

Abstract
A number of recent technical solutions have led to significant advances in G protein-coupled receptor (GPCR) structural biology. Apart from a detailed mechanistic view of receptor activation, the new structures have revealed novel ligand binding sites. Together, these insights provide avenues for rational drug design to modulate the activities of these important drug targets. The application of structural data to GPCR drug discovery ushers in an exciting era with the potential to improve existing drugs and discover new ones. In this review, we focus on technical solutions that have accelerated GPCR crystallography as well as some of the salient findings from structures that are relevant to drug discovery. Finally, we outline some of the approaches used in GPCR structure based drug design.
Keywords: drug design, drug discovery, G protein-coupled receptor (GPCR), membrane protein, X-ray crystallography
Introduction
The process of drug discovery from bench to market is a high risk long term investment that has been estimated to cost about $1.8 billion per drug (1). Such high costs coupled with the pressure of patent expiry dates and increased regulatory constraints have propelled the pharmaceutical industry to increase efficiency of research and development to reduce the attrition rate. A key area of focus has been improvements in the quality of compounds that are discovered in the early stages of the drug discovery pipeline. The main driver for this effort has been the general observation that the hits identified from cell-based high throughput screening (HTS)2 strategies are usually large lipophilic molecules that are difficult to optimize and carry a number of liabilities that significantly increase their failure rate (2). One of the main advances to tackle these issues has been the utilization of fragment-based drug discovery (FBDD) approaches that rely on screening small chemical fragments (100–250 Da). Because of their small size, a significantly larger portion of chemical space can be explored with fewer compounds when compared with HTS. In addition, initial hits from a fragment screen bind more efficiently to their target and represent excellent starting points for medicinal chemists to grow and optimize these into lead and candidate molecules (3). The initial fragment hits exhibit low affinity, so they need to be screened at high concentrations that make them incompatible with biological assays. Instead, biophysical assays are used in FBDD cascades, and in addition, these approaches are combined with structural information derived primarily from x-ray crystallography. The application of structure-based drug design (SBDD) allows medicinal chemists to rationally convert fragment hits into larger compounds with higher affinity while maintaining the efficiency of binding and drug-like properties. Historically, SBDD methods gained traction with soluble proteins such as enzymes as routine generation of structural data is facilitated by their high stability in purified form (4–6). This is in sharp contrast to membrane proteins such as G protein-coupled receptors (GPCRs) that have been refractory to SBDD due to the challenges associated with obtaining high quality crystals. The main problems in the field of GPCR structural biology stem from their low stability in purified form. This intrinsic instability is primarily due to their conformational flexibility as well as their hydrophobic nature. To overcome these issues, researchers have developed a number of tools and technologies that have collectively led to a significantly increased success rate in GPCR structure resolution. As a result, the application of SBDD to this class of medically important drug targets has become a real possibility.
Overview of Technology Developments Enabling GPCR Structures
Conventional protein crystallography requires highly purified and homogenous protein samples with accessible protein-protein interaction surfaces to allow formation of well ordered crystals. The hydrophobic nature of membrane proteins necessitates the addition of detergents for purification. Inclusion of detergent in purification has two consequences. Firstly, detergents remove the stabilizing effect of the membrane bilayer, leading to loss of structural integrity. The extent of this loss depends on the type of detergent used, and as a simple rule of thumb, there is an inverse correlation with detergent chain length. Detergents with shorter chain length result in increased loss of activity; conversely, longer chain detergents are more protective. Secondly and more importantly, detergent molecules reduce the hydrophilic surfaces required for crystallization. The extent of surface occlusion is also a function of detergent chain length, with shorter chain detergents forming smaller micelles when compared with detergents with longer chains. However, as outlined above, the application of the short chain detergents in protein purification is not generally conducive to purification of correctly folded and homogenous membrane proteins. One of the main advances made to overcome this challenge has been the advent of lipidic cubic phase (LCP) (7). The key feature of LCP is that the crystallogenesis step is carried out in a lipidic environment that offers a more protective surrounding for the membrane proteins. This method of crystallography has gained significant popularity, and the majority of non-rhodopsin structures have been solved using this method. Non-rhodopsin receptors that have been subjected successfully to the conventional method of vapor diffusion have invariably been significantly engineered to exhibit increased thermal stability and can therefore be readily purified in harsher detergents. A technological advance with the potential to significantly increase the success rate of LCP crystallography is the application of the x-ray free electron laser to crystals grown in LCP (8–10). Using this technique, it is possible to use small crystals for data collection by serial femtosecond crystallography. This method applies very intense and ultrashort x-ray pulses to thousands of microcrystals that are delivered to the intersection point with the beam using an injector. This method not only circumvents the need to grow sufficiently large crystals capable of withstanding radiation damage, it also reduces the total amount of protein required. Successful application of this method to GPCRs has been demonstrated by the recent structure resolution of the 5-hydroxytryptamine receptor 2B (5-HT2B), the smoothened receptor, and the angiotensin II type 1 receptor.
Recent developments in protein engineering have added another set of techniques that has greatly facilitated GPCR structural resolution. A routine protein engineering approach is to remove flexible extreme termini of receptors to reduce heterogeneity and increase chances of crystallogenesis. In addition, researchers routinely add fusion partners such as T4 lysozyme or apocytochrome b562RIL to either the N terminus or the second or third intracellular loops (11). Other fusion partners used include the catalytic domain of Pyrococcus abyssi glycogen synthase in the orexin 2 receptor (12) and rubredoxin in CCR5 (13). These fusion partners are selected because they are stable domains that crystallize readily, but more importantly, their N and C termini are in close proximity (less than 15 Å), thus allowing them to be inserted in the receptor loops without gross alteration of the receptor structure. However, in a number of cases, it appears that the addition of a fusion partner impacts the overall structure. In the cases of β2-adrenergic and adenosine A2A receptors, the addition of T4 lysozyme has been observed to increase the affinity of receptors for agonists when compared with wild type, indicating that the receptor conformation has been shifted toward the active form (14, 15). Comparison of the A2A structure in the absence of any fusion with the T4 fused structure provided structural explanation for this observation; the addition of T4 lysozyme appears to increase the outward movement of TM6 consistent with shifting the receptor conformation toward the active form (16). However, the structure of A2A with apocytochrome b562RIL was closer to the non-fused structure and did not exhibit the shift toward agonist conformation (17). These observations indicate that it is critical to understand the pharmacology of receptors following generation of fusion constructs. The addition of the fusion partner to the N terminus is one way to minimize the effect of fusion on receptor conformation while maintaining the beneficial effects of mediating improved crystal contacts (18, 19). In addition to fusion proteins, researchers have used antibody fragments to the same effect. Initially, mouse monoclonal antibody fragments raised against the cytoplasmic face of the receptor were used to increase the hydrophilic interaction surface and reduce flexibility (20). More recently, single chain camelid antibodies (nanobodies) have been used to great success in aiding GPCR crystallography. These antibodies are small (15 kDa), rigid, and easy to clone, express, and purify. As GPCR co-crystallization reagents, nanobodies have been primarily used to drive and stabilize the active receptor conformation because this conformation is generally more unstable, especially in the absence of G protein (21–23). In addition, a nanobody has been used to stabilize the ternary complex of an agonist-bound β2-adrenoreceptor in complex with the full heterotrimeric G protein (24).
Conformational thermostabilization has become another established protein engineering solution to aid GPCR crystallography. This approach relies on the identification of single point mutations that increase the thermal stability of detergent-solubilized receptors in a particular conformation. To generate a conformationally thermostabilized receptor, a detergent-compatible thermal stability assay needs to be set up. In its simplest form, a labeled ligand (usually a radio-labeled one) is used to measure receptor thermal stability in a detergent-compatible ligand binding assay. The application of ligand binding to measure stability not only allows screening of mutants in a high throughput manner, it also results in the biasing of the receptor population to a particular conformation. Initially, systematic scanning mutagenesis was used to generate the mutant library (25); however, molecular evolution approaches have now been developed that apply random mutagenesis screening strategies (26). Regardless of the approach used, these methods result in stabilization of receptors in a particular conformation, thus reducing the conformational heterogeneity, which increases the success of crystallization. As with receptors with fusions, it is critically important to thoroughly evaluate the pharmacology of the stabilized receptors to ensure that the effects of stabilization on conformation are consistent with the ligand pharmacology used in the process of stabilization (27). A key advantage of receptor conformational stabilization is that stabilized receptors do not rely on ligand-induced stability to maintain the conformation and structural integrity. Consequently and in contrast to wild-type receptors, stabilized receptors allow successful structural resolution in complex with weak binding ligands (16). This is a critical advantage, especially when structure determination is an integral part of an SBDD campaign where many early hits may not exhibit high affinity and structural information would be critical in their development.
Key Features of GPCR Structures Relevant to Drug Discovery
The application of the technical advances described above has led to a plethora of GPCR structures that have in a significant way paved the way for the application of SBDD approaches to this class of proteins. One of the key insights derived from the recent GPCR structural biology revolution is the understanding of receptor activation gained from analysis of structures in different conformations. In addition, a number of recent structures have revealed novel ligand binding sites outside of the main orthosteric binding pocket, which in combination with our structurally refined understanding of activation mechanism have opened up exciting possibilities for discovering drugs to modulate GPCRs. In this section, we focus on aspects of structural biology that are of relevance to drug discovery. As most of the structures resolved to date belong to receptors in class A, this section focuses primarily on this class of receptors. The residues are referred to in the Ballesteros-Weinstein numbering system (28).
The first sight of structural changes underpinning receptor activation came from the resolution of opsin in complex with the C-terminal fragment of the α-subunit of transducin (the light transduction heterotrimeric G protein) (29). When compared with the dark-state structure of rhodopsin (the inactive state), the opsin-Gαt peptide structure revealed that on activation, there is a large outward movement of TM6 that allows a large binding site to be created for the Gα subunit. This significant rearrangement upon activation was further corroborated by the structures of the agonist-bound β2-adrenergic receptor stabilized in the active conformation by a G protein-mimicking nanobody as well as the full ternary complex of this receptor with agonist and the G protein (21, 22, 24). These structures also revealed the outward movement of TM6, although the extent of this movement was larger than that observed in the opsin structure. Drawing on the information from these receptor systems and the wealth of receptor mutagenesis data, it is possible to propose a common activation mechanism (30). Central to this mechanism is a set of conserved residues in the core of the receptors consisting of Leu3.43, Phe6.44, and X6.40 where X is a bulky hydrophobic residue (e.g. valine, isoleucine, leucine, or methionine). Prior to activation, interactions of these residues maintain TM3 and TM6 in the inactive state. In this state, Leu3.43 is stabilized by X6.40 residue and Phe6.44, and following agonist binding, TM3 moves upwards, which results in stabilization of Leu3.43 against the conserved residue of Leu2.46, which in turn pushes up Asn7.49 of the highly conserved NPXXY motif. This upward movement of Asn7.49 allows Tyr7.53 to hydrogen-bond with Tyr5.58 via a water molecule, which facilitates its interaction with Arg3.50 of the so-called ionic lock motif of the (D/E)RY. The ionic lock has been proposed to maintain the TM3 and TM6 in the inactive conformation through a salt bridge between Arg3.50 and Glu6.30. Collectively, these structural changes result in the stabilization of the active conformation. Consistently with their proposed role in keeping the receptor in the inactive conformation, mutations that disrupt this network often result in constitutively active receptors (examples are discussed in Ref. 30).
Understanding how ligand binding will lead to receptor activation will be very important for utilizing structural information to support SBDD. However, given the varying nature of the agonist ligands as well as their different binding sites, it is difficult to provide a unifying route for activation originating from ligand binding. Each receptor system with its unique agonist molecule and binding site will effect the changes required for receptor activation differently. For example, in the case of the β2-adrenergic receptor, agonists form hydrogen bonds with two serine residues on TM5 (Ser5.42 and Ser5.46), which results in an inward movement of TM5 causing the conserved Pro5.50 to interact with and induce a different rotameric state at Ile3.40, which results in effect in the upward movement of TM3 and the key residue of Leu3.43 in the hydrophobic core. In addition, the rotameric change of Ile3.40 forces rotation of Phe6.44, which along with the upward shift of Leu3.43 completely destabilizes the hydrophobic core that leads to receptor activation (30). An interesting observation from the resolution of the related turkey β1-adrenergic receptor in complex with a range of full and partial agonists revealed how differential ligand efficacies might be rationalized from structural insight. Similar to β2-adrenergic receptor, a full agonist in complex with β1-adrenergic receptor forms hydrogen bonds with the two TM5 serines outlined above. In contrast, partial agonists such as salbutamol and dobutamine do not engage Ser5.46, which presumably results in weaker stabilization of the active conformation and thus leads to reduced levels of activity (31).
Although the collective knowledge gleaned from different structures is incredibly valuable and has advanced our knowledge of receptor biology significantly, it is wise to remember that crystal structures are in essence frozen snapshots. In addition, although different structures in different conformations can be used to generate a high tech version of a zoopraxiscope movie, the full spectrum of receptor activation is undoubtedly more complex. To compensate, researchers have used complementary approaches such as spectroscopic techniques and molecular dynamics simulations to get a more complete picture of the receptor activation mechanism. Using these approaches, it has been shown that there is a spectrum of conformations more complex than the simple active and inactive conformations. Specifically, computational approaches have provided evidence for metastable states that will be very difficult if not impossible to capture experimentally (32). Atomic-level simulations based on the available crystal structures of the β2-adrenergic receptor reveal that different sections of the receptor (the ligand binding site, G protein binding site, and connector region) exhibit weak allosteric coupling and can occupy different conformations independently (32). The consequence of this loose structural relationship is that ligand efficacy only needs modulation of the equilibrium between different conformations to achieve distinct pharmacological outcomes. These computational predictions have been experimentally validated using NMR and pulsed electron paramagnetic resonance spectroscopy, where an agonist-alone bound receptor exhibits substantial conformational heterogeneity and full activation is achieved in the presence of G protein. It appears that as agonist binding shifts the equilibrium toward active conformation, there is a concomitant increase in receptor conformation heterogeneity, which presumably allows the receptor to engage alternative signaling or regulatory proteins depending on the context and environment. Binding of G protein (or G protein mimetic) to the agonist-occupied receptor results in a reduction in receptor conformation heterogeneity and stabilization of the fully active conformation (33, 34).
In addition to the information regarding receptor activation, recent x-ray structures have revealed a wide diversity of unexpected binding sites not previously predicted by mutagenesis studies or pharmacology. It appears possible to activate or block activity of GPCRs by a number of different mechanisms other than mimicking or blocking the binding of the natural agonist ligand. The first example of this was the CRF1 structure (35) where the antagonist CP-376395 was found to bind deep within the transmembrane domain close to the intracellular side of the receptor. The binding site for the receptor has very restricted access, and it is possible that the ligand may enter the receptor through the membrane rather than through the entrance to the orthosteric peptide binding site even though this is an open cavity in the CRF1 receptor protein. CP-376395 has been noted to have a very slow on-rate (36), which may be due to ligand entry route.
Recent structures have revealed that multiple binding sites can be present on some of the receptors that offer further opportunities for drug design. The x-ray structures of the protease-activated PAR1 receptor and the related class A purinergic receptor P2Y12 both indicated the presence of multiple binding pockets within the transmembrane domain (37, 38). In P2Y12, pocket 1 is formed between TM3 and TM7, whereas pocket 2 is formed between TM1–3 and TM7 (Fig. 1). The antagonist AZD1283 is bound in pocket 1. This is similar to the position of vorapaxar binding to the PAR1 receptor. Modeling and mutagenesis studies suggest that some P2Y12 antagonists including those that bind covalently to the receptor may bind to pocket 2 rather than pocket 1 (38).
FIGURE 1.
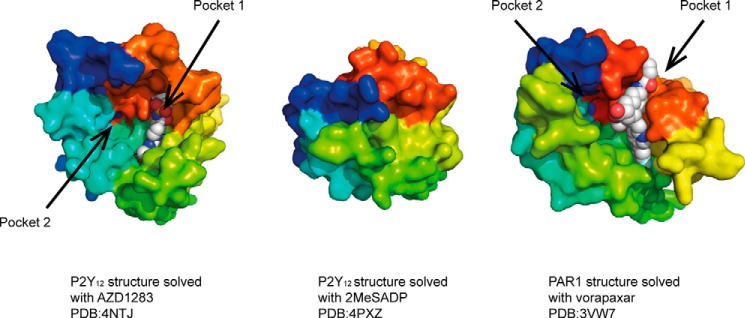
Surface representation of the extracellular face of P2Y12 and PAR1 receptors in complex with the indicated ligands. Receptors are depicted in rainbow spectrum starting with TM1 in blue and ending with TM7 in red. In the P2Y12 receptor, pocket 1 is formed between TM3 and TM7, whereas pocket 2 is formed between TM1–3 and TM7. P2Y12 structure with 2MeSADP shows closed lid conformation. For clarity, portions of the extracellular loops have been removed in the PAR1 structure.
Structures of P2Y12 have also been solved in complex with the full agonist 2MeSADP and the related 2MeSATP (39). These agonists bind to the same overall pocket as AZD1283 (i.e. pocket 1); however, they bind in a very different orientation, which is only partially overlapping. Of more significance and interest is that the agonist-bound structures show dramatic conformational rearrangements in the extracellular region of the receptor. In the agonist-bound form, the binding pocket appears to be occluded by a lid formed by the positively charged extracellular loops and the N terminus (Fig. 1). This closed conformation is facilitated by the negatively charged phosphate group of nucleotide agonist. In the absence of a negatively charged ligand, the binding pocket will remain open through charge repulsion of the arginine and lysine residues. Consistently, the P2Y12 structure in complex with the non-nucleotide antagonist AZD1283 shows an open pocket (Fig. 1). Such arrangement of positively charged residues and the rearrangement of the extracellular side in response to the binding of a negatively charged agonist molecule indicate the evolution of a specific mechanism for specific ligand recognition. The full functional implications of this charged network and structural rearrangement remain to be fully explored.
The structure of the P2Y1 receptor, another platelet receptor involved in platelet aggregation, has also revealed multiple binding sites (40). The nucleotide antagonist MRS2500 binds to a site at the top of the transmembrane domain between TM6 and TM7 but also involving the N terminus and extracellular loop 2. This site is distinct from the nucleotide binding site found in P2Y12, which sits deeper in the receptor. Interestingly, previous mutation studies on P2Y1 have indicated that some antagonists may bind deeper in the receptor at a site analogous to the P2Y12 receptor (Ref. 40 and references cited therein). The most unexpected finding of the P2Y1 structure is the binding site of the non-nucleotide antagonist BPTU, which was found to bind on the outside of the receptor on the lipid interface between TM1, TM2, and TM3 (Fig. 2). This binding site explains the unusual structure-activity relationship of related compounds where binding affinity appears to correlate with lipophilicity. BPTU acts as a negative allosteric modulator accelerating the dissociation of nucleotide agonists (40).
FIGURE 2.

Surface representation of GPR40, β2-adrenergic receptor and S1P1 receptor in complex with the indicated ligands. The top panel shows the extracellular faces of the receptors; in contrast to the β2-adrenergic receptor, the route to the ligand binding site GPR40 and S1P1 is occluded from top. The bottom panel shows the side view of the GPR40 and S1P1 receptors where the ligand binding site is clearly visible. It is thought that in these receptors with lipophilic ligands, the route to ligand binding is through the membrane bilayer.
The lipophilic agonist for the GPR40 (FFA1) receptor TAK875 also has an unusual binding site that is partly outside the helical bundle (41). This compound was found to bind partly in the perceived orthosteric binding site but extends through TM3 and TM4 to the lipid membrane. Similar to the CRF1 antagonist, this ligand is also considered to enter the receptor via the lipid bilayer rather than directly from the extracellular domain. An entry route from the lipid membrane has also been proposed based on the S1P1 structure between TM1 and TM7 because normal access to the pocket is excluded by the N terminus and extracellular loops (42) (Fig. 2). It seems likely that other lipid ligands may enter via similar routes, and indeed that has also been suggested for the cannabinoid receptor family, although this has not yet been confirmed by a structure (43).
Structure-based Design Techniques Applied to GPCRs
The availability of high resolution x-ray structures of GPCRs has provided the opportunity to apply structure-based methods to the design of drugs. Such methods are now well established for soluble enzyme targets but are only now being applied to membrane proteins such as GPCRs. Starting points for GPCR chemistry projects have until recently been cell-based HTS campaigns. However, for many targets of current interest, such as lipid and peptide receptors, these tend to have a poor hit rate. In addition, HTS methods bias ligand selection toward higher potency compounds, which often have high molecular weight and undesirable physicochemical properties. In the absence of structural information, lead optimization can be difficult. An alternative method is to use the detailed knowledge of the protein-ligand binding pocket to run a virtual screen whereby vast libraries (e.g. the ZINC database is a free database of ∼80 million commercially available compounds) are screened by docking compounds into models of the receptor and then scoring the fit using computational programs such as GLIDE (Schrödinger, LLC). The highest scoring hits are then selected for biochemical screening. Virtual screen hit rates of 3–10% have been reported for a range of GPCR targets including the adenosine A2A receptor (44), histamine H1 (45), and the chemokine receptor CXCR7 (46).
Characterizations of the binding site (in terms of size, shape, and physicochemical properties, such as lipophilicity and hydrogen bonding) are analyzed to design ligands that are optimized to achieve high affinity binding. It is now clear that water molecules within the receptor are an important consideration in drug design. The deep pockets of GPCRs are filled with water molecules. In the highest resolution structures, crystallographic waters can be resolved, and computational software programs such as WaterMap (Schrödinger, LLC) can be used to predict the positon of water molecules and determine their energetics. Water molecules that are trapped in lipophilic pockets have a high relative energy and have been called “unhappy waters” in comparison with “happy waters” present in bulk solvent. GPCR binding sites usually include a number of unhappy waters, and displacement of these by small molecule drugs is energetically favorable, contributing to potent and ligand-efficient binding. The location of water molecules within GPCR structures is important in understanding ligand binding, selectivity, and the design of new compounds (47).
Although the optimization of antagonist ligands is driven primarily by affinity, the application of structure-based approaches to the design of agonist ligands is more challenging. In this case, specific interactions must be made between the ligand and the receptor to trigger the conformational changes associated with receptor activation as described above. Compounds that bind preferentially to the agonist conformation will stabilize this form of the receptor and alter the equilibrium between agonist and antagonist forms. The availability of x-ray structures in the active conformation allow this to be modeled, although a deeper understanding of the structural basis of efficacy will require many more structures in complex with agonists of different levels of efficacy. For now, it is important to use structural information in conjunction with data obtained from cell-based functional assays to guide compound selection in agonist projects.
There is an increasing interest in the development of allosteric modulators as drugs directed at GPCRs. These may have improved selectivity and therapeutic index when compared with orthosteric ligands (48). Novel x-ray structures with allosteric modulators bound have now been solved for class A muscarinic M3 receptor (23), class B CRF1 receptor (35), and class C mGlu1 and mGlu5 receptors (49, 50). Of particular interest was the x-ray structure of the M3 muscarinic receptor simultaneously bound to the orthosteric agonist iperoxo and the positive allosteric modulator LY2119620 found in the extracellular vestibule of the receptor (23). The extracellular vestibule has been suggested as an entry point on the receptor for the binding of the endogenous ligand acetylcholine as well as drugs targeted at these receptors (51). For example, the orthosteric muscarinic antagonist tiotropium is predicted by molecular dynamics simulations to bind to an allosteric site in a metastable binding form on its way to bind to the orthosteric site in the receptor. Interestingly, differences in binding at the site between M2 and M3 receptors may contribute to the different kinetics that tiotropium shows at these receptor subtypes (51).
Biased agonists are another highly active area in the field of GPCR drug discovery (52). As yet, the structural basis of bias remains to be elucidated and will rely on solving structures in the presence of other signaling molecules such as β-arrestin as well as getting multiple co-structures with ligands showing different biases.
Drugs that have been identified using structure-based methods are now progressing to clinical trials (53). It is anticipated that as has been shown for soluble targets, the success rate of these compounds progressing through the different stages of development should be higher than those obtained by more empirical methods.
Author Contributions
All authors contributed to the planning, discussions, and writing of the manuscript.
This is the third article in the Thematic Minireview series “New Directions in G Protein-coupled Receptor Pharmacology.” All authors are full-time employees and shareholders of Heptares Therapeutics Limited.
- HTS
- high throughput screening
- FBDD
- fragment-based drug discovery
- SBDD
- structure-based drug design
- GPCR
- G protein-coupled receptor
- LCP
- lipidic cubic phase
- TM
- transmembrane
- 2MeSADP
- 2-methylthio-adenosine-5′-diphosphate
- 2MeSATP
- 2-methylthio-adenosine-5′-triphosphate.
REFERENCES
- 1. Paul S. M., Mytelka D. S., Dunwiddie C. T., Persinger C. C., Munos B. H., Lindborg S. R., Schacht A. L. (2010) How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nat. Rev. Drug Discov. 9, 203–214 [DOI] [PubMed] [Google Scholar]
- 2. Congreve M., Langmead C. J., Mason J. S., Marshall F. H. (2011) Progress in structure based drug design for G protein-coupled receptors. J. Med. Chem. 54, 4283–4311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Erlanson D. A. (2012) Introduction to fragment-based drug discovery. Top. Curr. Chem. 317, 1–32 [DOI] [PubMed] [Google Scholar]
- 4. Li J., Liu X., Li S., Wang Y., Zhou N., Luo C., Luo X., Zheng M., Jiang H., Chen K. (2013) Identification of novel small molecules as inhibitors of hepatitis C virus by structure-based virtual screening. Int. J. Mol. Sci. 14, 22845–22856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schindler T., Bornmann W., Pellicena P., Miller W. T., Clarkson B., Kuriyan J. (2000) Structural mechanism for STI-571 inhibition of Abelson tyrosine kinase. Science 289, 1938–1942 [DOI] [PubMed] [Google Scholar]
- 6. Wlodawer A., Vondrasek J. (1998) Inhibitors of HIV-1 protease: a major success of structure-assisted drug design. Annu. Rev. Biophys. Biomol. Struct. 27, 249–284 [DOI] [PubMed] [Google Scholar]
- 7. Caffrey M., Cherezov V. (2009) Crystallizing membrane proteins using lipidic mesophases. Nat. Protoc. 4, 706–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu W., Wacker D., Gati C., Han G. W., James D., Wang D., Nelson G., Weierstall U., Katritch V., Barty A., Zatsepin N. A., Li D., Messerschmidt M., Boutet S., Williams G. J., Koglin J. E., Seibert M. M., Wang C., Shah S. T. A., Basu S., Fromme R., Kupitz C., Rendek K. N., Grotjohann I., Fromme P., Kirian R. A., Beyerlein K. R., White T. A., Chapman H. N., Caffrey M., Spence J. C. H., Stevens R. C., Cherezov V. (2013) Serial femtosecond crystallography of G protein-coupled receptors. Science 342, 1521–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang H., Unal H., Gati C., Han G. W., Liu W., Zatsepin N. A., James D., Wang D., Nelson G., Weierstall U., Sawaya M. R., Xu Q., Messerschmidt M., Williams G. J., Boutet S., Yefanov O. M., White T. A., Wang C., Ishchenko A., Tirupula K. C., Desnoyer R., Coe J., Conrad C. E., Fromme P., Stevens R. C., Katritch V., Karnik S. S., Cherezov V. (2015) Structure of the angiotensin receptor revealed by serial femtosecond crystallography. Cell 161, 833–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weierstall U., James D., Wang C., White T. A., Wang D., Liu W., Spence J. C. H., Bruce Doak R., Nelson G., Fromme P., Fromme R., Grotjohann I., Kupitz C., Zatsepin N. A., Liu H., Basu S., Wacker D., Han G. W., Katritch V., Boutet S., Messerschmidt M., Williams G. J., Koglin J. E., Marvin Seibert M., Klinker M., Gati C., Shoeman R. L., Barty A., Chapman H. N., Kirian R. A., Beyerlein K. R., Stevens R. C., Li D., Shah S. T. A., Howe N., Caffrey M., Cherezov V. (2014) Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat. Commun. 5, 3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ghosh E., Kumari P., Jaiman D., Shukla A. K. (2015) Methodological advances: the unsung heroes of the GPCR structural revolution. Nat. Rev. Mol. cell Biol. 16, 69–81 [DOI] [PubMed] [Google Scholar]
- 12. Yin J., Mobarec J. C., Kolb P., Rosenbaum D. M. (2015) Crystal structure of the human OX2 orexin receptor bound to the insomnia drug suvorexant. Nature 519, 247–250 [DOI] [PubMed] [Google Scholar]
- 13. Tan Q., Zhu Y., Li J., Chen Z., Han G. W., Kufareva I., Li T., Ma L., Fenalti G., Li J., Zhang W., Xie X., Yang H., Jiang H., Cherezov V., Liu H., Stevens R. C., Zhao Q., Wu B. (2013) Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science 341, 1387–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rosenbaum D. M., Cherezov V., Hanson M. A., Rasmussen S. G. F., Thian F. S., Kobilka T. S., Choi H.-J., Yao X.-J., Weis W. I., Stevens R. C., Kobilka B. K. (2007) GPCR engineering yields high-resolution structural insights into β2-adrenergic receptor function. Science 318, 1266–1273 [DOI] [PubMed] [Google Scholar]
- 15. Jaakola V.-P., Griffith M. T., Hanson M. A., Cherezov V., Chien E. Y. T., Lane J. R., Ijzerman A. P., Stevens R. C. (2008) The 2.6 Å crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322, 1211–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Doré A. S., Robertson N., Errey J. C., Ng I., Hollenstein K., Tehan B., Hurrell E., Bennett K., Congreve M., Magnani F., Tate C. G., Weir M., Marshall F. H. (2011) Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 19, 1283–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chun E., Thompson A. A., Liu W., Roth C. B., Griffith M. T., Katritch V., Kunken J., Xu F., Cherezov V., Hanson M. A., Stevens R. C. (2012) Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure 20, 967–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thompson A. A., Liu W., Chun E., Katritch V., Wu H., Vardy E., Huang X.-P., Trapella C., Guerrini R., Calo G., Roth B. L., Cherezov V., Stevens R. C. (2012) Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 485, 395–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Siu F. Y., He M., de Graaf C., Han G. W., Yang D., Zhang Z., Zhou C., Xu Q., Wacker D., Joseph J. S., Liu W., Lau J., Cherezov V., Katritch V., Wang M.-W., Stevens R. C. (2013) Structure of the human glucagon class B G-protein-coupled receptor. Nature 499, 444–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rasmussen S. G. F., Choi H.-J., Rosenbaum D. M., Kobilka T. S., Thian F. S., Edwards P. C., Burghammer M., Ratnala V. R. P., Sanishvili R., Fischetti R. F., Schertler G. F. X., Weis W. I., Kobilka B. K. (2007) Crystal structure of the human β2 adrenergic G-protein-coupled receptor. Nature 450, 383–387 [DOI] [PubMed] [Google Scholar]
- 21. Rasmussen S. G. F., Choi H.-J., Fung J. J., Pardon E., Casarosa P., Chae P. S., Devree B. T., Rosenbaum D. M., Thian F. S., Kobilka T. S., Schnapp A., Konetzki I., Sunahara R. K., Gellman S. H., Pautsch A., Steyaert J., Weis W. I., Kobilka B. K. (2011) Structure of a nanobody-stabilized active state of the β2-adrenoceptor. Nature 469, 175–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ring A. M., Manglik A., Kruse A. C., Enos M. D., Weis W. I., Garcia K. C., Kobilka B. K. (2013) Adrenaline-activated structure of β2-adrenoceptor stabilized by an engineered nanobody. Nature 502, 575–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kruse A. C., Ring A. M., Manglik A., Hu J., Hu K., Eitel K., Hübner H., Pardon E., Valant C., Sexton P. M., Christopoulos A., Felder C. C., Gmeiner P., Steyaert J., Weis W. I., Garcia K. C., Wess J., Kobilka B. K. (2013) Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rasmussen S. G. F., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T. A., Lyons J. A., Caffrey M., Gellman S. H., Steyaert J., Skiniotis G., Weis W. I., Sunahara R. K., Kobilka B. K. (2011) Crystal structure of the β2-adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Serrano-Vega M. J., Magnani F., Shibata Y., Tate C. G. (2008) Conformational thermostabilization of the β1-adrenergic receptor in a detergent-resistant form. Proc. Natl. Acad. Sci. U.S.A. 105, 877–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Scott D. J., Plückthun A. (2013) Direct molecular evolution of detergent-stable G protein-coupled receptors using polymer encapsulated cells. J. Mol. Biol. 425, 662–677 [DOI] [PubMed] [Google Scholar]
- 27. Bennett K. A., Tehan B., Lebon G., Tate C. G., Weir M., Marshall F. H., Langmead C. J. (2013) Pharmacology and structure of isolated conformations of the adenosine A2A receptor define ligand efficacy. Mol. Pharmacol. 83, 949–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ballesteros J. A., Weinstein H. (1995) Integrated methods for the construction of three-dimensional models of structure-function relations in G protein-coupled receptors. Methods Neurosci. 25, 366–428, 10.1016/S1043-9471(05)80049-7 [DOI] [Google Scholar]
- 29. Scheerer P., Park J. H., Hildebrand P. W., Kim Y. J., Krauss N., Choe H.-W., Hofmann K. P., Ernst O. P. (2008) Crystal structure of opsin in its G-protein-interacting conformation. Nature 455, 497–502 [DOI] [PubMed] [Google Scholar]
- 30. Tehan B. G., Bortolato A., Blaney F. E., Weir M. P., Mason J. S. (2014) Unifying family A GPCR theories of activation. Pharmacol. Ther. 143, 51–60 [DOI] [PubMed] [Google Scholar]
- 31. Warne T., Moukhametzianov R., Baker J. G., Nehmé R., Edwards P. C., Leslie A. G. W., Schertler G. F. X., Tate C. G. (2011) The structural basis for agonist and partial agonist action on a β1-adrenergic receptor. Nature 469, 241–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dror R. O., Arlow D. H., Maragakis P., Mildorf T. J., Pan A. C., Xu H., Borhani D. W., Shaw D. E. (2011) Activation mechanism of the β2-adrenergic receptor. Proc. Natl. Acad. Sci. U.S.A. 108, 18684–18689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Manglik A., Kim T. H., Masureel M., Altenbach C., Yang Z., Hilger D., Lerch M. T., Kobilka T. S., Thian F. S., Hubbell W. L., Prosser R. S., Kobilka B. K. (2015) Structural insights into the dynamic process of β2-adrenergic receptor signaling. Cell 161, 1101–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nygaard R., Zou Y., Dror R. O., Mildorf T. J., Arlow D. H., Manglik A., Pan A. C., Liu C. W., Fung J. J., Bokoch M. P., Thian F. S., Kobilka T. S., Shaw D. E., Mueller L., Prosser R. S., Kobilka B. K. (2013) The dynamic process of β2-adrenergic receptor activation. Cell 152, 532–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hollenstein K., Kean J., Bortolato A., Cheng R. K. Y., Doré A. S., Jazayeri A., Cooke R. M., Weir M., Marshall F. H. (2013) Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature 499, 438–443 [DOI] [PubMed] [Google Scholar]
- 36. Ramsey S. J., Attkins N. J., Fish R., van der Graaf P. H. (2011) Quantitative pharmacological analysis of antagonist binding kinetics at CRF1 receptors in vitro and in vivo. Br. J. Pharmacol. 164, 992–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang C., Srinivasan Y., Arlow D. H., Fung J. J., Palmer D., Zheng Y., Green H. F., Pandey A., Dror R. O., Shaw D. E., Weis W. I., Coughlin S. R., Kobilka B. K. (2012) High-resolution crystal structure of human protease-activated receptor 1. Nature 492, 387–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang K., Zhang J., Gao Z. G., Zhang D., Zhu L., Han G. W., Moss S. M., Paoletta S., Kiselev E., Lu W., Fenalti G., Zhang W., Müller C. E., Yang H., Jiang H., Cherezov V., Katritch V., Jacobson K. A., Stevens R. C., Wu B., Zhao Q. (2014) Structure of the human P2Y12 receptor in complex with an antithrombotic drug. Nature 509, 115–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang J., Zhang K., Gao Z.-G., Paoletta S., Zhang D., Han G. W., Li T., Ma L., Zhang W., Müller C. E., Yang H., Jiang H., Cherezov V., Katritch V., Jacobson K. A., Stevens R. C., Wu B., Zhao Q. (2014) Agonist-bound structure of the human P2Y12 receptor. Nature 509, 119–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang D., Gao Z.-G., Zhang K., Kiselev E., Crane S., Wang J., Paoletta S., Yi C., Ma L., Zhang W., Han G. W., Liu H., Cherezov V., Katritch V., Jiang H., Stevens R. C., Jacobson K. A., Zhao Q., Wu B. (2015) Two disparate ligand-binding sites in the human P2Y1 receptor. Nature 520, 317–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Srivastava A., Yano J., Hirozane Y., Kefala G., Gruswitz F., Snell G., Lane W., Ivetac A., Aertgeerts K., Nguyen J., Jennings A., Okada K. (2014) High-resolution structure of the human GPR40 receptor bound to allosteric agonist TAK-875. Nature 513, 124–127 [DOI] [PubMed] [Google Scholar]
- 42. Hanson M. A., Roth C. B., Jo E., Griffith M. T., Scott F. L., Reinhart G., Desale H., Clemons B., Cahalan S. M., Schuerer S. C., Sanna M. G., Han G. W., Kuhn P., Rosen H., Stevens R. C. (2012) Crystal structure of a lipid G protein-coupled receptor. Science 335, 851–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hurst D. P., Grossfield A., Lynch D. L., Feller S., Romo T. D., Gawrisch K., Pitman M. C., Reggio P. H. (2010) A lipid pathway for ligand binding is necessary for a cannabinoid G protein-coupled receptor. J. Biol. Chem. 285, 17954–17964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Langmead C. J., Andrews S. P., Congreve M., Errey J. C., Hurrell E., Marshall F. H., Mason J. S., Richardson C. M., Robertson N., Zhukov A., Weir M. (2012) Identification of novel adenosine A2A receptor antagonists by virtual screening. J. Med. Chem. 55, 1904–1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. de Graaf C., Kooistra A. J., Vischer H. F., Katritch V., Kuijer M., Shiroishi M., Iwata S., Shimamura T., Stevens R. C., de Esch I. J., Leurs R. (2011) Crystal structure-based virtual screening for novel fragment-like ligands of the human histamine H1 receptor. J. Med. Chem. 54, 8195–8206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yoshikawa Y., Oishi S., Kubo T., Tanahara N., Fujii N., Furuya T. (2013) Optimized method of G-protein-coupled receptor homology modeling: Its application to the discovery of novel CXCR7 ligands. J. Med. Chem. 56, 4236–4251 [DOI] [PubMed] [Google Scholar]
- 47. Mason J. S., Bortolato A., Congreve M., Marshall F. H. (2012) New insights from structural biology into the druggability of G protein-coupled receptors. Trends Pharmacol. Sci. 33, 249–260 [DOI] [PubMed] [Google Scholar]
- 48. Conn P. J., Lindsley C. W., Meiler J., Niswender C. M. (2014) Opportunities and challenges in the discovery of allosteric modulators of GPCRs for treating CNS disorders. Nat. Rev. Drug Discov. 13, 692–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wu H., Wang C., Gregory K. J., Han G. W., Cho H. P., Xia Y., Niswender C. M., Katritch V., Meiler J., Cherezov V., Conn P. J., Stevens R. C. (2014) Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science 344, 58–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Doré A. S., Okrasa K., Patel J. C., Serrano-Vega M., Bennett K., Cooke R. M., Errey J. C., Jazayeri A., Khan S., Tehan B., Weir M., Wiggin G. R., Marshall F. H. (2014) Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature 511, 557–562 [DOI] [PubMed] [Google Scholar]
- 51. Dror R. O., Green H. F., Valant C., Borhani D. W., Valcourt J. R., Pan A. C., Arlow D. H., Canals M., Lane J. R., Rahmani R., Baell J. B., Sexton P. M., Christopoulos A., Shaw D. E. (2013) Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 503, 295–299 [DOI] [PubMed] [Google Scholar]
- 52. Violin J. D., Crombie A. L., Soergel D. G., Lark M. W. (2014) Biased ligands at G-protein-coupled receptors: Promise and progress. Trends Pharmacol. Sci. 35, 308–316 [DOI] [PubMed] [Google Scholar]
- 53. Congreve M., Andrews S. P., Doré A. S., Hollenstein K., Hurrell E., Langmead C. J., Mason J. S., Ng I. W., Tehan B., Zhukov A., Weir M., Marshall F. H. (2012) Discovery of 1,2,4-triazine derivatives as adenosine A2A antagonists using structure based drug design. J. Med. Chem. 55, 1898–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]