

Abstract
G-protein-coupled receptors (GPCRs) are frequent and fruitful targets for drug discovery and development, as well as being off-targets for the side effects of a variety of medications. Much of the druggable non-olfactory human GPCR-ome remains under-interrogated, and we present here various approaches that we and others have used to shine light into these previously dark corners of the human genome.
Keywords: chemical biology, functional genomics, G-protein-coupled receptor (GPCR), molecular pharmacology, neurological disease, chemical biology, cheminformatics, disease, druggable genome, molecular pharmacology
Introduction
G-protein-coupled receptors (GPCRs;2 7-transmembrane domain receptors) historically have represented both the most abundant and the most popular gene superfamily for therapeutic drug discovery and development, with perhaps 30–40% of approved drugs targeting the non-olfactory GPCRs (1, 2). Typical estimates are that, at any given time, between 20 and 40% of candidate medications target non-olfactory GPCRs as their canonical or principal molecular targets (2–4). Indeed, in 2014, of the 41 new molecular entities approved by the Food and Drug Administration (FDA) (http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugInnovation/ucm20025676.htm), 9 had GPCRs as their canonical sites of action (Table 1). Of the GPCRs targeted by known drugs, the Family A aminergic GPCRs are by far the most popular (2–4) with aripiprazole, which has multiple biogenic amine GPCR targets and a complex mode of action (5), being the bestseller in 2014. As the olfactory GPCRs do not yet represent therapeutic targets, we will focus on the non-olfactory GPCRs here.
TABLE 1.
New molecular entities targeting GPCRs in 2014
MOR, μ-opioid receptor.
| Drug name | GPCR | Indication | Ref. |
|---|---|---|---|
| Tasimelteon | MT1/MT2 melatonin receptor agonist | Non-24-hour sleep-wake disturbance | 60 |
| Droxidopa | Adrenergic GPCRs | Neurogenic orthostatic hypotension | 61, 62 |
| Albiglutide | GLP-1 glucagon-like receptor agonist | Type 2 diabetes | 63, 64 |
| Vorapaxar | PAR-1 thrombin receptor antagonist | Acute chest pain secondary to coronary artery disease | 65 |
| Olodaterol | β2-Adrenergic receptor agonist | Chronic obstructive pulmonary disease | 66 |
| Suvorexant | OX1/2 orexin receptor antagonist | Insomnia | 67 |
| Naloxegol | MOR-opioid receptor antagonist | Opioid-induced constipation | 68 |
| Dulaglutide | GLP-1 glucagon-like receptor agonist | Type 2 diabetes | 69 |
| Netupitant/palonosetron combination | NK1-neurokinin and 5-HT3 receptor antagonists (note 5-HT3 is a ligand-gated ion channel) | Chemotherapy-induced nausea | 70 |
Not only do GPCRs represent the principal therapeutic site of action of many approved and candidate medications, but GPCRs also represent prominent “off-targets” for severe and potentially life-threatening side effects. Of these, drugs with 5-HT2B serotonin receptor agonism have long been documented to induce severe, life-threatening valvular heart disease (6–8). Indeed, based on the potent 5-HT2B agonist activity of certain ergot derivatives used in treating Parkinson disease and migraine headaches (e.g. pergolide, cabergoline, and dihydroergotamine), we correctly predicted that these medications would also induce valvular heart disease (7, 8). Two of these drugs (pergolide and cabergoline) were withdrawn from the international market following large-scale trials demonstrating their life-threating side effects (8, 9). In follow-up studies, we surveyed 2200 FDA-approved and investigational medications, finding that 27 had potentially significant 5-HT2B agonism, of which 6 are currently FDA-approved (guanfacine, quinidine, xylometazoline, oxymetazoline, fenoldopam, and ropinirole) (10). Interestingly, of the 2200 drugs screened, around 30% displayed significant 5-HT2B antagonist activity (10), indicating that 5-HT2B receptors represent a “promiscuous target” for approved and candidate medications. Our discovery that ergotamine and other ergots displayed functional selectivity for β-arrestin over G-protein signaling at 5-HT2B receptors (10) led to the first structure-based explication of GPCR β-arrestin-biased signaling (11). The discovery that the 5-HT2B receptor was responsible for the side effects of the appetite-suppressing medications fenfluramine and dexfenfluramine (6–8) was thus a seminal finding of immense public health importance, which ensures that drugs under development will now be counter-screened against the 5-HT2B receptor for significant agonist activity before being advanced to clinical trials.
Simultaneously with the discovery that the side effects of fenfluramine were due to the 5-HT2B agonist activity of its main metabolite norfenfluramine (6–8), it became clear that its therapeutic (anorectic) actions were due to norfenfluramine's agonist activity at the closely related 5-HT2C receptor (12). This led to the prediction that 5-HT2C-selective agonists devoid of 5-HT2B agonist activity would represent safe and effective appetite suppressants (13) and the discovery of the 5-HT2C-preferring agonist lorcaserin, which was approved by the FDA as the first new obesity medication in nearly 20 years in 2012 (14–16). Taken together, this vignette underscores how an understanding of both on-target and off-target actions of drugs at a single subfamily of GPCRs, in this case the 5-HT2 serotonin receptor family, can be crucial for successful drug discovery efforts.
Chemical Informatics-based Approaches for Genome-wide GPCR-based Discovery
The discovery of small molecule drug-like compounds that interact with GPCRs in a number of ways (e.g. as orthosteric, allosteric, or biased ligands) is now relatively straightforward and will not be reviewed in any detail here as there are a number of excellent and recent review articles (17–19). As these are important concepts for GPCR drug discovery, however, they will be briefly defined. Thus, orthosteric ligands are those that occupy the site(s) of the native or natural ligand, whereas allosteric ligands occupy a site distinct from the orthosteric site (18, 19). Additionally, it is now appreciated that GPCRs signal via β-arrestin and that this signaling is frequently independent of canonical G-protein modes of signaling (17). Indeed, drugs that preferentially signal via β-arrestin are considered to be β-arrestin-biased (17). For the remainder of the review, we will focus on genome-wide approaches for GPCR-based discovery, highlighting both in silico and physical screening approaches for the discovery of novel small drug-like small molecules acting at GPCRs.
In silico approaches for discovering GPCR modulators typically take advantage of large chemical databases that annotate the biological properties of small molecules. Table 2 lists a few of the more popular and widely used databases. Essentially, these databases have large lists of chemical compound names and, typically, their chemical descriptors along with the biological activity associated with these compounds. Most commonly, as in the ChEMBL database and PDSP Ki database (KiDB), which rely mainly on published data, the activity is encoded as a Ki or EC50 value, whereas other databases (e.g. ChemBank and PubChem) provide the raw data as well as fitted data parameters. Utilizing the information from such databases, we and our collaborators have successfully predicted novel GPCR targets for known drugs (3, 20, 21) and have designed novel drugs targeting GPCRs entirely in silico (22). Importantly, in these exemplars of this overall approach, the GPCR-centric predictions were extensively validated both in vitro and in vivo in model organisms such as worms (23), zebrafish (24), mice (3, 22), and most remarkably, in humans (21).
TABLE 2.
Useful cheminformatic, chemical biology, and pharmacology databases
| Database acronyma | URL | Typeb | Ref. | Downloadablec |
|---|---|---|---|---|
| ChEMBL | https://www.ebi.ac.uk/chembl/ | C, T, ID | 71 | Y |
| KiDB; PDSP Ki Database | http://pdsp.med.unc.edu/kidb.php | C, T, ID (limited) | 72 | Y |
| Chembank | http://chembank.broadinstitute.org/ | C, T, ID | 73 | Y in part |
| SEA; Similarity Ensemble Approach | http://sea.bkslab.org/ | C, T, ID | 3, 20 | N but predictions can be made |
| PubChem | https://pubchem.ncbi.nlm.nih.gov/ | C, T, ID | 74 | Y |
| IUPHAR GPCR Database | http://www.guidetopharmacology.org/GRAC/ReceptorFamiliesForward?type=GPCR | C, T | Y |
a Where applicable, name comes after semicolon.
b C, chemical; T, target; ID, chemical identifiers.
c Y = yes; N = no.
All of these resources rely upon accurately curated, precise data and, of the cited resources, ChEMBL and KiDB would appear to be the most useful as the main source of their data is from peer-reviewed publications. ChEMBL historically has drawn its data from medicinal chemistry publications, although the most recent version of ChEMBL also incorporates large amounts of data from PubChem. KiDB obtains its data mainly from non-medicinal chemistry publications (e.g. biochemistry, cell biology, pharmacology, neuroscience, and so on). Examining ChEMBL, which is the largest of these resources, we find that a large number of GPCR targets are under-annotated with respect to both their biological function and the chemical matter with which they may interact (Fig. 1, A and B). As can be seen, at least 50% of the non-olfactory GPCRs in the human genome have had few publications associated with them based on a search of PubMed conducted in mid-2013. Additionally, more than 50% of the non-olfactory GPCRs in the human genome had few annotated small molecules (Table 2; GPCR Safari ChEMBL release 3.0). Indeed, of the 159 “orphan” GPCRs in the ChEMBL database, only 5 had annotated small molecules with documented bioactivity. Significantly, although ChEMBL is a curated database, it misidentifies the synthetic ligand 3-{4-[4-(2-cyanophenyl)-1-piperazinyl]butyl}-1H-indole-5-carboxamide as the natural ligand for GPR35 (https://www.ebi.ac.uk/chembl/sarfari/gpcrsarfari/report/protein/266), even though kynurenic acid has been proposed as a naturally occurring ligand for GPR35 (25, 26). This example of GPR35 being mis-annotated illustrates three important points: first, the need for careful expert curation; second, the fact that all of these databases contain a significant number of errors that could lead investigators astray; and third, the value of orthogonal (i.e. assays for which the readouts are independent) assays to validate “hits” and presumed active compounds.
FIGURE 1.

Many GPCRs are understudied. A, shown is a graph of PubMed publications for the non-olfactory GPCRs. For this graph, the time period studied was up to and including August 2013. For searching for publications referencing a particular GPCR, either the International Union of Basic and Clinical Pharmacology (IUPHAR)-approved name or the genome identifier was used, depending upon which resulted in the largest number of publications. B, many GPCRs have no identified chemical modulators. Shown is a graph of CheMBL compounds culled from GPCR SARfari (https://www.ebi.ac.uk/chembl/sarfari/gpcrsarfari) using version 3.00.
In Fig. 2, we show that most of the non-olfactory human GPCR-ome is un-interrogated with respect to the chemical matter as annotated in ChEMBL. The practical impact of this is that, when using a database such as ChEMBL for predicting on- and off-target actions of small molecules, most of the GPCR-ome is hidden from a cheminformatics perspective. GPCRs are not unique in that most of them are understudied, as a similar conclusion was reached for kinases a few years ago (27). Indeed, Isserlin et al. (28) have described what they have dubbed the “Harlow-Knapp (H-K) effect,” which they define as: “the propensity of the biomedical and pharmaceutical research communities to focus their activities, as quantified by the number of publications and patents, on a small fraction of the proteome.” Isserlin and colleagues (27, 28) noted that this was true for the targets they studied (kinases, nuclear hormone receptors, and ion channels) irrespective of whether they confined their bibliographic analysis to the “pre-genomic era” (i.e. prior to the publication of the draft human genome in 2000) or later dates (i.e. 2009). We performed a different type of analysis and re-interrogated the publication records for the druggable, non-olfactory GPCRs in 2014, and compared this with all publications predating 2013. As shown in Fig. 3, there was a similar although not identical trend, with most of the understudied GPCRs still being understudied and the more popular GPCRs continuing this trend.
FIGURE 2.

The GPCR tree is (mainly) un-interrogated. Shown in tree-based format are the data from Fig. 1B plotted on the GPCR tree (courtesy of V. Katritch and R. C. Stevens—Scripps/USC).
FIGURE 3.
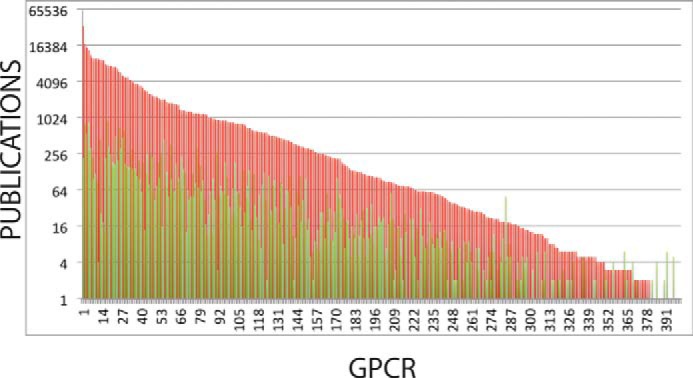
Understudied GPCRs continue to be understudied. Shown is a comparison of publications for individual GPCRs up to August 2013 (red) and for the entire year of 2014 (green; analysis performed March 2015). The vertical axis (number of publications) here is shown in log2 format to include both datasets on the same graph.
For resources available to interrogate GPCRs from a chemical standpoint, such as PubChem and ChemBank, these databases will essentially supply raw screening data with (in many but not all instances) confirmatory concentration-response curves from which estimates of potency and efficacy are derived. For example, PubChem lists screens for a large number of GPCRs and, from these screens, results for a handful of orphan GPCRs have been published in peer-reviewed journals (29–31). These published findings have led to the discovery that pamoic acid is a potent agonist for GPR35 via β-arrestin signaling (31), as well as the discovery of novel agonists and antagonists for GPR55 (29, 30).
As should be clear from the foregoing, cheminformatics-based approaches can be quite useful for predicting GPCR targets for both known drugs and other small, perhaps drug-like, molecules. Because the bulk of the GPCR-ome is relatively uncharted territory, i.e. because very few drug-like small molecules have been identified for a large number of human GPCRs, such studies are necessarily and unavoidably underpowered.
Physical Approaches for Interrogating the GPCR-ome
In the past, we and others have used both radioligand binding and functional assays to elucidate the ligand-based pharmacology of non-orphan GPCRs. This approach, which we dubbed “receptorome screening,” and which has been extensively described in prior reviews (17, 32–34), has led us to a number of important discoveries including: the identification of the κ-opioid receptor as the site of action of the widely abused hallucinogen salvinorin A (35); the discovery that the 5-HT2B serotonin receptor is the valvulopathy receptor (6); identification of the remarkably complex pharmacology of antipsychotic drugs (36); large-scale validation of cheminformatics predictions (3, 22); identification of GPCR as high affinity off-targets of kinase inhibitors (37–39); and large-scale validation of computationally docked and crystallography-confirmed binding poses (11, 40–48)
As radioligand-based approaches require radioligands with high specific activity and high affinity for their targets, they are not useful for the vast majority of GPCRs, for which such radioligands are unavailable. Additionally, the physical, informatics, and infrastructure requirements required to routinely screen more than a few GPCRs simultaneously using radioligand binding assays are beyond the resources of most academic and industrial laboratories. Fortunately, the National Institute of Mental Health's Psychoactive Drug Screening Program (NIMH-PDSP), which is housed in the authors' laboratory, provides screening as a free service to not-for-profit investigators, thereby making this resource available to a large part of the scientific community. Indeed, in the past 5 years, more than 500 investigators world-wide took advantage of the NIMH-PDSP for GPCR profiling of novel and candidate drug-like small molecules.
Functional screening methods are an alternative to radioligand binding-based approaches. Unfortunately, there are currently no published approaches suitable for interrogating the entire olfactory and non-olfactory GPCR-ome. Indeed, screening the entire druggable GPCR-ome is technically challenging due to the diverse G-protein-mediated signaling cascades used by GPCRs (e.g. Gs, Gi, Gq, or G12/13). In the past, forced coupling of Gs, Gi, and G12/13 G-proteins to a Gq-like Ca2+ readout has been frequently used (49, 50) to identify ligands for orphan and/or sparsely annotated GPCRs (17, 51). Approaches that rely on native coupling to known G-proteins have been successful in identifying novel and selective ligands for orphan GPCRs (52). Additionally, many GPCRs couple to G12 and G13. Interestingly, the G12/13-dependent shedding of a membrane-bound reporter protein (53) has been reported as a potential “universal” approach for both orphan and non-orphan GPCRs.
Other approaches have relied on platforms that take advantage of G-protein-independent β-arrestin recruitment because nearly all GPCRs induce arrestin translocation (54). Many methods have emerged to quantify GPCR-β-arrestin interactions, including high content screening (HCS) (55), bioluminescence resonance energy transfer (BRET) (56), and transcriptional activation following arrestin translocation (TANGO) (57). We have found the TANGO-based approach to be quite useful for chemical interrogation of GPCRs (11, 40, 43, 58, 59). Indeed, we have recently devised a genome-wide approach using a TANGO-based readout to screen nearly all of the druggable GPCR-ome in a facile, simultaneous, and parallel manner (47).
Conclusions and Recommendations
As we have shown, although GPCRs represent a useful and important target class for therapeutic drug discovery and biochemical study, most are under-interrogated. In part, this stems from the lack of robust and scalable ways to assess their activities. New technological platforms are becoming available that allow for unbiased interrogation of the druggable GPCR-ome (47), and when these are made freely available, they will likely begin to have a transformative effect on the study of GPCRs. Additionally, because of the “Harlow-Knapp effect,” many GPCRs will likely remain understudied despite their potential importance from both a basic science as well as a translational perspective.
Author Contributions
B. L. R. and W. K. K. conceived and wrote the paper. B. L. R. and W. K. K. performed the bibliographic analysis. Both authors approved the results and the final version of the manuscript.
Acknowledgments
We thank Seva Katritch and Ray Stevens for the use of an unannotated version of their GPCR tree.
This work was supported by National Institutes of Health Grants U01MH104974, U19MH82441, and RO1MH61887 and the NIMH Psychoactive Drug Screening Program Contract (to B. L. R.). This is the first article in the Thematic Minireview series “New Directions in G Protein-coupled Receptor Pharmacology.” The authors declare that they have no conflicts of interest with the contents of this article.
- GPCR
- G-protein-coupled receptor
- 5-HT2B
- 5-hydroxytryptamine receptor 2B
- TANGO
- transcriptional activation following arrestin translocation.
REFERENCES
- 1. Hopkins A. L., Groom C. R. (2002) The druggable genome. Nat. Rev. Drug. Discov. 1, 727–730 [DOI] [PubMed] [Google Scholar]
- 2. Overington J. P., Al-Lazikani B., Hopkins A. L. (2006) How many drug targets are there? Nat. Rev. Drug. Discov. 5, 993–996 [DOI] [PubMed] [Google Scholar]
- 3. Keiser M. J., Setola V., Irwin J. J., Laggner C., Abbas A. I., Hufeisen S. J., Jensen N. H., Kuijer M. B., Matos R. C., Tran T. B., Whaley R., Glennon R. A., Hert J., Thomas K. L., Edwards D. D., Shoichet B. K., Roth B. L. (2009) Predicting new molecular targets for known drugs. Nature 462, 175–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rask-Andersen M., Almén M. S., Schiöth H. B. (2011) Trends in the exploitation of novel drug targets. Nat. Rev. Drug. Discov. 10, 579–590 [DOI] [PubMed] [Google Scholar]
- 5. Shapiro D. A., Renock S., Arrington E., Chiodo L. A., Liu L. X., Sibley D. R., Roth B. L., Mailman R. (2003) Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology 28, 1400–1411 [DOI] [PubMed] [Google Scholar]
- 6. Rothman R. B., Baumann M. H., Savage J. E., Rauser L., McBride A., Hufeisen S. J., Roth B. L. (2000) Evidence for possible involvement of 5-HT2B receptors in the cardiac valvulopathy associated with fenfluramine and other serotonergic medications. Circulation 102, 2836–2841 [DOI] [PubMed] [Google Scholar]
- 7. Setola V., Hufeisen S. J., Grande-Allen K. J., Vesely I., Glennon R. A., Blough B., Rothman R. B., Roth B. L. (2003) 3,4-Methylenedioxymethamphetamine (MDMA, “Ecstasy”) induces fenfluramine-like proliferative actions on human cardiac valvular interstitial cells in vitro. Mol. Pharmacol. 63, 1223–1229 [DOI] [PubMed] [Google Scholar]
- 8. Roth B. L. (2007) Drugs and valvular heart disease. N. Engl. J. Med. 356, 6–9 [DOI] [PubMed] [Google Scholar]
- 9. Zanettini R., Antonini A., Gatto G., Gentile R., Tesei S., Pezzoli G. (2007) Valvular heart disease and the use of dopamine agonists for Parkinson's disease. N. Engl. J. Med. 356, 39–46 [DOI] [PubMed] [Google Scholar]
- 10. Huang X. P., Setola V., Yadav P. N., Allen J. A., Rogan S. C., Hanson B. J., Revankar C., Robers M., Doucette C., Roth B. L. (2009) Parallel functional activity profiling reveals valvulopathogens are potent 5-hydroxytryptamine2B receptor agonists: implications for drug safety assessment. Mol. Pharmacol. 76, 710–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wacker D., Wang C., Katritch V., Han G. W., Huang X. P., Vardy E., McCorvy J. D., Jiang Y., Chu M., Siu F. Y., Liu W., Xu H. E., Cherezov V., Roth B. L., Stevens R. C. (2013) Structural features for functional selectivity at serotonin receptors. Science 340, 615–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vickers S. P., Clifton P. G., Dourish C. T., Tecott L. H. (1999) Reduced satiating effect of d-fenfluramine in serotonin 5-HT2C receptor mutant mice. Psychopharmacology 143, 309–314 [DOI] [PubMed] [Google Scholar]
- 13. O'Connor K. A., Roth B. L. (2005) Finding new tricks for old drugs: an efficient route for public-sector drug discovery. Nat. Rev. Drug. Discov. 4, 1005–1014 [DOI] [PubMed] [Google Scholar]
- 14. Meltzer H. Y., Roth B. L. (2013) Lorcaserin and pimavanserin: emerging selectivity of serotonin receptor subtype-targeted drugs. J. Clin. Invest. 123, 4986–4991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thomsen W. J., Grottick A. J., Menzaghi F., Reyes-Saldana H., Espitia S., Yuskin D., Whelan K., Martin M., Morgan M., Chen W., Al-Shamma H., Smith B., Chalmers D., Behan D. (2008) Lorcaserin, a novel selective human 5-hydroxytryptamine2C agonist: in vitro and in vivo pharmacological characterization. J. Pharmacol. Exp. Ther. 325, 577–587 [DOI] [PubMed] [Google Scholar]
- 16. Smith S. R., Weissman N. J., Anderson C. M., Sanchez M., Chuang E., Stubbe S., Bays H., Shanahan W. R., and the Behavioral Modification and Lorcaserin for Overweight and Obesity Management (BLOOM) Study Group (2010) Multicenter, placebo-controlled trial of lorcaserin for weight management. N. Engl. J. Med. 363, 245–256 [DOI] [PubMed] [Google Scholar]
- 17. Allen J. A., Roth B. L. (2011) Strategies to discover unexpected targets for drugs active at G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 51, 117–144 [DOI] [PubMed] [Google Scholar]
- 18. Wootten D., Christopoulos A., Sexton P. M. (2013) Emerging paradigms in GPCR allostery: implications for drug discovery. Nat. Rev. Drug. Discov. 12, 630–644 [DOI] [PubMed] [Google Scholar]
- 19. Violin J. D., Crombie A. L., Soergel D. G., Lark M. W. (2014) Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol. Sci. 35, 308–316 [DOI] [PubMed] [Google Scholar]
- 20. Keiser M. J., Roth B. L., Armbruster B. N., Ernsberger P., Irwin J. J., Shoichet B. K. (2007) Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 25, 197–206 [DOI] [PubMed] [Google Scholar]
- 21. Lounkine E., Keiser M. J., Whitebread S., Mikhailov D., Hamon J., Jenkins J. L., Lavan P., Weber E., Doak A. K., Côté S., Shoichet B. K., Urban L. (2012) Large-scale prediction and testing of drug activity on side-effect targets. Nature 486, 361–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Besnard J., Ruda G. F., Setola V., Abecassis K., Rodriguiz R. M., Huang X. P., Norval S., Sassano M. F., Shin A. I., Webster L. A., Simeons F. R., Stojanovski L., Prat A., Seidah N. G., Constam D. B., Bickerton G. R., Read K. D., Wetsel W. C., Gilbert I. H., Roth B. L., Hopkins A. L. (2012) Automated design of ligands to polypharmacological profiles. Nature 492, 215–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lemieux G. A., Keiser M. J., Sassano M. F., Laggner C., Mayer F., Bainton R. J., Werb Z., Roth B. L., Shoichet B. K., Ashrafi K. (2013) In silico molecular comparisons of C. elegans and mammalian pharmacology identify distinct targets that regulate feeding. PLoS Biol. 11, e1001712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Laggner C., Kokel D., Setola V., Tolia A., Lin H., Irwin J. J., Keiser M. J., Cheung C. Y., Minor D. L., Jr., Roth B. L., Peterson R. T., Shoichet B. K. (2012) Chemical informatics and target identification in a zebrafish phenotypic screen. Nat. Chem. Biol. 8, 144–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang J., Simonavicius N., Wu X., Swaminath G., Reagan J., Tian H., Ling L. (2006) Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J. Biol. Chem. 281, 22021–22028 [DOI] [PubMed] [Google Scholar]
- 26. Jenkins L., Alvarez-Curto E., Campbell K., de Munnik S., Canals M., Schlyer S., Milligan G. (2011) Agonist activation of the G protein-coupled receptor GPR35 involves transmembrane domain III and is transduced via Gα13 and β-arrestin-2. Br. J. Pharmacol. 162, 733–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Edwards A. M., Isserlin R., Bader G. D., Frye S. V., Willson T. M., Yu F. H. (2011) Too many roads not taken. Nature 470, 163–165 [DOI] [PubMed] [Google Scholar]
- 28. Isserlin R., Bader G. D., A., E., Frye S., Willson T., Yu F. (2011) The human genome and drug discovery after a decade: roads (still) not taken. arXiv:1102.0448 [Google Scholar]
- 29. Kotsikorou E., Sharir H., Shore D. M., Hurst D. P., Lynch D. L., Madrigal K. E., Heynen-Genel S., Milan L. B., Chung T. D., Seltzman H. H., Bai Y., Caron M. G., Barak L. S., Croatt M. P., Abood M. E., Reggio P. H. (2013) Identification of the GPR55 antagonist binding site using a novel set of high-potency GPR55 selective ligands. Biochemistry 52, 9456–9469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kotsikorou E., Madrigal K. E., Hurst D. P., Sharir H., Lynch D. L., Heynen-Genel S., Milan L. B., Chung T. D., Seltzman H. H., Bai Y., Caron M. G., Barak L., Abood M. E., Reggio P. H. (2011) Identification of the GPR55 agonist binding site using a novel set of high-potency GPR55 selective ligands. Biochemistry 50, 5633–5647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao P., Sharir H., Kapur A., Cowan A., Geller E. B., Adler M. W., Seltzman H. H., Reggio P. H., Heynen-Genel S., Sauer M., Chung T. D., Bai Y., Chen W., Caron M. G., Barak L. S., Abood M. E. (2010) Targeting of the orphan receptor GPR35 by pamoic acid: a potent activator of extracellular signal-regulated kinase and β-arrestin2 with antinociceptive activity. Mol. Pharmacol. 78, 560–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Armbruster B. N., Roth B. L. (2005) Mining the receptorome. J. Biol. Chem. 280, 5129–5132 [DOI] [PubMed] [Google Scholar]
- 33. Jensen N. H., Roth B. L. (2008) Massively parallel screening of the receptorome. Comb. Chem. High Throughput Screen. 11, 420–426 [DOI] [PubMed] [Google Scholar]
- 34. Strachan R. T., Ferrara G., Roth B. L. (2006) Screening the receptorome: an efficient approach for drug discovery and target validation. Drug Discov. Today 11, 708–716 [DOI] [PubMed] [Google Scholar]
- 35. Roth B. L., Baner K., Westkaemper R., Siebert D., Rice K. C., Steinberg S., Ernsberger P., Rothman R. B. (2002) Salvinorin A: a potent naturally occurring nonnitrogenous κ-opioid selective agonist. Proc. Natl. Acad. Sci. U.S.A. 99, 11934–11939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roth B. L., Sheffler D. J., Kroeze W. K. (2004) Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat. Rev. Drug. Discov. 3, 353–359 [DOI] [PubMed] [Google Scholar]
- 37. Lin X., Huang X. P., Chen G., Whaley R., Peng S., Wang Y., Zhang G., Wang S. X., Wang S., Roth B. L., Huang N. (2012) Life beyond kinases: structure-based discovery of sorafenib as nanomolar antagonist of 5-HT receptors. J. Med. Chem. 55, 5749–5759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lin H., Sassano M. F., Roth B. L., Shoichet B. K. (2013) A pharmacological organization of G protein-coupled receptors. Nat. Methods 10, 140–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gregori-Puigjané E., Setola V., Hert J., Crews B. A., Irwin J. J., Lounkine E., Marnett L., Roth B. L., Shoichet B. K. (2012) Identifying mechanism-of-action targets for drugs and probes. Proc. Natl. Acad. Sci. U.S.A. 109, 11178–11183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Carlsson J., Coleman R. G., Setola V., Irwin J. J., Fan H., Schlessinger A., Sali A., Roth B. L., Shoichet B. K. (2011) Ligand discovery from a dopamine D3 receptor homology model and crystal structure. Nat. Chem. Biol. 7, 769–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Weiss D. R., Ahn S., Sassano M. F., Kleist A., Zhu X., Strachan R., Roth B. L., Lefkowitz R. J., Shoichet B. K. (2013) Conformation guides molecular efficacy in docking screens of activated β-2 adrenergic G protein coupled receptor. ACS Chem. Biol. 8, 1018–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang C., Wu H., Evron T., Vardy E., Han G. W., Huang X. P., Hufeisen S. J., Mangano T. J., Urban D. J., Katritch V., Cherezov V., Caron M. G., Roth B. L., Stevens R. C. (2014) Structural basis for Smoothened receptor modulation and chemoresistance to anticancer drugs. Nat. Commun. 5, 4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu H., Wacker D., Mileni M., Katritch V., Han G. W., Vardy E., Liu W., Thompson A. A., Huang X. P., Carroll F. I., Mascarella S. W., Westkaemper R. B., Mosier P. D., Roth B. L., Cherezov V., Stevens R. C. (2012) Structure of the human κ-opioid receptor in complex with JDTic. Nature 485, 327–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang C., Wu H., Katritch V., Han G. W., Huang X. P., Liu W., Siu F. Y., Roth B. L., Cherezov V., Stevens R. C. (2013) Structure of the human smoothened receptor bound to an antitumour agent. Nature 497, 338–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang C., Jiang Y., Ma J., Wu H., Wacker D., Katritch V., Han G. W., Liu W., Huang X. P., Vardy E., McCorvy J. D., Gao X., Zhou X. E., Melcher K., Zhang C., Bai F., Yang H., Yang L., Jiang H., Roth B. L., Cherezov V., Stevens R. C., Xu H. E. (2013) Structural basis for molecular recognition at serotonin receptors. Science 340, 610–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vardy E., Mosier P. D., Frankowski K. J., Wu H., Katritch V., Westkaemper R. B., Aubé J., Stevens R. C., Roth B. L. (2013) Chemotype-selective modes of action of κ-opioid receptor agonists. J. Biol. Chem. 288, 34470–34483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kroeze W. K., Sassano M. F., Huang X. P., Lansu K., McCorvy J. D., Giguère P. M., Sciaky N., Roth B. L. (2015) PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCR-ome. Nat. Struct. Mol. Biol. 22, 362–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Thompson A. A., Liu W., Chun E., Katritch V., Wu H., Vardy E., Huang X. P., Trapella C., Guerrini R., Calo G., Roth B. L., Cherezov V., Stevens R. C. (2012) Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 485, 395–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Coward P., Chan S. D., Wada H. G., Humphries G. M., Conklin B. R. (1999) Chimeric G proteins allow a high-throughput signaling assay of Gi-coupled receptors. Anal. Biochem. 270, 242–248 [DOI] [PubMed] [Google Scholar]
- 50. Coward P., Wada H. G., Falk M. S., Chan S. D., Meng F., Akil H., Conklin B. R. (1998) Controlling signaling with a specifically designed Gi-coupled receptor. Proc. Natl. Acad. Sci. U.S.A. 95, 352–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Beets I., Lindemans M., Janssen T., Verleyen P. (2011) Deorphanizing G protein-coupled receptors by a calcium mobilization assay. Methods Mol. Biol. 789, 377–391 [DOI] [PubMed] [Google Scholar]
- 52. Jin C., Decker A. M., Huang X. P., Gilmour B. P., Blough B. E., Roth B. L., Hu Y., Gill J. B., Zhang X. P. (2014) Synthesis, pharmacological characterization, and structure-activity relationship studies of small molecular agonists for the orphan GPR88 receptor. ACS Chem. Neurosci. 5, 576–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Inoue A., Ishiguro J., Kitamura H., Arima N., Okutani M., Shuto A., Higashiyama S., Ohwada T., Arai H., Makide K., Aoki J. (2012) TGFα shedding assay: an accurate and versatile method for detecting GPCR activation. Nat. Methods 9, 1021–1029 [DOI] [PubMed] [Google Scholar]
- 54. Lefkowitz R. J., Shenoy S. K. (2005) Transduction of receptor signals by β-arrestins. Science 308, 512–517 [DOI] [PubMed] [Google Scholar]
- 55. Barak L. S., Warabi K., Feng X., Caron M. G., Kwatra M. M. (1999) Real-time visualization of the cellular redistribution of G protein-coupled receptor kinase 2 and β-arrestin 2 during homologous desensitization of the substance P receptor. J. Biol. Chem. 274, 7565–7569 [DOI] [PubMed] [Google Scholar]
- 56. Angers S., Salahpour A., Joly E., Hilairet S., Chelsky D., Dennis M., Bouvier M. (2000) Detection of β2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET). Proc. Natl. Acad. Sci. U.S.A. 97, 3684–3689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Barnea G., Strapps W., Herrada G., Berman Y., Ong J., Kloss B., Axel R., Lee K. J. (2008) The genetic design of signaling cascades to record receptor activation. Proc. Natl. Acad. Sci. U.S.A. 105, 64–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. White K. L., Scopton A. P., Rives M. L., Bikbulatov R. V., Polepally P. R., Brown P. J., Kenakin T., Javitch J. A., Zjawiony J. K., Roth B. L. (2014) Identification of novel functionally selective κ-opioid receptor scaffolds. Mol. Pharmacol. 85, 83–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fenalti G., Giguere P. M., Katritch V., Huang X. P., Thompson A. A., Cherezov V., Roth B. L., Stevens R. C. (2014) Molecular control of δ-opioid receptor signalling. Nature 506, 191–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rajaratnam S. M., Polymeropoulos M. H., Fisher D. M., Roth T., Scott C., Birznieks G., Klerman E. B. (2009) Melatonin agonist tasimelteon (VEC-162) for transient insomnia after sleep-time shift: two randomised controlled multicentre trials. Lancet 373, 482–491 [DOI] [PubMed] [Google Scholar]
- 61. Bartholini J., Constantinidis J., Puig M., Tissot R., Pletscher A. (1975) The stereoisomers of 3,4-dihydroxyphenylserine as precursors of norepinephrine. J. Pharmacol. Exp. Ther. 193, 523–532 [PubMed] [Google Scholar]
- 62. Kaufmann H., Freeman R., Biaggioni I., Low P., Pedder S., Hewitt L. A., Mauney J., Feirtag M., Mathias C., on behalf of NOH301 Investigators (2014) Droxidopa for neurogenic orthostatic hypotension: a randomized, placebo-controlled, phase 3 trial. Neurology 83, 328–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bush M. A., Matthews J. E., De Boever E. H., Dobbins R. L., Hodge R. J., Walker S. E., Holland M. C., Gutierrez M., Stewart M. W. (2009) Safety, tolerability, pharmacodynamics and pharmacokinetics of albiglutide, a long-acting glucagon-like peptide-1 mimetic, in healthy subjects. Diabetes Obes. Metab. 11, 498–505 [DOI] [PubMed] [Google Scholar]
- 64. Matthews J. E., Stewart M. W., De Boever E. H., Dobbins R. L., Hodge R. J., Walker S. E., Holland M. C., Bush M. A., and the Albiglutide Study Group (2008) Pharmacodynamics, pharmacokinetics, safety, and tolerability of albiglutide, a long-acting glucagon-like peptide-1 mimetic, in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 93, 4810–4817 [DOI] [PubMed] [Google Scholar]
- 65. Becker R. C., Moliterno D. J., Jennings L. K., Pieper K. S., Pei J., Niederman A., Ziada K. M., Berman G., Strony J., Joseph D., Mahaffey K. W., Van de Werf F., Veltri E., Harrington R. A., and TRA-PCI Investigators (2009) Safety and tolerability of SCH 530348 in patients undergoing non-urgent percutaneous coronary intervention: a randomised, double-blind, placebo-controlled phase II study. Lancet 373, 919–928 [DOI] [PubMed] [Google Scholar]
- 66. Bouyssou T., Casarosa P., Naline E., Pestel S., Konetzki I., Devillier P., Schnapp A. (2010) Pharmacological characterization of olodaterol, a novel inhaled β2-adrenoceptor agonist exerting a 24-hour-long duration of action in preclinical models. J. Pharmacol. Exp. Ther. 334, 53–62 [DOI] [PubMed] [Google Scholar]
- 67. Cox C. D., Breslin M. J., Whitman D. B., Schreier J. D., McGaughey G. B., Bogusky M. J., Roecker A. J., Mercer S. P., Bednar R. A., Lemaire W., Bruno J. G., Reiss D. R., Harrell C. M., Murphy K. L., Garson S. L., Doran S. M., Prueksaritanont T., Anderson W. B., Tang C., Roller S., Cabalu T. D., Cui D., Hartman G. D., Young S. D., Koblan K. S., Winrow C. J., Renger J. J., Coleman P. J. (2010) Discovery of the dual orexin receptor antagonist [(7R)-4-(5-chloro-1,3-benzoxazol-2-yl)-7-methyl-1,4-diazepan-1-yl][5-methyl-2-(2H-1,2,3-triazol-2-yl)phenyl]methanone (MK-4305) for the treatment of insomnia. J. Med. Chem. 53, 5320–5332 [DOI] [PubMed] [Google Scholar]
- 68. Chey W. D., Webster L., Sostek M., Lappalainen J., Barker P. N., Tack J. (2014) Naloxegol for opioid-induced constipation in patients with noncancer pain. N. Engl. J. Med. 370, 2387–2396 [DOI] [PubMed] [Google Scholar]
- 69. Glaesner W., Vick A. M., Millican R., Ellis B., Tschang S. H., Tian Y., Bokvist K., Brenner M., Koester A., Porksen N., Etgen G., Bumol T. (2010) Engineering and characterization of the long-acting glucagon-like peptide-1 analogue LY2189265, an Fc fusion protein. Diabetes Metab. Res. Rev. 26, 287–296 [DOI] [PubMed] [Google Scholar]
- 70. Gralla R. J., Bosnjak S. M., Hontsa A., Balser C., Rizzi G., Rossi G., Borroni M. E., Jordan K. (2014) A phase III study evaluating the safety and efficacy of NEPA, a fixed-dose combination of netupitant and palonosetron, for prevention of chemotherapy-induced nausea and vomiting over repeated cycles of chemotherapy. Ann. Oncol. 25, 1333–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gaulton A., Bellis L. J., Bento A. P., Chambers J., Davies M., Hersey A., Light Y., McGlinchey S., Michalovich D., Al-Lazikani B., Overington J. P. (2012) ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 40, D1100–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Roth B. L., Lopez E., Patel S., Kroeze W. K. (2000) The multiplicity of serotonin receptors: uselessly diverse molecules or an embarrassment of riches? Neuroscientist 6, 252–262, 10.1177/107385840000600408 [DOI] [Google Scholar]
- 73. Seiler K. P., George G. A., Happ M. P., Bodycombe N. E., Carrinski H. A., Norton S., Brudz S., Sullivan J. P., Muhlich J., Serrano M., Ferraiolo P., Tolliday N. J., Schreiber S. L., Clemons P. A. (2008) ChemBank: a small-molecule screening and cheminformatics resource database. Nucleic Acids Res. 36, D351–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wang Y., Xiao J., Suzek T. O., Zhang J., Wang J., Bryant S. H. (2009) PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 37, W623–633 [DOI] [PMC free article] [PubMed] [Google Scholar]