

Background: PPARγ ligands are used to treat type 2 diabetes mellitus, but signaling by these drugs is incompletely understood.
Results: Rosiglitazone activation of GPR40 markedly enhanced PPARγ-dependent transcription through downstream effects on p38 MAPK, PGC1α, and EP300.
Conclusion: GPR40 and PPARγ can function as an integrated two-receptor signal transduction pathway.
Significance: Future drug development should consider the effects of prospective ligands at both receptors.
Keywords: E1A-binding protein p300 (P300), G protein-coupled receptor (GPCR), p38 MAPK, peroxisome proliferator-activated receptor (PPAR), peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1a)(PPARGC1A), sirtuin 1 (SIRT1), thiazolidinedione, G protein-coupled receptor 40 (GPR40), peroxisome proliferator-activated receptor gamma (PPARgamma)
Abstract
Peroxisome proliferator-activated receptor γ (PPARγ) ligands have been widely used to treat type 2 diabetes mellitus. However, knowledge of PPARγ signaling remains incomplete. In addition to PPARγ, these drugs also activate G protein-coupled receptor 40 (GPR40), a Gαq-coupled free fatty acid receptor linked to MAPK networks and glucose homeostasis. Notably, p38 MAPK activation has been implicated in PPARγ signaling. Here, rosiglitazone (RGZ) activation of GPR40 and p38 MAPK was found to boost PPARγ-induced gene transcription in human endothelium. Inhibition or knockdown of p38 MAPK or expression of a dominant negative (DN) p38 MAPK mutant blunted RGZ-induced PPARγ DNA binding and reporter activity in EA.hy926 human endothelial cells. GPR40 inhibition or knockdown, or expression of a DN-Gαq mutant likewise blocked activation of both p38 MAPK and PPARγ reporters. Importantly, RGZ induction of PPARγ target genes in primary human pulmonary artery endothelial cells (PAECs) was suppressed by knockdown of either p38 MAPK or GPR40. GPR40/PPARγ signal transduction was dependent on p38 MAPK activation and induction of PPARγ co-activator-1 (PGC1α). Silencing of p38 MAPK or GPR40 abolished the ability of RGZ to induce phosphorylation and expression of PGC1α in PAECs. Knockdown of PGC1α, its essential activator SIRT1, or its binding partner/co-activator EP300 inhibited RGZ induction of PPARγ-regulated genes in PAECs. RGZ/GPR40/p38 MAPK signaling also led to EP300 phosphorylation, an event that enhances PPARγ target gene transcription. Thus, GPR40 and PPARγ can function as an integrated two-receptor signal transduction pathway, a finding with implications for rational drug development.
Introduction
Obesity-associated type 2 diabetes mellitus has reached epidemic proportions in the United States and is a major risk factor for coronary artery disease and stroke (1). Thiazolidinediones (TZDs),4 synthetic insulin-sensitizing drugs that activate peroxisome proliferator-activated receptor γ (PPARγ), have been widely used to treat this disease (2, 3). In addition to lowering glucose levels (3), TZDs also lower blood pressure (4), improve lipid profiles (5), and reduce vascular inflammation (6). These effects in non-adipose tissues have raised the possibility that PPARγ ligands may be more broadly useful in vascular disorders, such as atherosclerosis (7), pulmonary arterial hypertension (8), and septic shock (9). However, side effects, including weight gain, fluid retention, congestive heart failure, and bone fractures, have been linked to TZDs (10). These adverse effects underscore our still incomplete understanding of PPARγ signaling and the need to develop safer, more effective PPARγ ligands (10).
The direct binding of TZDs and other ligands to PPARγ activates two major but distinct signal transduction pathways. Cis-activation drives transcription through ligand-dependent recruitment of co-activators, such as PPARγ co-activator-1α (PGC1α), PPARγ dimerization with the retinoid X receptor (11), and the binding of this complex to peroxisome proliferator response elements (PPREs) in the promoter region of target genes. Trans-repression of inflammatory response genes requires the covalent modification of PPARγ at lysine 395 by the small ubiquitin-like modifier with subsequent tethering of PPARγ to nuclear receptor co-repressor and histone deacetylase complexes at NFκB and AP-1 sites (12).
Besides these primary ligand-dependent pathways, adjunctive post-translational modifications of PPARγ and its co-activators also regulate PPARγ signaling. ERK and JNK directly phosphorylate PPARγ on serine 112 and inhibit its transcriptional activation (13–16). Cyclin-dependent kinase 5 phosphorylates PPARγ on serine 273, modulating the PPARγ transcriptional program (17). Furthermore, p38 MAPK phosphorylation of PGC1α (18–21) and CBP/EP300 (22) facilitate chromatin remodeling and PPARγ-dependent transcription. PGC1α, a key regulator of PPARγ, is additionally acetylated by GCN5 (23) and deacetylated by SIRT1 (24, 25), reducing and enhancing its co-transcriptional activity, respectively.
Although the anti-diabetic activity of TZDs was first reported in 1982 (26), recognition as ligand/agonists of the orphan nuclear receptor PPARγ came more than a decade later (27, 28). More recently, TZDs were found to bind to and activate Gαq protein-coupled receptor 40 (GPR40) (29–34), a cell membrane receptor associated with free fatty acid- and glucose-induced insulin secretion (35, 36), effects that overlap with those of PPARγ (37, 38). Importantly, GPR40 signaling causes rapid activation of ERK, p38 MAPK, and JNK (31, 33). Whereas we previously found that •NO activation of p38 MAPK initiated PPARγ signaling in human endothelial cells (39), others have also associated p38 MAPK with PPARγ-related effects in adipocytes (20, 40). Collectively, these results suggest that TZD signaling through GPR40 with subsequent activation of p38 MAPK might modulate PPARγ transcriptional activity in human endothelium.
This investigation sought to determine whether TZD activation of GPR40 and p38 MAPK influences PPARγ signaling in human endothelial cells and, if so, to explore the underlying mechanism. A two-receptor paradigm for PPARγ signal transduction is proposed with implications for the development of PPARγ therapeutics.
Experimental Procedures
Reagents
Rosiglitazone (RGZ), pioglitazone (PGZ), Nω-nitro-l-arginine methyl ester hydrochloride (l-NAME), and DETA NONOate (DTANO) were obtained from Cayman Chemical (Ann Arbor, MI). SB202190 (SB) was from EMD Chemicals (Gibbstown, NJ). GW1100 was purchased from OTAVA Ltd. (Ontario, Canada). PPRE reporter constructs containing three copies of PPRE upstream of the reporter gene 3-thymidine kinase-chloramphenicol acetyltransferase (PPRE-CAT) or 3-thymidine kinase-luciferase (PPRE-LUC), were kindly supplied by Dr. Ronald M. Evans (Salk Institute, La Jolla, CA) (41). Dr. Ae-Kyung Yi (University of Tennessee Health Science Center) provided the dominant negative p38 MAPK (DN-p38 MAPK) expression plasmid (42). The dominant-negative G-protein αq mutant (DN-Gαq; Q209L/D277N) expression plasmid was obtained from the UMR cDNA Resource Center (University of Missouri, Rolla. MO). This mutant has a lowered affinity for guanine nucleotides and an enhanced affinity for xanthine nucleotides, resulting in stable and specific complexes with cognate receptors that compete with endogenous wild-type G-proteins (43). Expression plasmids for PPARγ2 (catalogue no. 8895), EP300 (catalogue no. 23252), and PGC1α (catalogue no. 10974) were purchased from Addgene Inc. (Cambridge, MA). The GPR40 expression plasmid was from OriGene (Rockville, MD). FuGENE® 6 transfection reagent, plasmid pRL-TK expressing Renilla luciferase, used as an internal control for cell transfection, and the Dual-Luciferase® reporter assay system were from Promega (Madison, WI). Specific On-TARGETplus SMARTpool siRNA for p38α MAPK, GPR40, PGC1α, SIRT1, or EP300 and control siGENOME non-targeting siRNA were purchased from Dharmacon Inc. (Lafayette, CO). Specific GPR40 shRNA pool and its control plasmid were from Qiagen Inc. (Valencia, CA). Specific TaqMan® primers/probes were purchased from Applied Biosystems (Foster City, CA).
Cell Culture
EA.hy926 cells, a hybrid human endothelial cell line, were obtained from ATCC (Manassas, VA). A retrovirus-transfected HeLaS cell line, stably expressing FLAG-tagged PPARγ (HeLaS/F-PPARγ), was kindly provided by Dr. Kai Ge (National Institutes of Health, NIDDK, Bethesda, MD) (44). Both EA.hy926 and HeLaS/F-PPARγ cell lines were maintained in DMEM supplemented with 10% FBS, d-glucose (4.5 g/liter), l-glutamine (2 mmol/liter), sodium pyruvate (1 mmol/liter), penicillin (100 units/ml), and streptomycin (100 mg/ml). Primary human pulmonary artery endothelial cells (PAECs) were purchased from Lonza (Walkersville, MD) and used at passages 1–4. PAECs were cultured in endothelial growth medium 2 (EGM2TM) supplemented with growth factors (EGM2TM SingleQuot kit) from Lonza containing 2% FBS on flasks precoated with type I collagen (BD Biosciences). In experiments using TZDs, charcoal-stripped FBS was used instead of regular FBS. Phenol red-free DMEM was used in experiments using DTANO or l-NAME.
Reporter Gene Assay
EA.hy926 cells (2 × 105/2 ml/well) were seeded in 6-well plates 16 h prior to transfection with 100 ng of PPRE reporter (PPRE-CAT or PPRE-LUC), 100 ng of internal control pRL-TK, and 50 ng of PPARγ2 expression plasmid in the presence or absence of additional expression plasmids, including DN-p38 MAPK, DN-Gαq, GPR40, PGC1α, and EP300, as indicated. FuGENE® 6 transfection reagent was utilized at a ratio of 3 μl/μg of DNA. Twenty-four hours after transfection, cells were treated for an additional 24 h as indicated in the corresponding figure legends. Chloramphenicol acetyltransferase and luciferase activities were then measured using the CAT ELISA (Roche Diagnostics) and the Dual-Luciferase® reporter assay system (Promega), respectively. In reporter experiments with gene knockdown, cells were co-transfected with siRNA, shRNA, or their controls for 48 h, followed by 24-h stimulation. Non-targeting control or p38α MAPK siRNA was transfected using Nucleofector kits (Amaxa, Gaithersburg, MD), as described previously (39). GPR40 shRNA pool or its control plasmid was transfected using FuGENE® 6.
PAEC siRNA Silencing
PAECs (2 × 105/2 ml/well) were seeded in 6-well plates 16 h prior to transfection. GPR40, p38α MAPK, PGC1α, SIRT1, and EP300 siRNAs or non-targeting siRNA controls (30 nm) were transfected using DharmaFECT 1 (Dharmacon Inc.) in OPTI-MEM medium (Life Technologies, Inc.). Eight hours post-transfection, cells were washed once with PBS and cultured for 48 h in EGM2TM medium supplemented with growth factors and charcoal-stripped fetal calf serum, followed by treatment with RGZ for 24 h before measurement of PPARγ target gene mRNA or protein.
Detection of PPARγ Binding to Specific DNA Sequence
EA.hy926 cells were treated for 1 h with RGZ (10 μm) or vehicle control with or without SB (1 μm) pretreatment for 40 min, as indicated. Nuclear extracts (3–4 μg) were then prepared for TransAM® PPARγ ELISA (Active Motif, Carlsbad, CA), which detects human PPARγ1/2 binding to PPRE consensus sequence and does not cross-react with PPARα or PPARβ.
Western Blotting and Quantitative Real-time TaqMan® PCR
For Western blotting, samples (30 μg of whole cell lysates) were applied to a 4–12% Novex® Tris-glycine gel (Invitrogen) or a 4–15% Mini-PROTEAN® TGX gel (Bio-Rad) and subjected to electrophoretic separation. Separated protein was electrically transferred to a nitrocellulose membrane. The blot was blocked with 5% nonfat dry milk in PBS with 1% Tween® 20. Anti-phospho-p38 MAPK (pp38, catalog no. 4631), anti-total p38 MAPK (p38, catalog no. 9212), anti-PPARγ (catalog no. 2430), anti-EP300 (catalog no. D9B6; all from Cell Signaling Technology Inc., Danvers, MA), anti-GPR40 (catalog no. 3393-1; Epitomics, Burlingame, CA), and anti-SIRT1 (catalog no. 07-131; Millipore, Billerica, MA) primary antibodies were all used at a 1:1000 dilution. Anti-PGC1α (catalog no. 101707; Cayman Chemical) and anti-phospho-EP300 primary antibodies (anti-Ser(P)-1834; catalog no. PA5-12735; Thermo Scientific, Rockford, IL) were used at 1:200 and 1:500 dilutions, respectively. A secondary antibody, horseradish peroxidase-conjugated goat anti-rabbit IgG, was used at a 1:10,000 dilution. All antibodies were diluted in 5% nonfat dry milk in PBS with 1% Tween® 20. The protein bands were detected using SuperSignal® West Femto chemiluminescence substrate (Thermo Scientific). Densitometry analysis of blots was performed using Image LabTM software (Bio-Rad).
Total RNA was extracted using the RNeasy kit (Qiagen), and cDNA was synthesized with iScriptTM cDNA synthesis kits (Bio-Rad). TaqMan® PCR was performed with the Applied Biosystems ViiTM 7 instrument.
Immunoprecipitation
PAECs were transfected with various siRNAs, as indicated. Forty-eight hours post-transfection, cells were treated with RGZ (10 μm) or vehicle control for 1 and 4 h to examine effects on PGC1α phoshorylation and acetylation, respectively, before preparation of nuclear protein and whole cell lysates. Immunoprecipitation of nuclear protein (40 μg) was performed with anti-phosphoserine (catalog no. AB1603) and anti-phosphothreonine (catalog no. AB1607) antibodies from Millipore. Immunoprecipitation of whole cell lysates (500 μg) used anti-acetyl-lysine antibody (catalog no. 06-933; Millipore). After incubation with rotation at 4 °C overnight, Dynabeads® protein G (1.5 mg; Invitrogen) was added. Immunoprecipitates were then subjected to Western blotting with anti-PGC1α antibody (catalog no. 101707; Cayman Chemical).
Chromatin Immunoprecipitation (ChIP) Assay
PAECs were pretreated with SB (10 μm), GW1100 (20 μm), or vehicle control (CTRL) for 40 min and then incubated with RGZ (10 μm) or vehicle CTRL for another 24 h. The ChIP assay was carried out using the EZ ChIPTM kit (Millipore) as instructed by the manufacturer. Briefly, cells were cross-linked with 1% formaldehyde and then fragmented using a Misonix sonicator with microtip probe 4418 at a power setting of 4 and a 30% duty cycle. Sonication was performed three times for 10 s with a 50-s cooling on ice between pulses, shearing chromatin into 200–1000-bp fragments. Precipitation was carried out overnight at 4 °C with anti-PPARγ (catalog no. SC-7196; Santa Cruz Biotechnology, Inc., Dallas, TX), anti-EP300 (catalog no. PA1-848; Thermo Scientific), anti-PGC1α (catalog no. ab54481; Abcam Inc., Cambridge, MA), or control mouse IgG. Precipitated protein-DNA complexes were eluted, and cross-linkings were reversed for purification of DNA. PCR was performed using the primers 5′-TTTACTATTTCCACCAGCGGTCTC-3′ and 5′-TCCCCCACACAGCACATTACTG-3′ specific for the −355/−136 region of the human CD36 promoter that contains three potential PPREs. The PCR products were analyzed by electrophoresis on 2% agarose gels (E-Gel®, Invitrogen), stained with ethidium bromide, and quantified by densitometry.
Statistical Analysis
Data are presented as means ± S.E. For real-time PCR results, geometric means ± S.E. are plotted. All statistical analyses were carried out on log-transformed or Δ cycle threshold data (for real-time PCR) using JMP® version 11 (SAS® Institute Inc., Cary, NC). To analyze continuous dose and time effects, one-way analysis of variance models were used. To test the effects of nominal factors and their interactions, linear mixed models were used, which consider the correlation within each experiment. Non-significant factors were dropped from statistical models to calculate final p values. Post hoc contrasts were tested for our interested comparisons. All p values are two-sided and considered significant if p was <0.05.
Results
Ligand Activation of PPARγ Signaling in EA.hy926 Human Endothelial Cells Is p38 MAPK-dependent
Both RGZ and PGZ (10 μm for each), two clinically relevant TZD PPARγ ligands, activated p38 MAPK in EA.hy926 human endothelial cells (p ≤ 0.002 for both; Fig. 1, A and B, respectively). Phosphorylation of p38 MAPK occurred as early as 5 min with a maximal effect at 15 min. As expected, SB (1 μm), a specific p38 MAPK inhibitor, blocked RGZ-induced p38 MAPK phosphorylation (p < 0.001 for RGZ versus SB plus RGZ; p = 0.001 for an interaction between RGZ and SB; Fig. 1C). Therapeutic RGZ dosing in humans produces peak plasma concentrations of 1–2 μm (45). In kidney (HEK293T) cells, half-maximal stimulation of PPARγ-dependent transcription was calculated to occur at a RGZ concentration of 6.5 μm (46). Consistent with these results, dose-response testing in HeLaS/F-PPARγ cells found that 10 μm RGZ induced nearly maximal PPRE reporter activity (data not shown). Therefore, subsequent experiments were conducted with RGZ (10 μm) as a prototype for this class of PPARγ ligands. Endogenous PPARγ expression in EA.hy926 endothelial cells is relatively low; therefore, in all experiments directly assessing genomic PPARγ signaling, these cells were transfected with a PPARγ2 expression plasmid (Fig. 1D, inset). SB (1 μm), a specific p38 MAPK inhibitor that blocked RGZ-induced p38 MAPK phosphorylation (Fig. 1C), significantly blocked RGZ-induced PPARγ DNA binding (p = 0.009 for RGZ versus SB plus RGZ; Fig. 1D) and PPRE reporter gene activity (p = 0.04 for RGZ versus SB plus RGZ; Fig. 1E) in EA.hy926 cells. Furthermore, both p38α MAPK siRNA silencing (p < 0.001 for RGZ versus p38α siRNA plus RGZ; p = 0.03 for an interaction between RGZ and p38 siRNA; Fig. 1F) and overexpression of a DN-p38 MAPK mutant (p = 0.03 for RGZ versus DN-p38 plus RGZ; p = 0.04 for an interaction between RGZ and DN-p38; Fig. 1G) significantly repressed RGZ-induced PPRE reporter activity. The p38α MAPK siRNA used here was previously shown to effectively knockdown p38 MAPK protein in EA.hy926 cells (39).
FIGURE 1.

PPARγ ligands induce PPRE-driven transcription through p38 MAPK. A and B, RGZ and pioglitazone increased p38 MAPK phosphorylation in EA.hy926 cells (p < 0.001 for RGZ effect in A; p = 0.002 for PGZ effect in B). C, RGZ-induced p38 MAPK phosphorylation was blocked by SB, a specific p38 MAPK inhibitor (p < 0.001 for RGZ versus SB plus RGZ; p = 0.001 for an interaction between RGZ and SB). Phosphorylated p38 MAPK (pp38) and total p38 MAPK (p38) were measured by Western blotting. Cells were treated with SB (1 μm) for 40 min and then incubated with RGZ (10 μm) or vehicle CTRL for another 30 min in C. Densitometric results for A–C of three independent experiments and representative blots are shown. D, SB blocked RGZ-induced PPARγ DNA binding, as measured by the TranAM® ELISA (p = 0.009 for RGZ versus SB plus RGZ). EA.hy926 cells were first transfected with PPARγ2 expression plasmid (inset). At 48 h after transfection, cells were treated with SB (1 μm) for 40 min and then incubated with RGZ (10 μm) or vehicle CTRL for another 1 h. E, SB (1 μm added 40 min before RGZ or its control) reduced RGZ activation of a PPARγ reporter gene (p = 0.04 for RGZ versus SB plus RGZ). F and G, siRNA knockdown of p38α MAPK (p < 0.001 for RGZ versus p38 siRNA plus RGZ; p = 0.03 for an interaction between RGZ and p38 siRNA) or expression of a dominant negative p38 MAPK mutant (DN-p38 MAPK; p = 0.03 for RGZ versus DN-p38 plus RGZ; p = 0.04 for an interaction between RGZ and DN-p38) markedly suppressed RGZ activation of a PPARγ reporter gene. EA.hy926 cells were co-transfected with PPRE-CAT reporter and PPARγ2 expression plasmid with or without p38α MAPK siRNA (p38 siRNA), scrambled CTRL siRNA, a DN-p38 MAPK plasmid, or a control empty vector, as indicated. At 48 h after transfection, cells were treated with RGZ (10 μm) or CTRL for 24 h prior to the measurement of chloramphenicol acetyltransferase activity. Results are presented as means ± S.E. (error bars) of three independent experiments in E–G.
GPR40 Mediates RGZ Activation of p38 MAPK in EA.hy926 Human Endothelial Cells
Like RGZ, other PPARγ ligands, such as pioglitazone, ciglitazone, and troglitazone, have been shown to bind to Gαq-coupled GPR40 (30–32), resulting in rapid activation of p38 MAPK as well as other downstream signaling pathways (31, 33). Notably, GPR40, also known as free fatty acid receptor 1, has been implicated in fatty acid- and glucose-induced insulin secretion (35, 36), effects also regulated by PPARγ (37, 38). EA.hy926 endothelial cells expressed both GPR40 and PPARγ (Fig. 2A), but as noted above, endogenous PPARγ protein expression is relatively low. We next determined whether RGZ activates p38 MAPK via GPR40 in EA.hy926 cells.
FIGURE 2.

RGZ induces p38 MAPK phosphorylation through GPR40. A, EA.hy926 cells express GPR40 and PPARγ protein as measured by Western blotting; GPR40 and PPARγ expression is shown in a retrovirus-transfected HeLaS cell line, stably expressing PPARγ, as a positive control. A representative blot from four independent experiments is shown. B, GW1100, a GPR40 antagonist, blocked RGZ-induced p38 MAPK phosphorylation (pp38) as measured by Western blotting (p < 0.001 for RGZ versus GW1100 plus RGZ; p = 0.04 for an interaction between RGZ and GW1100). EA.hy926 cells were treated with GW1100 (20 μm) or vehicle CTRL for 40 min and then incubated with RGZ (10 μm) or CTRL for another 30 min. C, shRNA knockdown of GPR40 blocked RGZ-induced p38 MAPK phosphorylation (pp38; p = 0.006 for RGZ versus GPR40 shRNA plus RGZ; p = 0.004 for an interaction between RGZ and GPR40 shRNA). D, shRNA knockdown decreased GPR40 protein expression as measured by Western blotting (p = 0.007 for the main effect of GPR40 shRNA). EA.hy926 cells were transfected with specific GPR40 shRNA or CTRL shRNA. At 48 h after transfection, cells were treated with RGZ (10 μm) or CTRL for 30 min prior to Western blotting. E, l-NAME, a nitric-oxide synthase inhibitor, had no effect on RGZ-induced p38 MAPK phosphorylation (pp38) as measured by Western blotting (p = 0.57 for the main effect of l-NAME). EA.hy926 cells were treated with l-NAME (1 mm) or vehicle CTRL for 1 h and then incubated with RGZ (10 μm) or CTRL for another 30 min. F, DTANO (250 μm), a nitric oxide donor, time-dependently induced p38 MAPK phosphorylation in EA.hy926 cells (p = 0.008 for DTANO effect). G, GW1100 (20 μm), a specific GPR40 antagonist, did not affect DTANO-induced p38 MAPK phosphorylation in EA.hy926 cells (p = 0.55 for DTANO versus GW1100 plus DTANO). Cells were treated with GW1100 for 40 min and then incubated with DTANO (250 μm) or CTRL (degraded DTANO) for another 30 min. H, GPR40 shRNA did not affect DTANO-induced p38 MAPK phosphorylation, as measured by Western blotting (p = 0.61 for DTANO versus GPR40 shRNA plus DTANO). EA.hy926 cells were transfected as in C. At 48 h after transfection, cells were treated with DTANO (250 μm) or CTRL for 30 min. Densitometric results of independent experiments and a representative blot are shown in B (n = 4), C (n = 5), D and E (n = 3), F and G (n = 4), and H (n = 3). Error bars, S.E.
GW1100, a specific GPR40 antagonist, inhibited RGZ-induced p38 MAPK phosphorylation (p < 0.001 for RGZ versus GW1100 plus RGZ; p = 0.04 for an interaction between GW1100 and RGZ; Fig. 2B). Likewise, shRNA-mediated GPR40 silencing significantly blocked RGZ-induced p38 phosphorylation (p = 0.006 for RGZ versus GPR40 shRNA plus RGZ; p = 0.004 for an interaction between GPR40 shRNA and RGZ; Fig. 2C); shRNA knockdown decreased GPR40 protein expression about 50% in EA.hy926 cells (p = 0.007 for the main effect of GPR40 shRNA; Fig. 2D). These results demonstrate that GPR40 is necessary for RGZ activation of p38 MAPK in a human endothelial line.
Previously, we found that •NO activated p38 MAPK in EA.hy926 cells (39). Therefore, we next examined whether RGZ/GPR40/p38 MAPK signaling might be mediated through •NO as a second messenger. l-NAME, a •NO synthase inhibitor, had no effect on RGZ-induced p38 MAPK phosphorylation (p = 0.57 for the main effect of l-NAME; Fig. 2E). Conversely, •NO activation of p38 MAPK, which is probably related to its free radical biology (47), was not dependent on GPR40 in EA.hy926 cells. DTANO, a •NO donor, time-dependently induced p38 MAPK phosphorylation (p = 0.008; Fig. 2F); this phosphorylation was not altered by either GW1100, a specific GPR40 antagonist (p = 0.55 for DTANO versus GW1100 plus DTANO; Fig. 2G) or GPR40 knockdown (p = 0.61 for DTANO versus GPR40 shRNA plus DTANO; Fig. 2H).
RGZ Activation of GPR40 Enhances PPARγ/PPRE-driven Transcription in EA.hy926 Human Endothelial Cells
The above experiments in EA.hy926 human endothelial cells show that 1) PPARγ ligands activate p38 MAPK, 2) p38 MAPK modulates PPARγ transcriptional activity, and 3) PPARγ ligand activation of p38 MAPK occurs via GPR40 and is independent of •NO. Next, we tested more directly whether GPR40 signaling modulates ligand-activated PPARγ genomic signaling. Like p38 MAPK silencing and dominant negative mutant (Fig. 1, F and G), GW1100, a specific GPR40 antagonist, significantly repressed RGZ-induced PPRE reporter activity in EA.hy926 cells (p < 0.001 for RGZ versus GW1100 plus RGZ; p = 0.03 for an interaction between GW1100 and RGZ; Fig. 3A). In contrast, GPR40 knockdown reduced both basal and RGZ-stimulated PPRE reporter activity (p < 0.001 for GPR40 shRNA main effect; p = 0.32 for an interaction between RGZ and GPR40 shRNA; Fig. 3B). Overexpression of DN-Gαq, a mutant that competes with endogenous wild-type G-proteins for GPR40 coupling (43), also blocked both basal and RGZ-induced PPRE reporter activity (p < 0.001 for DN-Gαq mutant main effect; p = 0.88 for an interaction between RGZ and DN-Gαq; Fig. 3C). In addition, GPR40 overexpression enhanced both basal and RGZ-induced PPARγ transcriptional activity (p < 0.001 for GPR40 plasmid main effect; p = 0.11 for an interaction between RGZ and GPR40 plasmid; Fig. 3D). Consistent with our findings for RGZ-induced p38 MAPK activation (Fig. 2E), l-NAME (0–1 mm), a •NO synthase inhibitor, did not affect basal or RGZ-induced PPRE reporter activity (p = 0.25 for the main effect of l-NAME; Fig. 3E), further evidence that •NO plays no role in RGZ/GPR40/p38 MAPK/PPARγ signaling in EA.hy926 cells.
FIGURE 3.

RGZ induces PPRE-driven transcription through GPR40. A, GW1100, a specific GPR40 antagonist, blocked RGZ-induced PPARγ reporter gene activity in EA.hy926 cells (p < 0.001 for RGZ versus GW1100 plus RGZ). GW1100 (20 μm) or vehicle CTRL was added 40 min before RGZ (10 μm) or its vehicle CTRL. GPR40 shRNA knockdown (B) and expression of a Gαq dominant negative mutant (DN-Gαq) (C) inhibited both basal and RGZ-induced PPARγ reporter gene activity in EA.hy926 cells (p < 0.001 for GPR40 shRNA main effect in B; p < 0.001 for DN-Gαq main effect in C). D, GPR40 overexpression increased both basal and RGZ-induced PPARγ reporter gene activity in EA.hy926 cells (p < 0.001 for GPR40 plasmid main effect). E, l-NAME, a nitric-oxide synthase inhibitor, did not affect RGZ-induced PPARγ reporter gene activity in EA.hy926 cells (p = 0.25 for the main effect of l-NAME). l-NAME (0–1 mm) was added 1 h before RGZ (10 μm) or its CTRL. EA.hy926 cells were co-transfected with PPRE-LUC reporter and PPARγ2 expression plasmid with or without specific GPR40 shRNA, CTRL shRNA, a DN-Gαq plasmid, a GPR40 expression plasmid, or the appropriate control empty vector, as indicated. At 48 h after transfection, cells were treated with RGZ (10 μm) or vehicle CTRL for 24 h prior to the measurement of luciferase activity. Results are presented as means ± S.E. (error bars) of independent experiments in A–D (n = 5) and E (n = 3).
RGZ/GPR40/p38 MAPK Signaling Boosts the Transcription of PPARγ Target Genes in PAECs
The RGZ/GPR40/p38 MAPK/PPARγ signaling transduction pathway was further explored in PAECs. Similar to EA.hy926 cells, a hybrid human endothelial line, RGZ increased p38 MAPK phosphorylation ∼2-fold in PAECs (p < 0.001 for RGZ versus control within the control siRNA condition; Fig. 4A), and siRNA silencing of p38α blocked this effect (p = 0.43 for RGZ versus control within the p38 MAPK siRNA condition; p = 0.009 for an interaction between p38 MAPK siRNA and RGZ; Fig. 4A). Knockdown of p38α MAPK decreased total p38 MAPK protein about 80% (p < 0.001 for the main effect of p38α MAPK siRNA; Fig. 4A). PAECs expressed PPARγ protein, and neither RGZ (p = 0.55 for the main effect of RGZ; Fig. 4A) nor p38α MAPK knockdown (p = 0.10 for the main effect of p38α MAPK siRNA; Fig. 4A) significantly altered PPARγ expression. Furthermore, p38α MAPK siRNA silencing significantly inhibited RGZ-induced expression of CD36, CYP1A1, and FABP4, three PPARγ target genes (p < 0.001 for all three genes, RGZ versus RGZ plus p38α MAPK siRNA; Fig. 4B). Similar to effects in a PPARγ reporter system (Fig. 1F), significant interactions between RGZ and p38 MAPK knockdown were seen for CD36 (p < 0.001), CYP1A1 (p = 0.04), and FABP4 (p = 0.02). Table 1 shows all interactions (p values) between RGZ activation of PPARγ and siRNA silencing of GPR40 pathway components on the expression of these target genes in PAECs.
FIGURE 4.

RGZ induces PPARγ target genes through GPR40 and the downstream activation of p38 MAPK in PAECs. A, RGZ-induced p38α MAPK phosphorylation (p < 0.001 for RGZ versus CTRL within the CTRL siRNA condition). Knockdown of p38α MAPK blocked RGZ-induced p38 MAPK phosphorylation (p = 0.43 for RGZ versus CTRL within the p38α siRNA condition; p = 0.009 for an interaction between p38 MAPK siRNA and RGZ) and decreased total p38 MAPK protein (p < 0.001 for p38α siRNA main effect) but did not alter PPARγ protein expression (p = 0.10 for p38α siRNA main effect). B, p38α MAPK siRNA silencing significantly inhibited RGZ-induced expression of PPARγ target genes (p < 0.001 for RGZ versus RGZ plus p38α siRNA for all three genes); C, GPR40 siRNA knockdown reduced GPR40 protein expression (p = 0.002 for GPR40 siRNA main effect) without affecting PPARγ expression (p = 0.56 for GPR40 siRNA main effect). D, GPR40 siRNA knockdown suppressed RGZ-induced PPARγ target genes (p ≤ 0.005, RGZ versus RGZ plus GPR40 siRNA for all three genes). PAECs were transfected with specific siRNA or a scrambled CTRL siRNA. At 48 h after transfection, cells were treated with RGZ (10 μm) or vehicle CTRL for 30 min prior to the measurement of phosphorylated p38 MAPK (pp38), total p38 MAPK (p38), and PPARγ in A and for 24 h prior to the measurement of GPR40 and PPARγ in C by Western blotting. In B and D, 48 h after siRNA transfection, cells were treated with RGZ (10 μm) or vehicle CTRL for 24 h followed by measurement of PPARγ target genes by real-time PCR. Densitometry results of three independent experiments using different PAEC donors and a representative blot are shown in A and C. In B (n = 5) and D (n = 4), results are presented as geometric means ± S.E. (error bars) of independent experiments using different PAEC donors.
TABLE 1.
The PPARγ agonist RGZ and signaling through the GPR40 pathway function synergistically to induce PPARγ-regulated genes
| siRNA target |
p values for RGZ and siRNA silencing interactions |
|||
|---|---|---|---|---|
| CD36 | CYP1A1 | FABP4 | All three genes combined | |
| p38 MAPK | <0.001 | 0.04 | 0.02 | <0.001 |
| GPR40 | 0.04 | 0.02 | 0.11 | 0.03 |
| PGC1α | 0.005 | 0.18 | 0.006 | 0.03 |
| SIRT1 | <0.001 | 0.09 | 0.06 | <0.001 |
| EP300 | <0.001 | 0.007 | <0.001 | 0.008 |
| All siRNA silencing combined | <0.001 | 0.001 | <0.001 | <0.001 |
Like EA.hy926 cells, PAECs also express GPR40 as measured by Western blotting, and RGZ did not alter this expression (p = 0.30 for the main effect of RGZ; Fig. 4C). GPR40 siRNA significantly reduced GPR40 protein expression (p = 0.002 for the main effect of GPR40 siRNA; Fig. 4C) but had no effect on PPARγ expression in PAECs (p = 0.56 for the main effect of GPR40 siRNA; Fig. 4C). Similar to p38α MAPK knockdown, GPR40 siRNA significantly blocked the RGZ-induced PPARγ target genes CD36, CYP1A1, and FABP4 (p ≤ 0.005 for all three genes, RGZ versus RGZ plus GPR40 siRNA; Fig. 4D). Again, RGZ and GPR40 siRNA silencing appeared to interact at endogenous PPARγ target genes (Fig. 4D and Table 1). This was in contrast to the additive effects of RGZ and GPR40, knockdown, DN-Gαq mutant, or overexpression on a PPARγ reporter (Fig. 3). PAEC donor, passage number, and day of experiment varied between Fig. 4, B and D, possibly accounting for the observed variability in gene expression among the siRNA control conditions. These results further support the existence of a functional pathway in endothelial cells that connects GPR40 on the cell surface to nuclear PPARγ signaling, two receptors that can be activated by shared ligands.
PGC1α Transduces RGZ/GPR40/p38 MAPK Signals to PPARγ in PAECs
PGC1α is an essential co-activator of PPARγ. Activated p38 MAPK phosphorylates PGC1α and thereby activates and stabilizes PGC1α protein (18, 19, 48, 49). Therefore, we investigated the role of PGC1α in transducing GPR40/p38 MAPK signals to PPARγ in human PAECs. RGZ induced PGC1α phosphorylation (p = 0.001 for RGZ versus control within the control siRNA condition; Fig. 5A), and siRNA knockdown of either p38α MAPK (p = 0.45) or GPR40 abolished this effect (p = 0.60; Fig. 5A). RGZ also increased total PGC1α protein expression in PAECs (p = 0.01 within control siRNA; Fig. 5B), an effect that was similarly blocked by either p38α MAPK or GPR40 siRNA silencing (p ≥ 0.62 for both; Fig. 5B). Notably, RGZ increased total and phosphorylated PGC1α without altering their ratio (Fig. 5, A and B), and both effects may have contributed to PPARγ transcriptional activation.
FIGURE 5.

PGC1α transduces RGZ/GPR40/p38 MAPK signaling to PPARγ in PAECs. A, RGZ induced PGC1α phosphorylation, an effect blocked by siRNA knockdown of p38α MAPK or GPR40 (p = 0.001 for RGZ versus CTRL within CTRL siRNA; p ≥ 0.45, RGZ versus CTRL within p38α or GPR40 siRNA conditions). PAECs were transfected with specific siRNA or scrambled CTRL siRNA, as indicated. At 48 h after transfection, cells were treated with RGZ (10 μm) or vehicle CTRL for 1 h prior to the extraction of nuclear protein for immunoprecipitation with phosphothreonine and phosphoserine antibodies. Phospho-PGC1α (pPGC1α) in immunoprecipitates was detected by Western blotting with anti-PGC1α. B, RGZ induced PGC1α protein expression in PAECs, an effect blocked by siRNA knockdown of p38α MAPK or GPR40 (p = 0.01 for RGZ versus CTRL within CTRL siRNA; p ≥ 0.62, RGZ versus CTRL within p38α or GPR40 siRNA). Cells were transfected as in A. At 48 h after transfection, cells were treated with RGZ (10 μm) or vehicle CTRL for 4 h prior to the extraction of nuclear protein for measurement of PGC1α by Western blotting. C, RGZ slightly decreased (p < 0.001 for RGZ versus CTRL within CTRL siRNA), but SIRT1 siRNA increased PGC1α acetylation in PAECs (p < 0.001 for SIRT1 siRNA versus CTRL siRNA in the absence and presence of RGZ). At 48 h after transfection, PAECs were treated with RGZ (10 μm) or vehicle CTRL for 4 h prior to the extraction of whole cell lysates for immunoprecipitation with anti-acetyl-lysine antibodies. Acetyl-PGC1α (Ac-PGC1α) in immunoprecipitates was detected by Western blotting with anti-PGC1α. D, siRNA knockdown of PGC1α or SIRT1 inhibited RGZ-induced PPARγ target genes in PAECs (p < 0.001 for RGZ versus RGZ plus PGC1α or SIRT1 siRNA for CD36, CYP1A1, and FABP4). Results are presented as geometric means ± S.E. (error bars) of five independent experiments using different PAEC donors. E and F, specific siRNA for PGC1α and SIRT1 reduced their protein expression in PAECs, respectively (p ≤ 0.001, main effects of PGC1α and SIRT1 siRNA). Cells were transfected with siRNA for PGC1α or SIRT1 or scrambled CTRL siRNA. At 48 h after transfection, PAECs were treated with RGZ (10 μm) or vehicle CTRL for 24 h prior to isolation of total RNA for measurement of PPARγ target genes by real-time PCR or for 4 h prior to the preparation of nuclear protein or whole cell lysates for measurement of PGC1α and SIRT, respectively, by Western blotting. Densitometry results of independent experiments using different PAEC donors (top panels) and a representative blot (bottom panels) are shown in A and B (n = 4), C (n = 5), E (n = 3), and F (n = 4).
In contrast to the activation of PGC1α by p38 MAPK-mediated phosphorylation, acetylation inhibits PGC1α activity and therefore also plays a key regulatory role in the co-transcriptional activity of PGC1α (24, 25). SIRT1 binds to and deacetylates PGC1α both in vivo and in vitro and appears to be required for PGC1α activation (24, 25). Therefore, we evaluated the role of SIRT1 in RGZ/GPR40/p38 MAPK/PGC1α PPARγ signal transduction. RGZ decreased PGC1α acetylation in PAECs (p < 0.001 for RGZ versus control within the control siRNA condition; Fig. 5C), whereas SIRT1-specific siRNA increased PGC1α acetylation (p < 0.001 for SIRT1 siRNA versus control siRNA in the absence and presence of RGZ; Fig. 5C) and abolished the decrease in PGC1α acetylation seen with RGZ (p = 0.37 for RGZ versus control within the SIRT1 siRNA condition; Fig. 5C).
As expected and consistent with the importance of PGC1α as a PPARγ transcriptional co-activator, siRNA silencing of either PGC1α or SIRT1 significantly inhibited RGZ-induced PPARγ target genes (Fig. 5D, left and right panels, respectively) including CD36, CYP1A1, and FABP4 (p < 0.001 for all three genes, RGZ versus RGZ plus PGC1α or SIRT1 siRNA; Fig. 5D). As seen for p38 MAPK and GPR40 knockdown, the silencing of either PGC1α or SIRT1 profoundly reduced the ability of RGZ to induce PPARγ target genes (Fig. 5D and Table 1). Specific siRNA for PGC1α and SIRT1 effectively reduced PGC1α (p = 0.001 for the main effect of PGC1α siRNA; Fig. 5E) and SIRT1 (p < 0.001 for the main effect of SIRT1 siRNA; Fig. 5F) protein expression in PAECs, respectively. RGZ did not alter SIRT1 protein expression (p = 0.83 for the main effect of RGZ; Fig. 5F) or phosphorylation state (data not shown).
EP300 Contributes to the RGZ/GPR40/p38 MAPK/PGC1α/PPARγ Signaling Pathway
EP300, a general transcriptional co-activator with intrinsic histone acetyltransferase and chromatin remodeling activity, has been shown to interact with PGC1α and PPARγ, thereby enhancing PPARγ transcriptional activity (50, 51). Importantly, p38 MAPK has been demonstrated to directly phosphorylate EP300, thereby potentiating its acetyltransferase activity (22, 52). Therefore, the role of EP300 in the pathway described here was investigated in human endothelial cells. Consistent with the role of PGC1α in PPARγ signaling, PGC1α overexpression in EA.hy926 cells increased PPRE reporter activity (p < 0.001 for a plasmid dose effect in the absence of RGZ; Fig. 6A). A similar dose response was seen in the presence of RGZ, but the RGZ effect became smaller with increasing amounts of PGC1α plasmid (p < 0.001 for a negative interaction between RGZ and PGC1α; Fig. 6A), possibly because RGZ/p38 MAPK-dependent induction and phosphorylation of PGC1α was rendered less important by direct PGC1α overexpression.
FIGURE 6.

CBP/EP300 participation in RGZ/GPR40/p38 MAPK/PGC1α/PPARγ signaling. A–C, overexpression of PGC1α, CBP/EP300, or both enhanced PPARγ reporter gene activity. In A, PGC1α overexpression increased PPRE reporter activity in the absence of RGZ (p < 0.001 for a plasmid dose effect); the PGC1α effect became smaller in the presence of RGZ (p < 0.001 for a negative interaction between RGZ and PGC1α). In B, EP300 overexpression also increased both basal and RGZ-induced PPRE reporter activity (p = 0.003 for the main effect; p = 0.68 for an interaction between RGZ and EP300 plasmid). In C, PGC1α and EP300 additively enhanced PPRE reporter activity in the absence and presence of RGZ (p < 0.001 for PGC1α and EP300 main effects; p = 0.77 for an interaction between PGC1α and EP300). EA.hy926 cells were co-transfected with PPRE-LUC reporter; a PPARγ2 expression plasmid; and either a PGC1α expression plasmid, an EP300 expression plasmid, or both, as indicated. DNA amounts were balanced with the empty vector pcDNA3.1 plasmid. At 24 h after transfection, cells were treated with RGZ (10 μm) or vehicle CTRL for 24 h prior to the measurement of luciferase activity. D, RGZ induced EP300 phosphorylation in EA.hy926 cells (p = 0.01 for RGZ versus CTRL within CTRL), an effect blocked by SB, a specific p38 MAPK inhibitor, or GW1100, a specific GPR40 antagonist (p ≥ 0.19 for RGZ versus CTRL with SB or GW1100). Cells were treated with SB (10 μm), GW1100 (20 μm), or vehicle CTRL for 40 min and then incubated with RGZ (10 μm) or vehicle CTRL for another 15 min prior to the extraction of whole cell lysates for measurement of phosphorylated and total EP300 by Western blotting. Densitometry results of three independent experiments and a representative blot are shown. E, siRNA knockdown of EP300 inhibited PPARγ reporter gene activity (p < 0.001 for EP300 siRNA main effect; p = 0.53 for an interaction between EP300 siRNA and RGZ). EA.hy926 cells were first transfected with EP300 siRNA or scrambled CTRL siRNA for 48 h and then co-transfected with PPRE-LUC reporter and a PPARγ2 expression plasmid for 24 h, followed by an additional 24-h treatment of RGZ (10 μm) or vehicle CTRL prior to the measurement of luciferase activity. F, EP300 siRNA inhibited RGZ-induced PPARγ target genes (p < 0.001 for RGZ versus RGZ plus EP300 siRNA for all three genes) in PAECs. G, EP300 siRNA reduced EP300 mRNA in EA.hy926 cells and PAECs (p < 0.001 for the main effect of EP300 siRNA in both cell types). Cells were transfected with EP300 siRNA or scrambled CTRL siRNA for 48 h and then treated with RGZ (10 μm) or vehicle CTRL for 24 h prior to isolation of total RNA for measurement of PPARγ target genes and EP300 mRNA by real-time PCR. H, RGZ increased the binding of PPARγ (p < 0.001), PGC1α (p = 0.004), and EP300 (p = 0.04) to PPRE in the CD36 promoter (RGZ versus CTRL within the CTRL condition), an effect blocked by SB or GW1100 (p ≥ 0.18, RGZ versus CTRL for both inhibitors and all three proteins). PAECs were treated with SB (10 μm), GW1100 (20 μm), or vehicle control (CTRL) for 40 min and then incubated with RGZ (10 μm) or vehicle CTRL for another 24 h prior to ChIP assay with anti-PPARγ, anti-PGC1α, anti-EP300, or control IgG. Densitometry measurements were normalized to input DNA. Representative PCR results are shown for each pull-down. Panels represent the results of three (A–F) or four (G and H) independent experiments. For PAECs, different donors were used in performing replicates. Error bars, S.E.
EP300 overexpression also increased both basal and RGZ-induced PPRE reporter activity (p = 0.003 for EP300 main effect; p = 0.68 for an interaction between EP300 and RGZ; Fig. 6B). Although PGC1α overexpression had a stronger overall effect on PPRE reporter activity (p < 0.02 for PGC1α versus EP300 in both the absence and presence of RGZ; Fig. 6C), EP300 overexpression better preserved RGZ effect size and increased PPRE reporter activity additively with PGC1α in the presence of RGZ (p < 0.001 for PGC1α and EP300 main effects; p = 0.77 for an interaction between PGC1α and EP300; Fig. 6C). Notably, RGZ treatment of EA.hy926 cells increased EP300 phosphorylation in EA.hy926 cells (p = 0.01 for RGZ versus control within the control condition; Fig. 6D), an activating event blocked by either p38 MAPK (SB) or GPR40 (GW1100) inhibition (p ≥ 0.19 for RGZ versus control in the presence of SB or GW1100; Fig. 6D). Specific siRNA silencing of EP300 inhibited both basal and RGZ-induced PPRE reporter activity in EA.hy926 cells (p < 0.001 for EP300 siRNA main effect; p = 0.53 for an interaction between EP300 siRNA and RGZ; Fig. 6E). Likewise, siRNA knockdown of EP300 in PAECs significantly inhibited RGZ-induced PPARγ target genes, CD36, CYP1A1, and FABP4 (p < 0.001 for all three, RGZ versus RGZ plus EP300 siRNA; Fig. 6F). Similar to the knockdown of other GPR40 pathway components, EP300 silencing markedly interfered with the ability of RGZ/PPARγ signaling to induce CD36, CYP1A1, and FABP4 (Fig. 6F and Table 1). Efficient knockdown of EP300 mRNA expression was achieved in both EA.hy926 cells and PAECs (p < 0.001 for both; Fig. 6G).
RGZ-induced Binding of PPARγ, PGC1α, and EP300 to the Proximal CD36 Promoter; Dependence on p38 MAPK and GPR40
PGC1α and EP300 remodel and open chromatin for active transcription. Therefore, RGZ/GPR40/p38 MAPK signaling may be critical for certain genes and in some cell types for PPARγ transcriptional activation. ChIP assays in human PAECs demonstrated that RGZ increased the binding of endogenous PPARγ (p < 0.001), PGC1α (p = 0.004), and EP300 (p = 0.04) to native PPREs in the proximal CD36 promoter (comparing RGZ with control in the absence of SB or GW1100; Fig. 6H). These effects on PPARγ, PGC1α, and EP300 binding were all abolished by either p38 MAPK (SB) or GPR40 (GW1100) inhibition (p ≥ 0.18 for RGZ versus control in the presence of SB or GW1100; p ≤ 0.03 for interactions between RGZ and SB or GW1100 on the binding of PPARγ, PGC1α, and EP300; Fig. 6H).
Discussion
Our results demonstrate that GPR40 and PPARγ can function together as an integrated two-receptor signal transduction pathway. Besides the direct activation of its canonical receptor PPARγ, RGZ also required GPR40 to optimally propagate a PPARγ nuclear signal in human endothelium (Fig. 7). GPR40 and PPARγ appeared to function at least additively and sometimes synergistically to initiate PPARγ genomic responses, depending on the transcriptional context. This conclusion is based on the following: 1) RGZ activated p38 MAPK; 2) PPARγ DNA binding and reporter activity was at least partially p38 MAPK-dependent; 3) GPR40 inhibition or knockdown blocked RGZ activation of p38 MAPK; 4) inhibition of GPR40 signaling, including use of an antagonist, GPR40 silencing, or expression of a DN-Gαq mutant, suppressed, whereas GPR40 overexpression further increased, RGZ-induced PPRE reporter activity; 5) RGZ activation of p38 MAPK and the PPRE reporter was independent of •NO synthase, and •NO activation of p38 MAPK was likewise GPR40-independent; and importantly, 6) in human primary PAECs, knockdown of p38 MAPK or GPR40 substantially reduced the ability of RGZ to induce PPARγ target genes.
FIGURE 7.
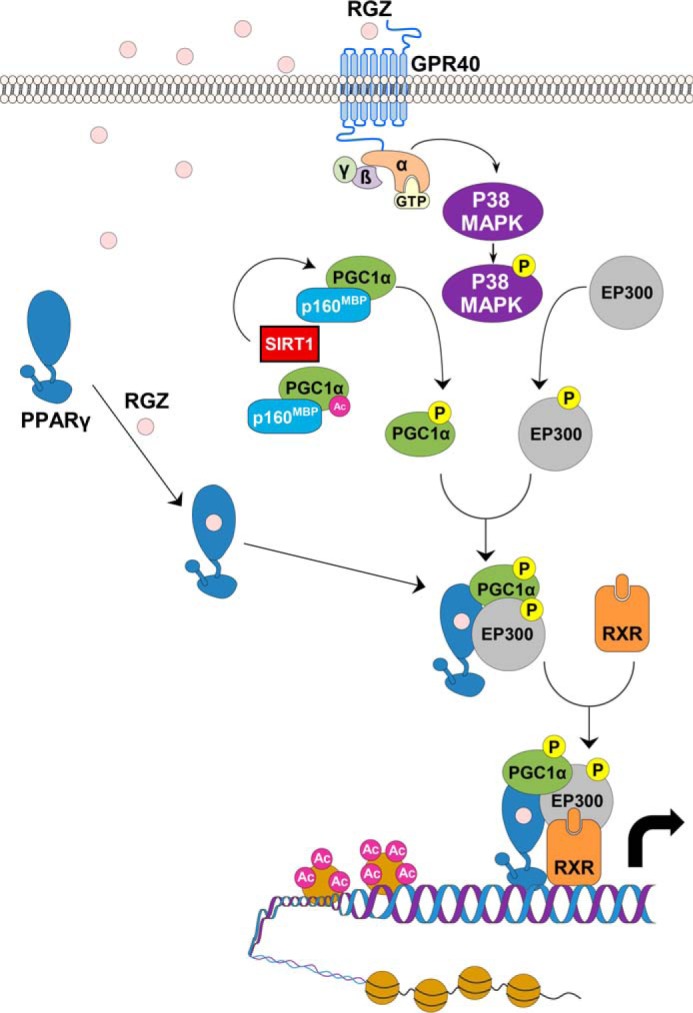
Proposed GPR40 and PPARγ integrated signal transduction pathway in human endothelial cells. In the classical PPARγ signaling pathway, RGZ binds directly to PPARγ, inducing a conformational change that results in its dissociation from co-repressors (not depicted), such as nuclear co-repressor and histone deacetylases, and the recruitment of co-activators, including PGC1α and EP300. However, as shown here, RGZ and other PPARγ ligands also bind to and activate GPR40 on the cell surface, resulting in p38 MAPK phosphorylation, which in turn phosphorylates and thereby activates both PGC1α and EP300. As noted, PGC1α deacetylation by SIRT1 is also essential for its activation. Phosphorylation releases PGC1α from its repressor p160MBP and leads to a conformational change, permitting PGC1α to dock with PPARγ and recruit newly activated EP300. EP300 is a histone acetyltransferase that remodels local chromatin and enhances gene transcription. The activated PPARγ complex thus heterodimerizes with retinoid X receptor, which binds to peroxisome proliferator response elements in accessible promoters, inducing the transcription of target genes.
In addition to these findings, RGZ treatment of PAECs increased the phosphorylation and expression of PGC1α, a key co-activator of PPARγ (50). Knockdown of either p38α MAPK or GPR40 abolished these effects of RGZ on PGC1α. RGZ also modestly decreased PGC1α acetylation, an activating event; knockdown of SIRT1, a deacetylase, eliminated this effect and increased PGC1α acetylation. As expected, given its essential role in the formation of a PPARγ transcriptional activation complex (50), knockdown of PGC1α or its essential activator SIRT1 inhibited RGZ-induced PPARγ target gene expression in PAECs. Finally, EP300, an acetylase that docks with PGC1α and remodels chromatin to optimize the transcription of PPARγ target genes (50, 53), was also phosphorylated and activated by p38 MAPK. Like PGC1α, EP300 appeared to play an important downstream role in RGZ/GPR40/p38 MAPK modulation of PPARγ signaling. Collectively, these experiments demonstrate that p38 MAPK, PGC1α, and EP300 link GPR40 to downstream PPARγ genomic signaling. Binding to and activating both GPR40 and PPARγ appears to be a common feature of several PPARγ agonists (29–34). This direct connection between GPR40 signaling and PPARγ transcriptional activation argues that the effects of these ligands on human endothelium might be best understood as a cognate two-receptor system, integrated by p38 MAPK, PGC1α, and EP300.
Activation of p38 MAPK increases transcription of PPARγ-regulated genes in endothelial cells (39) and adipocytes (20, 21, 54). TZDs (20, 55–57) have been long known to activate p38 MAPK independent of PPARγ in various cell types, including adipocytes, astrocytes, and epithelial cells. However, the potential role of GPR40 was not appreciated at the time of these early studies, and the underlying mechanisms seemed to be cell type-dependent (20, 55–57). Reactive oxygen species were implicated in astrocytes (55) and adipocytes (20), whereas endoplasmic reticulum stress was implicated in liver epithelial cells (57). Here, RGZ activation of p38 MAPK was directly tied to GPR40 in human endothelium. Both the GPR40 antagonist GW1100 and GPR40 gene silencing significantly blocked RGZ-induced p38 MAPK phosphorylation. This finding is consistent with recent reports that TZDs bind to and activate GPR40 in bronchial epithelial cells (32), osteocytes (33), and GPR40-transfected HEK293 cells (31, 58), causing the rapid phosphorylation of p38 MAPK (31, 33, 58).
As already noted, p38 MAPK has been shown to phosphorylate the PPARγ co-activator PGC1α (18–21). Phosphorylation disrupts PGC1α binding to its repressor, p160 Myb-binding protein (p160MBP), freeing it to dock with PPARγ (19, 53). Docking with PPARγ changes the conformation of PGC1α and allows binding of EP300, a histone acetyltransferase essential for PGC1α/PPARγ-dependent gene transcription (19, 50, 53, 59). Moreover, p38 MAPK phosphorylation of PGC1α increases its half-life (18). Furthermore, the p38 MAPK/ATF2 pathway induces PGC1α mRNA transcription (48, 49), increasing PGC1α protein expression and further enhancing PGC1α/PPARγ-mediated gene transcription. In addition to PGC1α, p38 MAPK also phosphorylates EP300, potentiating its histone acetyltransferase activity (22, 52), an additional mechanism by which p38 MAPK contributes to the transcriptional activation of PPARγ. Consistent with these previous findings, PGC1α and EP300 were shown here to connect RGZ/GPR40/p38 MAPK transmembrane signaling to the downstream transcriptional activation of RGZ/PPARγ in the cell nucleus.
Like phosphorylation, reversible acetylation is another key modulator of PGC1α (24, 25). So far, only two proteins have been unequivocally shown to regulate the reversible acetylation of PGC1α, the acetyltransferase GCN5 (23) and the NAD+-dependent deacetylase SIRT1 (24, 25). GCN5 acetylates and inhibits PGC1α activity (23), whereas SIRT1 deacetylates and enhances PGC1α activity and in turn induces transcription of its target genes (24, 25). Overexpression of Sirt1 in the liver of mice induces gluconeogenic genes under the control of PGC1α, whereas Sirt1 knockdown attenuates this effect (60). Also, in skeletal muscle, Sirt1 is required for PGC1α-mediated induction of the fatty acid oxidation pathway (61). In the present study, RGZ was seen to modestly decrease PGC1α acetylation in endothelial cells; SIRT1 knockdown eliminated this effect, increasing PGC1α acetylation. These results suggest that PGC1α phosphorylation might facilitate its deacetylation by SIRT1. It was previously reported that JNK1 phosphorylates SIRT1 and promotes its enzymatic activity in HEK293T cells (62); p38 MAPK has been reported to increase SIRT1 expression in neurons (63) and decrease it in chondrocytes (64). In our study, RGZ did not affect either SIRT1 phosphorylation or expression in endothelial cells. However, consistent with its essential role in activating PGC1α via deacetylation, SIRT1 knockdown significantly inhibited RGZ-induced PPARγ target gene expression in endothelial cells.
Although the effects of RGZ/PPARγ and RGZ/GPR40 were mostly additive in PPRE reporter assays, the dual activation of both receptors appeared interdependent and synergistic at PPARγ-regulated target genes in the chromatin microenvironment (see Table 1 for a summary of interactions between RGZ and GPR40 pathway siRNA silencing). Across all three PPARγ-regulated genes combined, interaction testing between RGZ and each siRNA target indicated that RGZ activation of GPR40 and PPARγ function synergistically (Table 1). Likewise, across all siRNA silencing combined, each PPARγ-regulated gene, CD36 (p < 0.001), CYP1A1 (p = 0.001) and FABP4 (p < 0.001), demonstrated significant interactions between RGZ-induced PPARγ responses and signaling through the GPR40 pathway (Table 1). Largely additive effects on a PPRE reporter plasmid and evidence for GPR40 and PPARγ interdependence at endogenous genes might be explained by the ability of the proposed signaling cascade to actively remodel chromatin. Consistent with this concept, p38 MAPK or GPR40 inhibition both markedly blocked the RGZ-induced binding of PPARγ, PGC1α, and EP300 to a PPRE-rich site in the proximal promoter of CD36, a prototypic PPARγ target gene.
TZDs, synthetic ligands of PPARγ, including RGZ, ciglitazone, troglitazone, and pioglitazone, have all been shown to activate GPR40 with subsequent signal transduction through stress kinases (29–34). RGZ compared with pioglitazone (two TZDs used to treat type 2 diabetes mellitus) produces a more potent and prolonged activation of ERK1/2 (31), a stress kinase pathway associated with vascular inflammation (65). These differences in the potency and sustainability of ERK1/2 activation could potentially explain some of the efficacy and safety differences among existing synthetic PPARγ ligands. Different from TZDs, 15-deoxy-Δ12,14-prostaglandin J2, a natural PPARγ ligand (27), did not appear to activate GPR40 in bronchial epithelial cells (32). Conversely, agonists selective for GPR40 have been described with little or no effect on PPARγ activity (29, 66). Therefore, it may be possible to design drugs that activate PPARγ independently of GPR40 or selectively activate GPR40/p38 MAPK while circumventing GPR40/ERK activation. Such selective agents or biased ligands (67, 68) might arguably be less inflammatory and thus have better risk/benefit profiles in patients with vascular disease. In addition, unexplored effects through other unidentified cognate GPR and nuclear receptor pairs, as exemplified by GPR40/PPARγ, could explain important safety and efficacy differences among nuclear receptor-directed drugs.
Author Contributions
S. W. and R. L. D. conceived the study, designed experiments, analyzed and interpreted data, and wrote the paper. S. W. performed transfections, Western blots, PPARγ target gene real-time PCR experiments, and the chromatin immunoprecipitation assays. K. S. A. identified the role played by EP300, helped design key experiments related to acetylation, maintained the pulmonary artery endothelial cell cultures, and revised drafts of the manuscript. J. M. E., E. J. D., and G. A. F. contributed to the scientific concept, provided technical advice, raised critical questions, and revised drafts of the manuscript. E. J. D. also lent important expertise in nuclear receptor signaling. J. Y. W. performed the experiments shown in Fig. 2, B–D. A. P. started the study and performed the experiments shown in Fig. 1. R. C. and J. S. performed the statistical analyses. All authors read, edited, and approved the final version of the manuscript.
Acknowledgments
We thank Drs. Ronald M. Evans and Ae-Kyung Yi for kindly sharing plasmids and Kai Ge for providing the HeLaS/F-PPARγ cell line. We also appreciate the help of Kelly Byrne in editing and formatting the manuscript and figures.
This work was supported, in whole or in part, by National Institutes of Health intramural funds. The authors declare that they have no conflicts of interest with the contents of this article.
- TZDs
- thiazolidinediones
- PPARγ
- peroxisome proliferator-activated receptor γ
- PGC1α
- PPARγ co-activator-1α
- PPRE
- peroxisome proliferator response elements
- CBP
- CREB-binding protein
- CREB
- cAMP-response element-binding protein
- GPR40
- G-protein-coupled receptor 40
- RGZ
- rosiglitazone
- PGZ
- pioglitazone
- L-NAME
- Nω-nitro-l-arginine methyl ester hydrochloride
- DTANO
- DETA NONOate
- SB
- SB202190
- CAT
- chloramphenicol acetyltransferase
- LUC
- luciferase
- DN-p38
- dominant negative p38 MAPK
- DN-Gαq
- dominant negative G-protein αq
- PAEC
- primary human pulmonary artery endothelial cell
- CTRL
- control.
REFERENCES
- 1. Buse J. B., Ginsberg H. N., Bakris G. L., Clark N. G., Costa F., Eckel R., Fonseca V., Gerstein H. C., Grundy S., Nesto R. W., Pignone M. P., Plutzky J., Porte D., Redberg R., Stitzel K. F., Stone N. J., American Heart Association, and American Diabetes Association (2007) Primary prevention of cardiovascular diseases in people with diabetes mellitus: a scientific statement from the American Heart Association and the American Diabetes Association. Circulation 115, 114–126 [DOI] [PubMed] [Google Scholar]
- 2. Yki-Järvinen H. (2004) Thiazolidinediones. N. Engl. J. Med. 351, 1106–1118 [DOI] [PubMed] [Google Scholar]
- 3. McGuire D. K., Inzucchi S. E. (2008) New drugs for the treatment of diabetes mellitus: part I: thiazolidinediones and their evolving cardiovascular implications. Circulation 117, 440–449 [DOI] [PubMed] [Google Scholar]
- 4. Ketsawatsomkron P., Pelham C. J., Groh S., Keen H. L., Faraci F. M., Sigmund C. D. (2010) Does peroxisome proliferator-activated receptor-γ (PPARγ) protect from hypertension directly through effects in the vasculature? J. Biol. Chem. 285, 9311–9316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goldberg R. B., Kendall D. M., Deeg M. A., Buse J. B., Zagar A. J., Pinaire J. A., Tan M. H., Khan M. A., Perez A. T., Jacober S. J., and GLAI Study Investigators (2005) A comparison of lipid and glycemic effects of pioglitazone and rosiglitazone in patients with type 2 diabetes and dyslipidemia. Diabetes Care 28, 1547–1554 [DOI] [PubMed] [Google Scholar]
- 6. Wang N., Verna L., Chen N. G., Chen J., Li H., Forman B. M., Stemerman M. B. (2002) Constitutive activation of peroxisome proliferator-activated receptor-γ suppresses pro-inflammatory adhesion molecules in human vascular endothelial cells. J. Biol. Chem. 277, 34176–34181 [DOI] [PubMed] [Google Scholar]
- 7. Erdmann E., Spanheimer R., Charbonnel B., and PROactive Study Investigators (2010) Pioglitazone and the risk of cardiovascular events in patients with Type 2 diabetes receiving concomitant treatment with nitrates, renin-angiotensin system blockers, or insulin: results from the PROactive study (PROactive 20). J. Diabetes 2, 212–220 [DOI] [PubMed] [Google Scholar]
- 8. Hansmann G., de Jesus Perez V. A., Alastalo T. P., Alvira C. M., Guignabert C., Bekker J. M., Schellong S., Urashima T., Wang L., Morrell N. W., Rabinovitch M. (2008) An antiproliferative BMP-2/PPARγ/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J. Clin. Invest. 118, 1846–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reddy R. C., Narala V. R., Keshamouni V. G., Milam J. E., Newstead M. W., Standiford T. J. (2008) Sepsis-induced inhibition of neutrophil chemotaxis is mediated by activation of peroxisome proliferator-activated receptor-γ. Blood 112, 4250–4258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kahn B. B., McGraw T. E. (2010) Rosiglitazone, PPARγ, and type 2 diabetes. N. Engl. J. Med. 363, 2667–2669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Spiegelman B. M. (1998) PPAR-γ: adipogenic regulator and thiazolidinedione receptor. Diabetes 47, 507–514 [DOI] [PubMed] [Google Scholar]
- 12. Pascual G., Fong A. L., Ogawa S., Gamliel A., Li A. C., Perissi V., Rose D. W., Willson T. M., Rosenfeld M. G., Glass C. K. (2005) A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature 437, 759–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hu E., Kim J. B., Sarraf P., Spiegelman B. M. (1996) Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARγ. Science 274, 2100–2103 [DOI] [PubMed] [Google Scholar]
- 14. Adams M., Reginato M. J., Shao D., Lazar M. A., Chatterjee V. K. (1997) Transcriptional activation by peroxisome proliferator-activated receptor γ is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J. Biol. Chem. 272, 5128–5132 [DOI] [PubMed] [Google Scholar]
- 15. Camp H. S., Tafuri S. R. (1997) Regulation of peroxisome proliferator-activated receptor γ activity by mitogen-activated protein kinase. J. Biol. Chem. 272, 10811–10816 [DOI] [PubMed] [Google Scholar]
- 16. Rangwala S. M., Rhoades B., Shapiro J. S., Rich A. S., Kim J. K., Shulman G. I., Kaestner K. H., Lazar M. A. (2003) Genetic modulation of PPARγ phosphorylation regulates insulin sensitivity. Dev. Cell 5, 657–663 [DOI] [PubMed] [Google Scholar]
- 17. Choi J. H., Banks A. S., Estall J. L., Kajimura S., Boström P., Laznik D., Ruas J. L., Chalmers M. J., Kamenecka T. M., Blüher M., Griffin P. R., Spiegelman B. M. (2010) Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARγ by Cdk5. Nature 466, 451–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Puigserver P., Rhee J., Lin J., Wu Z., Yoon J. C., Zhang C. Y., Krauss S., Mootha V. K., Lowell B. B., Spiegelman B. M. (2001) Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARγ coactivator-1. Mol. Cell 8, 971–982 [DOI] [PubMed] [Google Scholar]
- 19. Fan M., Rhee J., St-Pierre J., Handschin C., Puigserver P., Lin J., Jäeger S., Erdjument-Bromage H., Tempst P., Spiegelman B. M. (2004) Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to PGC-1α: modulation by p38 MAPK. Genes Dev. 18, 278–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Teruel T., Hernandez R., Benito M., Lorenzo M. (2003) Rosiglitazone and retinoic acid induce uncoupling protein-1 (UCP-1) in a p38 mitogen-activated protein kinase-dependent manner in fetal primary brown adipocytes. J. Biol. Chem. 278, 263–269 [DOI] [PubMed] [Google Scholar]
- 21. Cao W., Daniel K. W., Robidoux J., Puigserver P., Medvedev A. V., Bai X., Floering L. M., Spiegelman B. M., Collins S. (2004) p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol. Cell. Biol. 24, 3057–3067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bratton M. R., Frigo D. E., Vigh-Conrad K. A., Fan D., Wadsworth S., McLachlan J. A., Burow M. E. (2009) Organochlorine-mediated potentiation of the general coactivator p300 through p38 mitogen-activated protein kinase. Carcinogenesis 30, 106–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lerin C., Rodgers J. T., Kalume D. E., Kim S. H., Pandey A., Puigserver P. (2006) GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1α. Cell Metab. 3, 429–438 [DOI] [PubMed] [Google Scholar]
- 24. Nemoto S., Fergusson M. M., Finkel T. (2005) SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1α. J. Biol. Chem. 280, 16456–16460 [DOI] [PubMed] [Google Scholar]
- 25. Rodgers J. T., Lerin C., Haas W., Gygi S. P., Spiegelman B. M., Puigserver P. (2005) Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 434, 113–118 [DOI] [PubMed] [Google Scholar]
- 26. Sohda T., Mizuno K., Tawada H., Sugiyama Y., Fujita T., Kawamatsu Y. (1982) Studies on antidiabetic agents. I. Synthesis of 5-[4-(2-methyl-2-phenylpropoxy)-benzyl]thiazolidine-2,4-dione (AL-321) and related compounds. Chem. Pharm. Bull. 30, 3563–3573 [DOI] [PubMed] [Google Scholar]
- 27. Forman B. M., Tontonoz P., Chen J., Brun R. P., Spiegelman B. M., Evans R. M. (1995) 15-Deoxy-δ12,14-prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ. Cell 83, 803–812 [DOI] [PubMed] [Google Scholar]
- 28. Lehmann J. M., Moore L. B., Smith-Oliver T. A., Wilkison W. O., Willson T. M., Kliewer S. A. (1995) An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor γ (PPARγ). J. Biol. Chem. 270, 12953–12956 [DOI] [PubMed] [Google Scholar]
- 29. Zhou C., Tang C., Chang E., Ge M., Lin S., Cline E., Tan C. P., Feng Y., Zhou Y. P., Eiermann G. J., Petrov A., Salituro G., Meinke P., Mosley R., Akiyama T. E., Einstein M., Kumar S., Berger J., Howard A. D., Thornberry N., Mills S. G., Yang L. (2010) Discovery of 5-aryloxy-2,4-thiazolidinediones as potent GPR40 agonists. Bioorg. Med. Chem. Lett. 20, 1298–1301 [DOI] [PubMed] [Google Scholar]
- 30. Kotarsky K., Nilsson N. E., Flodgren E., Owman C., Olde B. (2003) A human cell surface receptor activated by free fatty acids and thiazolidinedione drugs. Biochem. Biophys. Res. Commun. 301, 406–410 [DOI] [PubMed] [Google Scholar]
- 31. Smith N. J., Stoddart L. A., Devine N. M., Jenkins L., Milligan G. (2009) The action and mode of binding of thiazolidinedione ligands at free fatty acid receptor 1. J. Biol. Chem. 284, 17527–17539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gras D., Chanez P., Urbach V., Vachier I., Godard P., Bonnans C. (2009) Thiazolidinediones induce proliferation of human bronchial epithelial cells through the GPR40 receptor. Am. J. Physiol. Lung Cell. Mol. Physiol. 296, L970–L978 [DOI] [PubMed] [Google Scholar]
- 33. Mieczkowska A., Baslé M. F., Chappard D., Mabilleau G. (2012) Thiazolidinediones induce osteocyte apoptosis by a G protein-coupled receptor 40-dependent mechanism. J. Biol. Chem. 287, 23517–23526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stoddart L. A., Brown A. J., Milligan G. (2007) Uncovering the pharmacology of the G protein-coupled receptor GPR40: high apparent constitutive activity in guanosine 5′-O-(3-[35S]thio)triphosphate binding studies reflects binding of an endogenous agonist. Mol. Pharmacol. 71, 994–1005 [DOI] [PubMed] [Google Scholar]
- 35. Itoh Y., Kawamata Y., Harada M., Kobayashi M., Fujii R., Fukusumi S., Ogi K., Hosoya M., Tanaka Y., Uejima H., Tanaka H., Maruyama M., Satoh R., Okubo S., Kizawa H., Komatsu H., Matsumura F., Noguchi Y., Shinohara T., Hinuma S., Fujisawa Y., Fujino M. (2003) Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 422, 173–176 [DOI] [PubMed] [Google Scholar]
- 36. Alquier T., Peyot M. L., Latour M. G., Kebede M., Sorensen C. M., Gesta S., Ronald Kahn C., Smith R. D., Jetton T. L., Metz T. O., Prentki M., Poitout V. (2009) Deletion of GPR40 impairs glucose-induced insulin secretion in vivo in mice without affecting intracellular fuel metabolism in islets. Diabetes 58, 2607–2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim H. S., Noh J. H., Hong S. H., Hwang Y. C., Yang T. Y., Lee M. S., Kim K. W., Lee M. K. (2008) Rosiglitazone stimulates the release and synthesis of insulin by enhancing GLUT-2, glucokinase and BETA2/NeuroD expression. Biochem. Biophys. Res. Commun. 367, 623–629 [DOI] [PubMed] [Google Scholar]
- 38. Yang C., Chang T. J., Chang J. C., Liu M. W., Tai T. Y., Hsu W. H., Chuang L. M. (2001) Rosiglitazone (BRL 49653) enhances insulin secretory response via phosphatidylinositol 3-kinase pathway. Diabetes 50, 2598–2602 [DOI] [PubMed] [Google Scholar]
- 39. Ptasinska A., Wang S., Zhang J., Wesley R. A., Danner R. L. (2007) Nitric oxide activation of peroxisome proliferator-activated receptor γ through a p38 MAPK signaling pathway. FASEB J. 21, 950–961 [DOI] [PubMed] [Google Scholar]
- 40. Hata K., Nishimura R., Ikeda F., Yamashita K., Matsubara T., Nokubi T., Yoneda T. (2003) Differential roles of Smad1 and p38 kinase in regulation of peroxisome proliferator-activating receptor γ during bone morphogenetic protein 2-induced adipogenesis. Mol. Biol. Cell 14, 545–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kliewer S. A., Umesono K., Noonan D. J., Heyman R. A., Evans R. M. (1992) Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature 358, 771–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yeo S. J., Gravis D., Yoon J. G., Yi A. K. (2003) Myeloid differentiation factor 88-dependent transcriptional regulation of cyclooxygenase-2 expression by CpG DNA: role of NF-κB and p38. J. Biol. Chem. 278, 22563–22573 [DOI] [PubMed] [Google Scholar]
- 43. Yu B., Gu L., Simon M. I. (2000) Inhibition of subsets of G protein-coupled receptors by empty mutants of G protein α subunits in Go, G11, and G16. J. Biol. Chem. 275, 71–76 [DOI] [PubMed] [Google Scholar]
- 44. Ge K., Cho Y. W., Guo H., Hong T. B., Guermah M., Ito M., Yu H., Kalkum M., Roeder R. G. (2008) Alternative mechanisms by which mediator subunit MED1/TRAP220 regulates peroxisome proliferator-activated receptor γ-stimulated adipogenesis and target gene expression. Mol. Cell. Biol. 28, 1081–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chu K. M., Hu O. Y., Pao L. H., Hsiong C. H. (2007) Pharmacokinetics of oral rosiglitazone in Taiwanese and post hoc comparisons with Caucasian, Japanese, Korean, and mainland Chinese subjects. J. Pharm. Pharm. Sci. 10, 411–419 [DOI] [PubMed] [Google Scholar]
- 46. Camp H. S., Li O., Wise S. C., Hong Y. H., Frankowski C. L., Shen X., Vanbogelen R., Leff T. (2000) Differential activation of peroxisome proliferator-activated receptor-γ by troglitazone and rosiglitazone. Diabetes 49, 539–547 [DOI] [PubMed] [Google Scholar]
- 47. Maciag A. E., Holland R. J., Robert Cheng Y. S., Rodriguez L. G., Saavedra J. E., Anderson L. M., Keefer L. K. (2013) Nitric oxide-releasing prodrug triggers cancer cell death through deregulation of cellular redox balance. Redox. Biol. 1, 115–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Akimoto T., Pohnert S. C., Li P., Zhang M., Gumbs C., Rosenberg P. B., Williams R. S., Yan Z. (2005) Exercise stimulates Pgc-1α transcription in skeletal muscle through activation of the p38 MAPK pathway. J. Biol. Chem. 280, 19587–19593 [DOI] [PubMed] [Google Scholar]
- 49. Bordicchia M., Liu D., Amri E. Z., Ailhaud G., Dessì-Fulgheri P., Zhang C., Takahashi N., Sarzani R., Collins S. (2012) Cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. J. Clin. Invest. 122, 1022–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Puigserver P., Adelmant G., Wu Z., Fan M., Xu J., O'Malley B., Spiegelman B. M. (1999) Activation of PPARγ coactivator-1 through transcription factor docking. Science 286, 1368–1371 [DOI] [PubMed] [Google Scholar]
- 51. Gelman L., Zhou G., Fajas L., Raspé E., Fruchart J. C., Auwerx J. (1999) p300 interacts with the N- and C-terminal part of PPARγ2 in a ligand-independent and -dependent manner, respectively. J. Biol. Chem. 274, 7681–7688 [DOI] [PubMed] [Google Scholar]
- 52. Wang Q. E., Han C., Zhao R., Wani G., Zhu Q., Gong L., Battu A., Racoma I., Sharma N., Wani A. A. (2013) p38 MAPK- and Akt-mediated p300 phosphorylation regulates its degradation to facilitate nucleotide excision repair. Nucleic Acids Res. 41, 1722–1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Handschin C., Spiegelman B. M. (2006) Peroxisome proliferator-activated receptor γ coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev. 27, 728–735 [DOI] [PubMed] [Google Scholar]
- 54. Maekawa T., Jin W., Ishii S. (2010) The role of ATF-2 family transcription factors in adipocyte differentiation: antiobesity effects of p38 inhibitors. Mol. Cell. Biol. 30, 613–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lennon A. M., Ramaugé M., Dessouroux A., Pierre M. (2002) MAP kinase cascades are activated in astrocytes and preadipocytes by 15-deoxy-Δ(12–14)-prostaglandin J(2) and the thiazolidinedione ciglitazone through peroxisome proliferator activator receptor γ-independent mechanisms involving reactive oxygenated species. J. Biol. Chem. 277, 29681–29685 [DOI] [PubMed] [Google Scholar]
- 56. Duan S. Z., Ivashchenko C. Y., Russell M. W., Milstone D. S., Mortensen R. M. (2005) Cardiomyocyte-specific knockout and agonist of peroxisome proliferator-activated receptor-γ both induce cardiac hypertrophy in mice. Circ. Res. 97, 372–379 [DOI] [PubMed] [Google Scholar]
- 57. Gardner O. S., Shiau C. W., Chen C. S., Graves L. M. (2005) Peroxisome proliferator-activated receptor γ-independent activation of p38 MAPK by thiazolidinediones involves calcium/calmodulin-dependent protein kinase II and protein kinase R: correlation with endoplasmic reticulum stress. J. Biol. Chem. 280, 10109–10118 [DOI] [PubMed] [Google Scholar]
- 58. Sugawara Y., Nishii H., Takahashi T., Yamauchi J., Mizuno N., Tago K., Itoh H. (2007) The lipid raft proteins flotillins/reggies interact with Gαq and are involved in Gq-mediated p38 mitogen-activated protein kinase activation through tyrosine kinase. Cell. Signal. 19, 1301–1308 [DOI] [PubMed] [Google Scholar]
- 59. Wallberg A. E., Yamamura S., Malik S., Spiegelman B. M., Roeder R. G. (2003) Coordination of p300-mediated chromatin remodeling and TRAP/mediator function through coactivator PGC-1α. Mol. Cell 12, 1137–1149 [DOI] [PubMed] [Google Scholar]
- 60. Rodgers J. T., Puigserver P. (2007) Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc. Natl. Acad. Sci. U.S.A. 104, 12861–12866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gerhart-Hines Z., Rodgers J. T., Bare O., Lerin C., Kim S. H., Mostoslavsky R., Alt F. W., Wu Z., Puigserver P. (2007) Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 26, 1913–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nasrin N., Kaushik V. K., Fortier E., Wall D., Pearson K. J., de Cabo R., Bordone L. (2009) JNK1 phosphorylates SIRT1 and promotes its enzymatic activity. PloS One 4, e8414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang L., Zhang L., Chen Z. B., Wu J. Y., Zhang X., Xu Y. (2009) Icariin enhances neuronal survival after oxygen and glucose deprivation by increasing SIRT1. Eur. J. Pharmacol. 609, 40–44 [DOI] [PubMed] [Google Scholar]
- 64. Hong E. H., Lee S. J., Kim J. S., Lee K. H., Um H. D., Kim J. H., Kim S. J., Kim J. I., Hwang S. G. (2010) Ionizing radiation induces cellular senescence of articular chondrocytes via negative regulation of SIRT1 by p38 kinase. J. Biol. Chem. 285, 1283–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fernandez-Pisonero I., Duenas A. I., Barreiro O., Montero O., Sanchez-Madrid F., Garcia-Rodriguez C. (2012) Lipopolysaccharide and sphingosine-1-phosphate cooperate to induce inflammatory molecules and leukocyte adhesion in endothelial cells. J. Immunol. 189, 5402–5410 [DOI] [PubMed] [Google Scholar]
- 66. Tanaka H., Yoshida S., Oshima H., Minoura H., Negoro K., Yamazaki T., Sakuda S., Iwasaki F., Matsui T., Shibasaki M. (2013) Chronic treatment with novel GPR40 agonists improve whole-body glucose metabolism based on the glucose-dependent insulin secretion. J. Pharmacol. Exp. Ther. 346, 443–452 [DOI] [PubMed] [Google Scholar]
- 67. Rajagopal S., Rajagopal K., Lefkowitz R. J. (2010) Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat. Rev. Drug Discov. 9, 373–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. DeWire S. M., Violin J. D. (2011) Biased ligands for better cardiovascular drugs: dissecting G-protein-coupled receptor pharmacology. Circ. Res. 109, 205–216 [DOI] [PubMed] [Google Scholar]