent signals were collected for each cycle. The data were then analyzed using Applied Biosystems' real-time PCR analysis software SDS-2.2. For relative mRNA quantification of all of the genes, SYBR Green real-time RT-PCR was used with normalization to the housekeeping gene S16 as an internal control. The primer sequences were as follows: GATA2 for, 5′-TGGGCTCTACCACAAGATGA-3′, and rev, 5′-GCCATAAGGTGGTGGTTGTC-3′; GATA3 for, 5′-TCAGCCCACCACCCCATTAC-3′, and rev, 5′-GTTGCCCCGCAGTTCACA-3′; PPARγ for, 5′-ACACAATGCTGGCCTCCTTG-3′, and rev, 5′-AAAAGGCTTTCGCAGGCTCT-3′; C/EBPα for, 5′-ACACAATGCTGGCCTCC TTG-3′, and rev, 5′-AAAAGGCTTTCGCAGGCT CT-3′; CtBP1 for, 5′-AGAAGAGGACGTTGAAGCCA-3′, and rev, 5′-ATCCTGAACCTGTACCGACG-3′; CtBP2 for, 5′-ATAGCAAGTCCTGCAGGGTG-3′, and rev, 5′-GTGGAACAGATCCGTGAGGT-3′; and β-catenin for, 5′-GACCACAAGCAGAGTGCTGA-3′, and rev, 5′-TGAAAGGTTTCTGAGAGTCCAA-3′.
Western Blotting
The cells were lysed in radioimmune precipitation assay buffer supplemented with protease and phosphatase inhibitors (Sigma-Aldrich). The lysates were sonicated for 1 min and centrifuged at 14,000 × g for 10 min at 4 °C. The lysates were resolved with 4–20% Tris/glycine SDS-PAGE and transferred to nitrocellulose membranes. Horseradish peroxidase-conjugated secondary antibody and the Amersham Biosciences ECL advance Western blotting detection system (GE Healthcare Life Sciences) were used. The integrated density was measured with the NIH ImageJ system and normalized with β-actin.
Immunofluorescence Staining
Immunofluorescence was carried out as described elsewhere (39). In brief, 3T3-L1 preadipocytes were cultured on cell culture chamber slides (Nalge Nunc International) with or without AICAR treatment for 24 h. After washing with PBS and treating with 4% paraformaldehyde for 15 min, the slides were washed with PBS 3 times, treated with 5% Triton X-100 for 10 min, and blocked with 1% BSA for 30 min. The cell culture slides were incubated with the primary antibody for β-catenin overnight at 4 °C. After staining with fluorescence-conjugated secondary antibody (Santa Cruz Biotechnology), the slides were mounted using vector DAPI mounting medium (Vector Laboratories Inc., Burlingame, CA). The samples were examined, captured, and quantified under an Axio Observer Z1 inverted confocal microscope equipped with a Zeiss LSM 5 Live DuoScan laser scanning system (Carl Zeiss MicroImaging).
Chromatin Immunoprecipitation (ChIP) Assay
ChIP analysis was carried out as described elsewhere (39). In brief, after cells were lysed, the nuclei were isolated, cross-linked with 1% formaldehyde, and lysed in a lysis buffer (1% SDS, 10 mm EDTA, and 50 mm Tris, pH 8.1), and the cross-linked chromatin DNA was sheared by sonication on ice to an average length of ∼500 bp. The sonicated supernatant was diluted 10-fold in a ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mm EDTA, 16.7 mm Tris-HCl, pH 8.1, and 167 mm NaCl). Chromatin that had been precleared using protein G-agarose (Upstate) was incubated with the specific antibody or IgG as the control at 4 °C overnight. The resultant immunoprecipitations were recovered by incubation with the protein G-agarose (Upstate) at 4 °C for 2 h followed by low speed centrifugation. The washed pellets were reverse cross-linked. The DNA used for quantitative PCR analysis was extracted with phenol/chloroform/isoamyl alcohol (25:24:1) precipitated with ethanol. The PCR primer sequences used for the ChIP assay were as follows: PPARγ promoter for, 5′-ACCAAGTCTTGCCAAAGCAG-3′, and rev, 5′-TCTTGCAAAAGATTGGTTGG-3′; non-promoter control for, 5′-TGTGCCAGGCTCTGTGTTTA-3′, and rev, 5′-TCAGAAATCCACCTGCCTCT-3′; GATA3 promoter for, 5′-GTTTGGGAAAGCAAGCAGAG-3′, and rev, 5′-TCAGCTTAGGGGCTATGTGC-3′; non-promoter control for, 5′-AGGCTGGCTCTCCTCAGTTT-3′, and rev, 5′-ATTCGCTGCCTCTAGACCTG-3′.
Co-immunoprecipitation Assay
The cell nuclear extract, prepared in radioimmune precipitation assay buffer (50 mm Tris-HCl, pH 7.5,100 mm NaCl, 2 mm EDTA, 1% Nonidet P-40, 1 mm EGTA, and protease inhibitor) and 100 mg of protein extract, was precleared with the control IgG (Santa Cruz Biotechnology) and protein A/G beads (Upstate). The precleared extract was then incubated with its specific antibody or preimmune serum and protein A/G beads in 1.5 ml of immunoprecipitation buffer (0.5% Nonidet P-40, 10 mm Tris-HCl, 150 mm NaCl, 2 mm EDTA, 10% glycerol, and protease inhibitor) at 4 °C for 4–6 h. After a brief centrifugation, the pellets were washed in immunoprecipitation buffer 4–5 times at 4 °C for 10 min, and the immunoprecipitated protein complexes were analyzed by Western blot analysis using the specific antibodies.
TCF/LEF Luciferase Reporter Assay
The 3T3-L1 preadipocytes were transfected with TOPFlash plasmids, transfection grade T-cell factor (Tcf) reporter plasmids that contained three copies of the Tcf binding site (AGATCAAAGGG) upstream of the thymidine kinase (TK) minimal promoter and luciferase open reading frame. A FOPFlash plasmid was used as a negative control. This plasmid contains two full copies (AGGCCAAAGGG) and one incomplete copy (GCCAAAGGG) of the Tcf binding site (mutated) followed by three copies in the reverse orientation (CCCCTTTGGCCT) upstream of the TK minimal promoter and Luciferase open reading frame. After 16 h of transfection, the cells were stimulated by AICAR (0.5 mm) for 24 h together with or without AMPK knockdown or β-catenin knockdown. The cells were then washed twice with PBS and lysed for 15 min. The lysates were collected by centrifugation at 13,000 rpm, and 20 μl of lysate was used to measure the luciferase reporter activity. The luciferase activity was normalized to Renilla luciferase activity from the co-transfected internal control plasmid, pRL-TK. A Dual-Luciferase assay was performed using the Dual-Luciferase reporter assay system (Promega).
RNAi
For the knockdown experiments, siRNAs specific for GATA3 (SMARTpool of several siRNAs selected by Dharmacon), CtBP (SMARTpool of several siRNAs selected by Dharmacon), AMPK (SMARTpool of several siRNAs selected by Dharmacon), β-catenin (AAACATAATGAGGACCTACAC), and the negative control siRNA (5′-AAUUCUCCGAACGUGUCACGU-3′) (Thermo Scientific Dharmacon) were transfected into the 3T3-L1 adipocytes via a DharmaFECT transfection reagent.
Statistical Analyses
The values are expressed as mean ± S.E. A two-tailed non-paired Student's t test was used to compare pairs of groups. Any statistical differences between three or more groups were evaluated using a one-way analysis of variance (ANOVA) with Dunnet's multiple comparison post hoc tests at α = 0.05. A p value of <0.05 was regarded as statistically significant.
Results
AICAR Increases GATA3 Expression, Which Inhibits Adipogenesis
AICAR, an activator of AMPK, and GATA factors including GATA2 and GATA3 inhibit adipocyte differentiation. To determine whether there is a relationship between the two anti-adipogenic effects, we first examined the GATA factor expression. Surprisingly, we observed that GATA3 but not GATA2 was up-regulated by AICAR stimulation in 3T3-L1 preadipocytes at the RNA and protein levels (Fig. 1, A and B). GATA factors inhibit adipogenesis by suppressing PPARγ gene expression via directly binding to the GATA site on the PPARγ promoter (2). To examine whether AICAR-stimulated GATA3 expression affects the binding of GATA3 at the PPARγ promoter, we performed a ChIP analysis. The result showed that AICAR increased GATA3 binding at the PPARγ2 promoter but not in the negative control region of the PPARγ gene (Fig. 1C), suggesting that AICAR may induce the expression of GATA factors, especially GATA3, making them bind to the adipogenesis-required gene promoter and regulating their expression. The adipogenesis key marker genes PPARγ and C/EBPα were consistently down-regulated by AICAR stimulation. However, the knockdown of GATA3 (Fig. 1D), at least in part, attenuated the down-regulation induced by AICAR stimulation (Fig. 1E), suggesting that AICAR-inhibited adipogenesis is mediated by GATA3. Next, we investigated the impact of GATA3 knockdown on AICAR-inhibited adipogenesis. As shown in Fig. 1F, the GATA3 knockdown was able to partially rescue AICAR-inhibited adipogenesis, as measured by increased lipid accumulation compared with AICAR-treated adipocytes only. This suggests that GATA3 acts as a downstream factor to mediate the AICAR effects in adipocytes. Given that AICAR needs to activate AMPK to exert its function, we next examined whether AICAR-stimulated GATA3 expression is mediated by activated AMPK. We knocked down AMPKαin 3T3-L1 cells and found that AICAR-induced GATA3 expression was impaired by AMPK knockdown (Fig. 1, G and H), indicating that AICAR activates AMPK to increase GATA3 expression and that this increased GATA3 expression inhibits adipocyte differentiation.
AICAR Phosphorylates GSK3β, Which Stabilizes β-Catenin and Stimulates Wnt/β-Catenin Activation
Next, we investigated the mechanism through which AICAR increases GATA3 expression. Surprisingly, we observed that Wnt/β-catenin signaling is involved in regulating the expression of GATA3 by AICAR. Wnt/β-catenin has been reported to prevent the differentiation of 3T3-L1preadipocytes (15, 16) and to inhibit key transcription factors in adipogenesis including PPARγ and C/EBPα. We examined whether Wnt/β-catenin mediates AICAR-induced GATA3 expression. First, we investigated whether AICAR regulates β-catenin expression. As Fig. 2, A and B, shows, AICAR stimulation increased β-catenin expression at both the RNA and protein levels. In contrast, AMPKα knockdown decreased the effect of AICAR on β-catenin expression (Fig. 2, A and B), suggesting that AICAR-activated AMPK signaling regulates β-catenin expression in preadipocytes. Importantly, AICAR increased the nuclear β-catenin content, but AMPK knockdown blocked the increase (Fig. 2C). The activation of Wnt/β-catenin signaling by AICAR was further confirmed by immunofluorescence staining analyses that showed increased nuclear localization of β-catenin in the 3T3-L1 cells (Fig. 2D). These data further verified that AMPK activation may stabilize the β-catenin and activate the Wnt/β-catenin signaling pathway in preadipocytes. To further elucidate the mechanism by which AICAR-activated AMPK stimulates Wnt/β-catenin signaling activation, we then assessed whether AICAR could phosphorylate (Ser9) to inactivate GSK3β, a component of β-catenin degradation complex. We examined both the total and phosphorylated GSK3β proteins and found, as shown in Fig. 2E, that the phosphorylation of GSK3β proteins was increased by AICAR stimulation at the indicated times, but this did not increase the total GSK3β proteins. Importantly, AMPK knockdown impaired the effect of AICAR on increased GSK3β phosphorylation. These data suggest that AICAR-activated AMPK may inactivate GSK3β by increasing its phosphorylation and thus inhibit β-catenin degradation, which leads to the accumulation of β-catenin in the nucleus and the activation of Wnt/β-catenin signaling and its downstream effects. Supporting this interpretation, we found that the anti-adipogenic effect of AICAR was impaired by the knockdown of β-catenin (Fig. 2F) as reflected by the Oil Red O staining of the intracellular lipids, indicating that the anti-adipogenic effect of AICAR may be mediated by Wnt/β-catenin signaling. To investigate whether AICAR-induced GATA3 expression is also mediated by Wnt/β-catenin signaling, we first used the Wnt antagonist Dkk1 to treat 3T3-L1 preadipocytes. The results showed that the AICAR-induced increase in GATA3 RNA and protein levels was impaired by DKK1 treatment (Fig. 2, G and H), suggesting that β-catenin-mediated AICAR effects on anti-adipogenesis may be partially a result of the regulation of GATA factors, especially GATA3 expression. Notably, the increased GATA3 binding at the PPARγ2 promoter by AICAR was also partially abolished by the DKK1 treatment (Fig. 2I). These data support our hypothesis that Wnt/β-catenin may modulate the adipocyte differentiation process by mediating AICAR-induced GATA3 expression in preadipocytes.
β-Catenin Activation Induces GATA3 Expression, Which Inhibits Adipogenesis
As our findings suggested that disrupted β-catenin signaling by DKK1 inhibits GATA3 expression, we next investigated whether β-catenin directly regulates GATA3 expression. We knocked down the β-catenin using short hairpin RNA (shRNA) specific to β-catenin. As shown in Fig. 3, A and B, the mRNA and protein levels of β-catenin were significantly reduced in these cells. We then observed that GATA3 expression was down-regulated by the knockdown of β-catenin (Fig. 3C). In contrast, β-catenin activation by LiCl, which is a potent activator of Wnt/β-catenin signaling, up-regulated GATA3 expression (Fig. 3D). Importantly, the knockdown of β-catenin impaired AICAR induced GATA3 expression (Fig. 3E). These data suggest that β-catenin acts downstream of AICAR to directly regulate GATA3 expression. To address the role of β-catenin in the GATA3-induced anti-adipogenesis effect, we overexpressed GATA3 in 3T3-L1 cells (Fig. 3F). As shown in Fig. 3, G–I, when the cells were induced to differentiate by using a differentiation mixture, the ectopic expression of GATA3 led to decreased lipid accumulation and reduced expression of the adipogenic markers PPARγ and C/EBPα. However, the knockdown of β-catenin, at least in part, attenuated the inhibition effect of GATA3 on adipogenesis as indicated by the Oil Red O staining and the expression of PPARγ and C/EBPα at the RNA and protein levels (Fig. 3, G–I). In addition, GATA3 binding at the PPARγ promoter was decreased by the knockdown of β-catenin but increased by LiCl-activated β-catenin (Fig. 3I). These data strongly suggest that the anti-adipogenic effect of the Wnt/β-catenin signaling pathway is due to the direct modulation of GATA3 expression.
AMPK Increases β-Catenin/TCF Transcriptional Activity, Which Regulates GATA3 Expression
To investigate the mechanism by which β-catenin mediates AICAR-induced GATA3 expression, we first performed a ChIP assay to detect whether the GATA3 expression was regulated by β-catenin binding to the GATA3 promoter. Interestingly, we observed that β-catenin did bind at the GATA3 promoter (Fig. 4A) but not in the negative control sequence (Non-pro). Importantly, β-catenin activation by LiCl treatment increased binding at the GATA3 promoter, and knocking down of β-catenin decreased the binding of β-catenin at the GATA3 promoter (Fig. 4A). Similarly, with AICAR stimulation, the binding of β-catenin at the GATA3 promoter also increased (Fig. 4A), supporting our hypothesis that the AICAR activation of β-catenin signaling up-regulates GATA3 expression by facilitating the stabilization of β-catenin at the GATA3 promoter. In contrast, β-catenin knockdown decreased its binding at the GATA3 promoter. These data further confirm the underlying mechanism through which β-catenin directly regulates GATA3 expression in preadipocytes. As β-catenin does not bind to DNA by itself and transcription factors TCF/LEF family can interact with and recruit β-catenin to regulatory sites of the Wnt target gene (13), we also assessed the binding of TCF at the GATA3 promoter. With AICAR stimulation, the binding of TCF at the GATA3 promoter was found to increase (Fig. 4B), suggesting that the binding of TCF to the GATA3 promoter contributes to the β-catenin-mediated transcription activation of GATA3. To directly assess whether AICAR-activated AMPK regulates β-catenin/TCF transcriptional activity in 3T3-L1 preadipocytes, we performed TCF/LEF luciferase reporter assays using the TCF-dependent luciferase reporter (containing three copies of the TCF binding site), TOPFlash. As shown in Fig. 4C, AICAR increased TOPFlash luciferase activity, but AMPK knockdown impaired this effect. Importantly, β-catenin knockdown also abolished AICAR-enhanced TOPFlash activity. Taken together, these data indicate that AICAR-activated AMPK contributes to β-catenin/TCF transcriptional activity and that AICAR induced up-regulation of GATA3 represents a specific effect of AICAR on β-catenin/TCF transcriptional activity.
Some reports suggest that TCF may interact with some co-repressors to inhibit Wnt/β-catenin downstream gene expression when the Wnt/β-catenin signal is not activated (12, 14, 40). Therefore, we hypothesize that AICAR stimulation may activate β-catenin to bind competitively with co-repressors to TCF through overlapping binding sites; the β-catenin binding thus displaces the co-repressors, leading to the relieving of transcriptional repression. Interestingly, CtBP, a transcription co-repressor, had a weak binding at the GATA3 promoter compared with the control region. However, it was observed that CtBP binding at the GATA3 promoter was abolished by AICAR stimulation (Fig. 4D), whereas the disruption of β-catenin signaling by DKK1 increased the binding of CtBP at the GATA3 promoter (Fig. 4D). Collectively, these data suggest that CtBP may inhibit GATA3 expression in the absence of Wnt/β-catenin by binding to the GATA3 promoter, but the activation of the Wnt/β-catenin signaling pathway can relieve the CtBP repression effect via releasing the CtBP binding, leading to recruited β-catenin binding at the GATA3 promoter by TCF. Notably, CtBP does not bind DNA by itself. The unknown factors that recruit CtBP to the GATA3 promoter warrant further investigation.
CtBP Inhibits the Interaction of β-Catenin and TCF, Thus Repressing GATA3 Expression
To investigate the effect of CtBP on GATA3 expression, we knocked down the CtBP in the 3T3-L1 preadipocytes. As shown in Fig. 5A, the knockdown of CtBP transcriptionally enhanced GATA3 expression. The GATA3 protein level also mirrored the mRNA level (Fig. 5B). To test our hypothesis that β-catenin and CtBP interact competitively with TCF to bind to the GATA3 promoter to exert inverse functions, we performed an immunoprecipitation experiment. Surprisingly, as Fig. 5C shows, we did not observe any interaction between CtBP and TCF. In contrast, a weak interaction between CtBP and β-catenin was detected. These data are consistent with previous reports of no detectable association observed between TCF and CtBP in vivo in mammalian cells (32). However, the interaction detected between CtBP and β-catenin indicates that CtBP may associate with β-catenin to inhibit the interaction of β-catenin with TCF, leading to the repression of Wnt/β-catenin target genes. A similar study reports that CtBP interacts with β-catenin via the adapter, adenomatous polyposis coli, a factor that promotes the destruction of β-catenin in the cytoplasm, and this decreases the availability of the free nuclear β-catenin required for binding to TCF and to activate Wnt/β-catenin downstream target gene expression (32). Interestingly, with AICAR stimulation, the interaction between CtBP and β-catenin was blocked and the interaction between β-catenin and TCF increased (Fig. 5C). These data suggest that AICAR may facilitate β-catenin stabilization and disassociation from the CtBP repressor and thus increase the interaction of β-catenin and TCF, leading to the positive regulation of Wnt/β-catenin target gene expression. In support of view, the knockdown of CtBP promoted the interaction between β-catenin and TCF (Fig. 5D). Importantly, CtBP knockdown increased TOPFlash activity (Fig. 5E), suggesting that the disassociation of CtBP from its interaction with β-catenin enhances the β-catenin/TCF transcriptional activity and thus facilitates downstream target gene expression. CtBP knockdown consistently increased the binding of β-catenin and TCF at the GATA3 promoter in preadipocytes (Fig. 4, A and B). Furthermore, GATA3 and β-catenin expression were greatly down-regulated upon the differentiation of 3T3-L1 cells into adipocytes (Fig. 5F). We then investigated the changes of β-catenin binding at the GATA3 promoter before and after adipocyte differentiation. We observed that β-catenin binding dramatically declined with adipocyte differentiation (Fig. 5G), whereas the CtBP repressor binding at the GATA3 promoter was enhanced with adipocyte differentiation (Fig. 5H). Collectively, these data suggest that Wnt/β-catenin signaling positively regulates GATA3 expression in preadipocytes via TCF recruiting of β-catenin to the GATA3 promoter and that AICAR can activate Wnt/β-catenin signaling to promote the Wnt/β-catenin signaling effect. Together these processes lead to an anti-adipogenic effect. In contrast, CtBP negatively regulates GATA3 expression, and this inhibition may explain why GATA3 expression is down-regulated in differentiated adipocytes.
FIGURE 5.
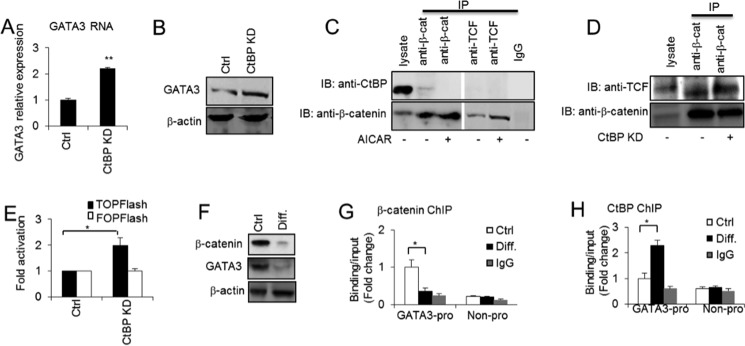
CtBP inhibits GATA3 expression by inhibiting the interaction of β-catenin and TCF. A and B, GATA3 expression was determined at the RNA and protein levels in 3T3-L3 preadipocytes without or with CtBP knockdown (KD) by RT-PCR (A) and Western blotting (B). C, co-immunoprecipitations between endogenous CtBP and β-catenin or TCF-4 and between endogenous β-catenin and TCF-4 in 3T3-L1 preadipocytes without or with AICAR (0.5 mm) treatment for 48 h. D, co-immunoprecipitations between endogenous β-catenin and TCF-4 in 3T3-L1 preadipocytes without or with CtBP knockdown. IP, immunoprecipitation; IB, immunoblotting. E, TCF/LEF luciferase reporter assays in 3T3-L1 preadipocytes without or with CtBP knockdown. F, β-catenin and GATA3 protein levels were determined by Western blotting in 3T3-L1 preadipocytes (Ctrl) and differentiated adipocytes at day 9 (Diff.). G, ChIP assay of β-catenin binding at the GATA3 promoter in 3T3-L1 preadipocytes and differentiated adipocytes at day 9. H, ChIP assay of the CtBP binding at the GATA3 promoter in 3T3-L1 preadipocytes and differentiated adipocytes at day 9. Statistics were performed using Student's t test, and bar graphs are mean ± S.E. In vitro data are the means of three independent experiments. *, p < 0.05; **, p < 0.01.
As our data suggested that CtBP regulates GATA3 expression in 3T3-L1 preadipocytes, we investigated the effect of CtBP on adipogenesis. CtBP1 and CtBP2 mRNA levels did not significantly change with adipocyte differentiation, although a decreased trend was observed, as shown in Fig. 6A. The total CtBP protein level did not significantly change (Fig. 6B). When CtBP was knocked down, the levels of the adipogenic markers PPARγ and C/EBPα were not significantly affected (Fig. 6, C and D). These data suggest that CtBP is sustained throughout adipogenesis and that it acts as a repressor in preadipocytes that regulates GATA3 expression. Further studies will help to illustrate the precise role of CtBP in adipogenesis.
FIGURE 6.
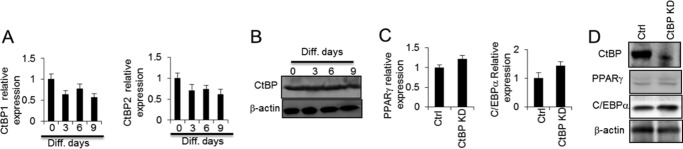
CtBP is expressed during adipogenesis. A, Ctbp1 and Ctbp2 mRNA levels were determined throughout adipogenesis at the indicated differentiation (Diff.) days of 3T3-L1 adipocytes by RT-PCR. B, CtBP protein level was determined by Western blotting throughout adipogenesis. C and D, PPARγ and C/EBPα level were determined in 3T3-L1 adipocytes without or with CtBP knockdown (KD). Ctrl, control. Statistics were performed using Student's t test, and bar graphs are mean ± S.E. In vitro data are the means of three independent experiments. *, p < 0.05.
Discussion
In this study, we have provided evidence to suggest that in preadipocytes, AMPK activation by AICAR increases GATA3 expression through inactivating GSK3β, thus activating the Wnt/β-catenin signaling pathway and enhancing the β-catenin/TCF transcriptional activity and binding at the GATA3 promoter. The increased GATA3 expression ultimately leads to inhibited adipogenesis. However, the anti-adipogenic effect of GATA3 is attenuated by the disruption of Wnt/β-catenin signaling. The AMPK-induced GATA3 expression is also mediated by decreased binding of the CtBP repressor at the GATA3 promoter and the abolition of the interaction between CtBP and β-catenin. With adipocyte differentiation, the disassociation of β-catenin/TCF from the GATA3 promoter and increased CtBP binding at the GATA3 promoter may explain, at least in part, the robust reduction of GATA3 levels during this process.
Modulation of early preadipocyte differentiation is one strategy to inhibit adipogenesis and trigger white adipose tissue remodeling, thus reducing adiposity (41). Multiple transcription factors can attenuate adipogenesis. GATA2 and GATA3 expressed in preadipocytes have been reported to inhibit adipogenesis by directly binding to the PPARγ promoter, which suppresses its basal activity or the antagonism of C/EBPs activity (2, 3). PPARγ and C/EBPs are master regulators of adipogenesis. As a sensor of energy, AMPK has been shown to reduce fat accumulation and increase glucose tolerance, insulin sensitivity, and mitochondrial biogenesis (42–47). The AMPK activator AICAR can inhibit 3T3-L1 adipocyte differentiation by blocking the expression of C/EBPα and PPARγ (36). In our study, surprisingly, we found that AICAR increased GATA3 expression but not GATA2 expression in 3T3-L1 preadipocytes. Importantly, treatment with AICAR increased GATA3 binding to the PPARγ promoter, and the knockdown of AMPK attenuated AICAR-increased GATA3 expression, suggesting that the AICAR effect is mediated by the AMPK pathway. The binding of GATA3 at the PPARγ promoter suppresses PPARγ expression and thus inhibits adipogenesis (2), suggesting that AMPK inhibits adipogenesis by inhibiting its master regulators. It is likely that there is cross-talk among these different pathways, which collectively repress adipogenesis.
Wnt binding to Frizzled (FZD1) receptors and to low density lipoprotein receptor-related protein (LRP) co-receptors leads to the hypophosphorylation of β-catenin, which results in the stabilization and accumulation of β-catenin in the cytoplasm and its translocation into the nucleus where it binds to TCF/LEF transcription factors and activates Wnt target genes (48, 49). The Wnt/β-catenin pathway is one of the extracellular signaling pathways known to regulate adipogenesis (48, 49). The ectopic expression of Wnt in preadipocytes activates Wnt/β-catenin signaling and represses adipogenesis (15, 16, 50). Furthermore, treatment with pharmacological agents that activate canonical Wnt signaling can block adipocyte differentiation (16), whereas the inhibition of Wnt signaling in preadipocytes can induce adipocyte differentiation. Understanding how to activate the Wnt/β-catenin signaling pathway may provide some potential avenues for the treatment of obesity-related metabolic complications. This study has demonstrated that AICAR stimulation in preadipocytes can increase the nuclear accumulation of β-catenin and activate Wnt/β-catenin signaling, leading to the inhibition of adipocyte differentiation. The AICAR effect is mediated by AMPK, as AMPK knockdown attenuates the AICAR effect of increasing and stabilizing the β-catenin. This finding is consistent with other studies that found that AICAR-induced AMPK activation inhibits adipogenesis in 3T3-L1 adipocytes via the Wnt/β-catenin pathway (51). Importantly, for the first time, we demonstrated that AICAR-activated AMPK may inactivate GSK3β by phosphorylation. As GSK3β targets the β-catenin to degradation complex, the inactivation of GSK3β by AICAR allows β-catenin to escape degradation and thus accumulate in the nucleus. However, whether AMPK directly phosphorylates GSK3β or AMPK stimulates other kinases to phosphorylate GSK3β remains unclear. In addition, we revealed that the AICAR-mediated increase in GATA3 expression is also impaired by the disruption of the Wnt/β-catenin signaling pathway. These results identify GATA3 as a novel downstream target of Wnt/β-catenin and suggest that there is cross-talk between AMPK and the Wnt/β-catenin signaling pathway in preadipocytes. In fact, previous studies have shown that AMPK regulates cell differentiation and development by promoting β-catenin expression, stabilizing β-catenin, and enhancing β-catenin/TCF-mediated transcription (34, 35). Additionally, AICAR and activated AMPK inhibit adipocyte differentiation by down-regulating the expression of adipogenic factors in vitro and reducing adipose tissue content in diet-induced obesity (DIO) mice (37, 46, 52). Because of the important role of AMPK and β-catenin signaling not only in adipocyte differentiation but also in cell differentiation and tissue development, the cross-talk between AMPK and Wnt/β-catenin likely has important physiological and developmental implications.
One of the mechanisms through which Wnt/β-catenin signaling inhibits adipogenesis is thought to be the prevention of the induction of the master adipogenic regulators C/EBPα and PPARγ during preadipocyte differentiation (15). However, the underlying mechanism through which β-catenin regulates the expression of these adipogenic regulators is still unclear. Our study has shown that β-catenin increases GATA3 expression, which inhibits PPARγ and C/EBPα expression. In our study, the knockdown of β-catenin decreased GATA3 expression in preadipocytes and attenuated the GATA3-mediated inhibition of adipocyte differentiation. The GATA3-caused reduction in the expression of key adipogenic regulators was also impaired by the knockdown of β-catenin. GATA3 binding at the PPARγ promoter was increased by the activation of β-catenin signaling but decreased by the knockdown of β-catenin. These data suggest that the effect of β-catenin in adipocytes is mediated by GATA3 and that the inhibited expression of key adipogenic regulators by β-catenin also occurs through the regulation of GATA3 expression. Our results provide insights into the mechanisms through which Wnt/β-catenin signaling functions to block adipogenesis and to regulate adipogenic regulators by modulating its new target, GATA3. Furthermore, the recruitment of β-catenin by TCF to the promoter of GATA3 also regulates the expression of GATA3. We observed that the activation of β-catenin signaling, either by AICAR or LiCl stimulation, enhanced the binding of β-catenin at the GATA3 promoter, but the disruption of β-catenin signaling reduced the binding of β-catenin at the GATA3 promoter. These data taken together demonstrate that transcription factors such as GATA3 and signaling pathways, including the AMPK and Wnt/β-catenin signaling pathways, coordinately and strictly regulate adipocyte differentiation via their cross-talk.
CtBP is able to interact with gene-specific transcriptional factors and recruit several known epigenetic modifying enzymes such as LSD1, HDACs, and G9a, among others, to the target genes (18, 53). CtBP is also involved in the Wnt/β-catenin signaling pathway. First, some studies report that CtBP inhibits Wnt target genes by interacting with TCFs (30, 31). However, some later studies did not find any interaction between CtBP and TCF but did find that some adaptors, such as APC (adenomatous polyposis coli), mediate the interaction between β-catenin and CtBP, and thus CtBP lowers the availability of free β-catenin for binding to TCF, leading to suppressed Wnt target genes (32, 33). Some other reports suggest that CtBP can act as a repressor of Wnt target genes in dimers and an activator of Wnt target gene in monomers (26, 54). Here, we found that CtBP can inhibit GATA3 expression in the absence of Wnt signaling activation in preadipocytes. When Wnt/β-catenin signaling was activated, CtBP was released from the interaction with β-catenin and GATA3 promoter, leading to increased interaction between β-catenin and TCF at the GATA3 promoter and induced GATA3 expression. It is possible that CtBP is recruited to the GATA3 promoter by some unknown factors in the absence of Wnt/β-catenin activation and thatβ-catenin activation releases the CtBP from the GATA3 promoter and promotes β-catenin and TCF interaction, leading to increased GATA3 expression. Supporting our interpretation, our data demonstrated that AMPK signaling activation by AICAR can stimulate β-catenin-dependent TCF/LEF transcriptional activity by increasing β-catenin/TCF binding and releasing CtBP binding at downstream target binding sites, thus promoting target anti-adipogenic gene expression such as GATA3. These data suggest that AMPK, Wnt/-catenin/TCF, and CtBP collectively regulate GATA3 expression in preadipocytes and thus affect adipocyte differentiation. The blocked expression of β-catenin in adipocyte differentiation results in decreased binding of β-catenin and increased CtBP binding at the GATA3 promoter, which facilitates the down-regulation of GATA3. However, although CtBP inhibits the expression of GATA3, which is a repressor of adipogenesis, the effect of CtBP on adipocyte differentiation is not positive. Our data indicated that CtBP expression was sustained throughout adipogenesis, and the knockdown of CtBP had no significant effect on the expression of key adipogenic marker genes. Further study will help to illustrate the detailed function of CtBP during adipocyte differentiation.
In conclusion, our study identifies a new regulatory mechanism for adipocyte differentiation: AMPK activation stimulates Wnt/β-catenin signaling by inactivating GSK3β to inhibit β-catenin degradation. This results in the activation of β-catenin/TCF transcriptional activity and downstream target gene expression such as GATA3 and relieves the co-repressor CtBP from the β-catenin/TCF binding site. Collectively, this leads to the suppressed expression of adipogenic regulators and the anti-adipogenic effect.
Author Contributions
L. W. and L. J. D. designed the study and wrote the paper. L. W. designed, performed, and analyzed the experiments shown in Figs. 1, 2, 3, 5, and 6. L. J. D. performed and analyzed the ChIP and immunoprecipitation experiments shown in Figs. 4 and 5. Both authors reviewed the results and approved the final version of the manuscript.
This work was supported by Grants FDCT 025/2014/A1 and FDCT 088/2014/A2 from the Science and Technology Development Fund (FDCT) of Macao SAR (to L. J. D.) and by Multi-year Research Grants MYRG2015-00037-FHS and MYRG2015-00167-FHS from the University of Macau (to L. J. D.). The authors declare that they have no conflicts of interest with the contents of this article.
- PPARγ
- peroxisome proliferator-activated receptor γ
- C/EBP
- CCAAT/enhancer-binding protein
- AICAR
- 5-aminoimidazole-4-carboxamide ribonucleotide
- DKK1
- Dickkopf 1
- AMPK
- AMP-activated protein kinase
- CtBP
- C-terminal-binding protein
- TCF
- T-cell factor
- LEF
- lymphoid-enhancing factor
- TK
- thymidine kinase
- ANOVA
- analysis of variance.
REFERENCES
- 1. Rosen E. D., MacDougald O. A. (2006) Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 7, 885–896 [DOI] [PubMed] [Google Scholar]
- 2. Tong Q., Dalgin G., Xu H., Ting C. N., Leiden J. M., Hotamisligil G. S. (2000) Function of GATA transcription factors in preadipocyte-adipocyte transition. Science 290, 134–138 [DOI] [PubMed] [Google Scholar]
- 3. Tong Q., Tsai J., Tan G., Dalgin G., Hotamisligil G. S. (2005) Interaction between GATA and the C/EBP family of transcription factors is critical in GATA-mediated suppression of adipocyte differentiation. Mol. Cell. Biol. 25, 706–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cadigan K. M., Nusse R. (1997) Wnt signaling: a common theme in animal development. Genes Dev. 11, 3286–3305 [DOI] [PubMed] [Google Scholar]
- 5. Primus A., Freeman G. (2004) The cnidarian and the canon: the role of Wnt/β-catenin signaling in the evolution of metazoan embryos. BioEssays 26, 474–478 [DOI] [PubMed] [Google Scholar]
- 6. Daniels D. L., Weis W. I. (2005) β-Catenin directly displaces Groucho/TLE repressors from Tcf/Lef in Wnt-mediated transcription activation. Nat. Struct. Mol. Biol. 12, 364–371 [DOI] [PubMed] [Google Scholar]
- 7. Ding Y., Dale T. (2002) Wnt signal transduction: kinase cogs in a nano-machine? Trends Biochem. Sci. 27, 327–329 [DOI] [PubMed] [Google Scholar]
- 8. Daniels D. L., Eklof Spink K., Weis W. I. (2001) β-Catenin: molecular plasticity and drug design. Trends Biochem. Sci. 26, 672–678 [DOI] [PubMed] [Google Scholar]
- 9. Behrens J., Jerchow B. A., Würtele M., Grimm J., Asbrand C., Wirtz R., Kühl M., Wedlich D., Birchmeier W. (1998) Functional interaction of an axin homolog, conductin, with β-catenin, APC, and GSK3β. Science 280, 596–599 [DOI] [PubMed] [Google Scholar]
- 10. Sharma M., Chuang W. W., Sun Z. (2002) Phosphatidylinositol 3-kinase/Akt stimulates androgen pathway through GSK3β inhibition and nuclear β-catenin accumulation. J. Biol. Chem. 277, 30935–30941 [DOI] [PubMed] [Google Scholar]
- 11. Wu D., Pan W. (2010) GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem. Sci. 35, 161–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Roose J., Molenaar M., Peterson J., Hurenkamp J., Brantjes H., Moerer P., van de Wetering M., Destrée O., Clevers H. (1998) The Xenopus Wnt effector XTcf-3 interacts with Groucho-related transcriptional repressors. Nature 395, 608–612 [DOI] [PubMed] [Google Scholar]
- 13. Roose J., Clevers H. (1999) TCF transcription factors: molecular switches in carcinogenesis. Biochim. Biophys. Acta 1424, M23–M37 [DOI] [PubMed] [Google Scholar]
- 14. Barolo S., Posakony J. W. (2002) Three habits of highly effective signaling pathways: principles of transcriptional control by developmental cell signaling. Genes Dev. 16, 1167–1181 [DOI] [PubMed] [Google Scholar]
- 15. Ross S. E., Hemati N., Longo K. A., Bennett C. N., Lucas P. C., Erickson R. L., MacDougald O. A. (2000) Inhibition of adipogenesis by Wnt signaling. Science 289, 950–953 [DOI] [PubMed] [Google Scholar]
- 16. Bennett C. N., Ross S. E., Longo K. A., Bajnok L., Hemati N., Johnson K. W., Harrison S. D., MacDougald O. A. (2002) Regulation of Wnt signaling during adipogenesis. J. Biol. Chem. 277, 30998–31004 [DOI] [PubMed] [Google Scholar]
- 17. Longo K. A., Wright W. S., Kang S., Gerin I., Chiang S. H., Lucas P. C., Opp M. R., MacDougald O. A. (2004) Wnt10b inhibits development of white and brown adipose tissues. J. Biol. Chem. 279, 35503–35509 [DOI] [PubMed] [Google Scholar]
- 18. Shi Y., Sawada J., Sui G., Affar el B., Whetstine J. R., Lan F., Ogawa H., Luke M. P., Nakatani Y., Shi Y. (2003) Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 422, 735–738 [DOI] [PubMed] [Google Scholar]
- 19. Subramanian T., Chinnadurai G. (2003) Association of class I histone deacetylases with transcriptional corepressor CtBP. FEBS Lett. 540, 255–258 [DOI] [PubMed] [Google Scholar]
- 20. Zhao L. J., Subramanian T., Chinnadurai G. (2006) Changes in C-terminal binding protein 2 (CtBP2) corepressor complex induced by E1A and modulation of E1A transcriptional activity by CtBP2. J. Biol. Chem. 281, 36613–36623 [DOI] [PubMed] [Google Scholar]
- 21. Wang L., Zhou H., Wang Y., Cui G., Di L. J. (2015) CtBP maintains cancer cell growth and metabolic homeostasis via regulating SIRT4. Cell Death Dis. 6, e1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Di L. J., Byun J. S., Wong M. M., Wakano C., Taylor T., Bilke S., Baek S., Hunter K., Yang H., Lee M., Zvosec C., Khramtsova G., Cheng F., Perou C. M., Miller C. R., Raab R., Olopade O. I., Gardner K. (2013) Genome-wide profiles of CtBP link metabolism with genome stability and epithelial reprogramming in breast cancer. Nat. Commun. 4, 1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Di L. J., Fernandez A. G., De Siervi A., Longo D. L., Gardner K. (2010) Transcriptional regulation of BRCA1 expression by a metabolic switch. Nat. Struct. Mol. Biol. 17, 1406–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bajpe P. K., Heynen G. J., Mittempergher L., Grernrum W., de Rink I. A., Nijkamp W., Beijersbergen R. L., Bernards R., Huang S. (2013) The corepressor CTBP2 is a coactivator of retinoic acid receptor/retinoid X receptor in retinoic acid signaling. Mol. Cell. Biol. 33, 3343–3353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Paliwal S., Ho N., Parker D., Grossman S. R. (2012) CtBP2 promotes human cancer cell migration by transcriptional activation of Tiam1. Genes Cancer 3, 481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bhambhani C., Chang J. L., Akey D. L., Cadigan K. M. (2011) The oligomeric state of CtBP determines its role as a transcriptional co-activator and co-repressor of Wingless targets. EMBO J. 30, 2031–2043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stossi F., Madak-Erdogan Z., Katzenellenbogen B. S. (2009) Estrogen receptor α represses transcription of early target genes via p300 and CtBP1. Mol. Cell. Biol. 29, 1749–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim J. H., Cho E. J., Kim S. T., Youn H. D. (2005) CtBP represses p300-mediated transcriptional activation by direct association with its bromodomain. Nat. Struct. Mol. Biol. 12, 423–428 [DOI] [PubMed] [Google Scholar]
- 29. Zhao L. J., Subramanian T., Zhou Y., Chinnadurai G. (2006) Acetylation by p300 regulates nuclear localization and function of the transcriptional corepressor CtBP2. J. Biol. Chem. 281, 4183–4189 [DOI] [PubMed] [Google Scholar]
- 30. Valenta T., Lukas J., Korinek V. (2003) HMG box transcription factor TCF-4's interaction with CtBP1 controls the expression of the Wnt target Axin2/Conductin in human embryonic kidney cells. Nucleic Acids Res. 31, 2369–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brannon M., Brown J. D., Bates R., Kimelman D., Moon R. T. (1999) XCtBP is a XTcf-3 co-repressor with roles throughout Xenopus development. Development 126, 3159–3170 [DOI] [PubMed] [Google Scholar]
- 32. Hamada F., Bienz M. (2004) The APC tumor suppressor binds to C-terminal binding protein to divert nuclear β-catenin from TCF. Dev. Cell 7, 677–685 [DOI] [PubMed] [Google Scholar]
- 33. Sierra J., Yoshida T., Joazeiro C. A., Jones K. A. (2006) The APC tumor suppressor counteracts beta-catenin activation and H3K4 methylation at Wnt target genes. Genes Dev. 20, 586–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhao J., Yue W., Zhu M. J., Sreejayan N., Du M. (2010) AMP-activated protein kinase (AMPK) cross-talks with canonical Wnt signaling via phosphorylation of β-catenin at Ser-552. Biochem. Biophys. Res. Commun. 395, 146–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhao J. X., Yue W. F., Zhu M. J., Du M. (2011) AMP-activated protein kinase regulates β-catenin transcription via histone deacetylase 5. J. Biol. Chem. 286, 16426–16434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Habinowski S. A., Witters L. A. (2001) The effects of AICAR on adipocyte differentiation of 3T3-L1 cells. Biochem. Biophys. Res. Commun. 286, 852–856 [DOI] [PubMed] [Google Scholar]
- 37. Giri S., Rattan R., Haq E., Khan M., Yasmin R., Won J. S., Key L., Singh A. K., Singh I. (2006) AICAR inhibits adipocyte differentiation in 3T3L1 and restores metabolic alterations in diet-induced obesity mice model. Nutr. Metab. (Lond.) 3, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang W., Hong Y. H., Shen X. Q., Frankowski C., Camp H. S., Leff T. (2001) Regulation of transcription by AMP-activated protein kinase: phosphorylation of p300 blocks its interaction with nuclear receptors. J. Biol. Chem. 276, 38341–38344 [DOI] [PubMed] [Google Scholar]
- 39. Wang L., Jia Y., Rogers H., Wu Y. P., Huang S., Noguchi C. T. (2012) GATA-binding protein 4 (GATA-4) and T-cell acute leukemia 1 (TAL1) regulate myogenic differentiation and erythropoietin response via cross-talk with Sirtuin1 (Sirt1). J. Biol. Chem. 287, 30157–30169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cavallo R. A., Cox R. T., Moline M. M., Roose J., Polevoy G. A., Clevers H., Peifer M., Bejsovec A. (1998) Drosophila Tcf and Groucho interact to repress Wingless signalling activity. Nature 395, 604–608 [DOI] [PubMed] [Google Scholar]
- 41. Marchildon F., St-Louis C., Akter R., Roodman V., Wiper-Bergeron N. L. (2010) Transcription factor Smad3 is required for the inhibition of adipogenesis by retinoic acid. J. Biol. Chem. 285, 13274–13284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bergeron R., Ren J. M., Cadman K. S., Moore I. K., Perret P., Pypaert M., Young L. H., Semenkovich C. F., Shulman G. I. (2001) Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am. J. Physiol. Endocrinol. Metab. 281, E1340–E1346 [DOI] [PubMed] [Google Scholar]
- 43. Narkar V. A., Downes M., Yu R. T., Embler E., Wang Y. X., Banayo E., Mihaylova M. M., Nelson M. C., Zou Y., Juguilon H., Kang H., Shaw R. J., Evans R. M. (2008) AMPK and PPARδ agonists are exercise mimetics. Cell 134, 405–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Viollet B., Andreelli F., Jørgensen S. B., Perrin C., Geloen A., Flamez D., Mu J., Lenzner C., Baud O., Bennoun M., Gomas E., Nicolas G., Wojtaszewski J. F., Kahn A., Carling D., Schuit F. C., Birnbaum M. J., Richter E. A., Burcelin R., Vaulont S. (2003) The AMP-activated protein kinase α2 catalytic subunit controls whole-body insulin sensitivity. J. Clin. Invest. 111, 91–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hardie D. G. (2011) AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 25, 1895–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang L., Di L., Noguchi C. T. (2014) AMPK is involved in mediation of erythropoietin influence on metabolic activity and reactive oxygen species production in white adipocytes. Int. J. Biochem. Cell Biol. 54, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang L., Jia Y., Rogers H., Suzuki N., Gassmann M., Wang Q., McPherron A. C., Kopp J. B., Yamamoto M., Noguchi C. T. (2013) Erythropoietin contributes to slow oxidative muscle fiber specification via PGC-1alpha and AMPK activation. Int. J. Biochem. Cell Biol. 45, 1155–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Christodoulides C., Lagathu C., Sethi J. K., Vidal-Puig A. (2009) Adipogenesis and WNT signalling. Trends Endocrinol. Metab. 20, 16–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Prestwich T. C., Macdougald O. A. (2007) Wnt/β-catenin signaling in adipogenesis and metabolism. Curr. Opin. Cell Biol. 19, 612–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Moldes M., Zuo Y., Morrison R. F., Silva D., Park B. H., Liu J., Farmer S. R. (2003) Peroxisome-proliferator-activated receptor γ suppresses Wnt/β-catenin signalling during adipogenesis. Biochem. J. 376, 607–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lee H., Kang R., Bae S., Yoon Y. (2011) AICAR, an activator of AMPK, inhibits adipogenesis via the WNT/β-catenin pathway in 3T3-L1 adipocytes. Int. J. Mol. Med. 28, 65–71 [DOI] [PubMed] [Google Scholar]
- 52. Vingtdeux V., Chandakkar P., Zhao H., Davies P., Marambaud P. (2011) Small-molecule activators of AMP-activated protein kinase (AMPK), RSVA314 and RSVA405, inhibit adipogenesis. Mol. Med. 17, 1022–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhao L. J., Subramanian T., Vijayalingam S., Chinnadurai G. (2007) PLDLS-dependent interaction of E1A with CtBP: regulation of CtBP nuclear localization and transcriptional functions. Oncogene 26, 7544–7551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fang M., Li J., Blauwkamp T., Bhambhani C., Campbell N., Cadigan K. M. (2006) C-terminal-binding protein directly activates and represses Wnt transcriptional targets in Drosophila. EMBO J. 25, 2735–2745 [DOI] [PMC free article] [PubMed] [Google Scholar]