

Background: BBS9 is a component of the octameric BBSome, a complex that transports membrane proteins to the primary cilium.
Results: BBS9 has four folded domains, based on structure prediction; the N-terminal domain is a β-propeller.
Conclusion: BBS9 likely acts as a scaffold component in the BBSome coat.
Significance: The structural data help in understanding a mutation that causes Bardet-Biedl syndrome.
Keywords: analytical ultracentrifugation, chromatography, cilia, crystal structure, genetic disease, protein translocation
Abstract
The Bardet-Biedl syndrome protein complex (BBSome) is an octameric complex that transports membrane proteins into the primary cilium signaling organelle in eukaryotes and is implicated in human disease. Here we have analyzed the 99-kDa human BBS9 protein, one of the eight BBSome components. The protein is composed of four structured domains, including a β-stranded N-terminal domain. The 1.8 Å crystal structure of the 46-kDa N-terminal domain reveals a seven-bladed β-propeller. A structure-based homology search suggests that it functions in protein-protein interactions. We show that the Bardet-Biedl syndrome-causing G141R mutation in BBS9 likely results in misfolding of the β-propeller. Although the C-terminal half of BBS9 dimerizes in solution, the N-terminal domain only does so in the crystal lattice. This C-terminal dimerization interface might be important for the assembly of the BBSome.
Introduction
The primary cilium is a singular, microtubule-based protrusion from the plasma membrane found in nearly all vertebrate cell types. The microtubule bundle, termed the axoneme, forms the structural scaffold of this finger-like projection and is surrounded by the lipid bilayer except at its base, where it originates from the mother centriole in the cytoplasm (1). Unlike motile cilia, the role of the primary cilium is not locomotion but rather signal transduction and, as a consequence, is essential to Hedgehog and non-canonical Wnt signaling pathways (2). Despite being continuous with the plasma membrane, the membrane of the primary cilium has a unique composition of proteins, namely an enrichment of specific G-protein-coupled receptors involved in signaling (1, 3). How this organelle maintains the integrity and specificity of its compartment remains an active area of investigation, although a septin diffusion barrier has been shown to play a critical role (4, 5).
One such component to the establishment and maintenance of the unique protein composition of the ciliary membrane is the BBSome,2 an immunoprecipitation-stable, eight-membered protein complex comprising BBS1, BBS2, BBS4, BBS5, BBS7, BBS8, BBS9, and BBIP10 (6, 7). The genes coding for seven of these proteins were originally identified based on their mutation in a rare, autosomal recessive genetic disorder termed Bardet-Biedl syndrome (BBS (8)). BBS is a pleiotropic disease characterized by mental retardation, polycystic kidneys, obesity, vision problems, and hypogonadism (1, 8). Evidence for BBSome function was seen in BBS2 and BBS4 knock-out mice, where a G-protein-coupled receptor resident of the ciliary membrane, somatostatin receptor type 3, fails to localize to the primary cilium in neurons (9). The BBSome has been shown to localize to the ciliary membrane and to coat liposomes in an Arl6-GTP-dependent manner through thin section electron microscopy, leading to the hypothesis that the complex forms a membrane protein transport coat similar to COPI or COPII (10). Consistent with this hypothesis, proteins of the BBSome and the coat-forming elements of COPI, COPII, and clathrin share a similar domain composition and architecture (10): namely, N-terminal β-propeller and C-terminal appendage domains, as is the case for BBS1, BBS2, BBS7, and BBS9, and α-solenoid domains, as in BBS4 and BBS8 (10). Recently, an additional role of the BBSome in the assembly and recycling of intraflagellar transport particles, which move cargo along the axoneme via microtubule motors, has been identified through a loss-of-function mutation of BBS1 in Caenorhabditis elegans (11).
Given the structural homology to vesicle coat proteins on a domain level, we set out to assess whether and to what degree this homology held true on the atomic level. To this end, another group has already reported the first high-resolution crystal structure of a BBS protein, the BBS1 N-terminal β-propeller from Chlamydomonas reinhardtii in complex with the Arl6-GTPase (12). Here we present the high-resolution crystal structure of the BBS9 N-terminal β-propeller from Homo sapiens and explain the structural basis of the G141R point mutation that causes BBS in humans (13). BBS9 knockdown in zebrafish leads to defects in ciliogenesis and neuronal development that the G141R mutant fails to rescue (14).
Experimental Procedures
Cloning
The H. sapiens BBS9 open reading frame was PCR-amplified with BamHI (5′) and NotI (3′) restriction site overhangs from cDNA (Open Biosystems) and subcloned into a modified pETDuet-1 vector, containing an N-terminal His10-Arg8-ScSUMO-3C cleavage site tag, via restriction digest. Subsequent inverse PCRs were performed on this vector to remove the coding region for either the N-terminal domain (HsBBS91–407) or the C-terminal half of the protein (HsBBS9405–887). The resulting linearized vectors were recircularized by enzymatic treatment with T4 phosphonucleotide kinase (New England Biolabs) and T4 ligase (New England Biolabs). HsBBS91–407 G141R and all other point mutations were generated via site-directed mutagenesis.
Protein Expression and Purification
LOBSTR(DE3)-RIL Escherichia coli (Ref. 15, kerafast) cells transformed with the His10-Arg8-ScSUMO-3C-HsBBS91–407 pETDuet-1 vector were grown in LB medium at 37 °C to an optical density of 0.65. Cultures were shifted to 18 °C for 30 min prior to induction with 0.2 mm isopropyl-β-d-1-thiogalactopyranoside. Expression was carried out for 16 h, after which the cultures were pelleted by centrifugation at 6,000 × g for 6 min. Cell pellets were resuspended in lysis buffer (50 mm potassium phosphate, pH 8.0, 500 mm NaCl, 30 mm imidazole, pH 8.0, and 3 mm β-mercaptoethanol) at 4 °C and were lysed by passage through a cell disruptor (Constant Systems) at 25 kpsi. After lysis, 1 mm PMSF and 250 units of TurboNuclease (Eton Biosciences) were added to the lysate, which was subsequently centrifuged at 9,500 × g for 25 min to separate soluble and insoluble fractions. Nickel-Sepharose 6 Fast Flow beads (GE Healthcare) were washed twice with lysis buffer prior to addition to the soluble fraction. Nickel binding was carried out at 4 °C for 1 h. After binding, the beads were pelleted, at 2,000 × g, and washed three times with lysis buffer. Six column volumes of elution buffer (250 mm imidazole, pH 8.0, 150 mm NaCl, and 3 mm β-mercaptoethanol) were used to elute the His10-Arg8-ScSUMO-3C-HsBBS91–407. Following elution, the protein was diluted in half with 10 mm potassium phosphate, pH 8.0, 1 mm DTT, and 0.1 mm EDTA (buffer A) and loaded onto an SP Sepharose Fast Flow cation exchange column (1 or 5 ml depending on prep size, GE healthcare). Following a 2-column volume wash with buffer A, bound protein was eluted with a 15-column volume gradient of 10 mm potassium phosphate, pH 8.0, 1 mm DTT, 0.1 mm EDTA, and 1 m NaCl (buffer B). The peak corresponding to bound His10-Arg8-ScSUMO-3C-HsBBS91–407 was pooled, following verification by SDS-PAGE. The N-terminal tag was cut with human rhinovirus 3C (3C) protease, added in a 1:100 ratio, and dialyzed overnight into 10 mm potassium phosphate, pH 8.0, 150 mm NaCl, 1 mm DTT, and 0.1 mm EDTA. The dialyzed sample was run over a second SP Sepharose Fast Flow column, and the flow-through fraction was collected. After the flow-through was concentrated, the sample was run over an Superdex S200 gel filtration column equilibrated in 10 mm Tris/HCl, pH 7.4, 150 mm NaCl, 1 mm DTT, and 0.1 mm EDTA (GF buffer 1). The peak corresponding to HsBBS91–407was pooled and concentrated after SDS-PAGE analysis. Full-length HsBBS91–887 was identically expressed and purified. HsBBS9405–887 was identically expressed and purified, with the exception that gel filtration was performed in 10 mm HEPES, pH 7.4, 150 mm NaCl, 1 mm Tris(2-carboxyethyl)phosphine, and 0.1 mm EDTA (GF buffer 2). Selenomethionine(SeMet)-derivatized HsBBS91–407 was expressed as described previously (16) and purified identically to native HsBBS91–407, with the exception that 10 mm β-mercaptoethanol was used in the nickel purification and 5 mm DTT was used in subsequent steps. The HsBBS91–407 point mutant purifications were performed identically to the wild type HsBBS91–407 nickel purification.
Limited Proteolysis
22 μl of 0.3 mg/ml HsBBS91–887 was digested with 2 μl of trypsin, at one of six different concentrations, in GF buffer 1. Trypsin was used at 0.1 mg/ml or diluted 1:3, 1:9, 1:27, 1:81, or 1:243 with GF buffer 1 from the 0.1 mg/ml stock. After a 30-min incubation at room temperature, the reactions were quenched with 6 μl of SDS-PAGE loading dye and boiled for 3 min. SDS-PAGE was performed on these samples, and a band of ∼40 kDa was produced upon treatment with 1:243 diluted trypsin that was stable through treatment with 1:9 diluted trypsin. This band was excised, placed in 50% methanol, and subjected to LC-MS/MS.
Crystallization
Initial crystals of native HsBBS91–407, set up at 7.2 mg/ml, were obtained via vapor diffusion with 10% w/v PEG 4000, 0.1 m MES, pH 6.5, and 0.2 m MgCl2 as part of the PEGII suite (Qiagen) in a 96-well, sitting drop tray. Single, albeit multilayered, three-dimensional crystals were observed after 2 days at 18 °C. Crystals were transferred into a cryoprotectant solution containing the crystallization condition with 20% v/v PEG 200 and cryocooled in liquid nitrogen. SeMet-derivatized HsBBS91–407 crystals, set up at 6.8 mg/ml, were obtained in 10% w/v PEG 4000, 0.1 m trisodium citrate, pH 6.1, and 0.15 m ammonium sulfate in 24-well, hanging drop plates at 18 °C. When compared with the native crystals, the SeMet crystals were optically superior (i.e. they appeared singular and unlayered) and grew larger to a final size of 0.1 mm. The SeMet crystals were transferred into a cryoprotectant solution containing the crystallization condition with 20% v/v glycerol and cryocooled in liquid nitrogen.
Structure Determination
Datasets were collected at the Advanced Photon Source user end station 24. A complete native dataset was collected to 2.4 Å, and a complete SeMet dataset, at the selenium peak wavelength, was collected to 1.8 Å. Data reduction was performed in HKL2000 (17). Molecular replacement attempts were performed in phaser-MR, part of the PHENIX suite (18). The phase problem was solved using single anomalous dispersion, and selenium positions were determined in HYSS, run as part of the PHENIX AutoSol program, for the SeMet dataset. The anomalous data extended to 2.5 Å, as assessed by phenix.xtriage. The initial model was built using PHENIX AutoBuild with the structure factors from the SeMet dataset because it was superior to the native data. Subsequent manual building was performed in Coot (19), and phenix.refine was used for refinement.
Structure Analysis
The HsBBS91–407 non-crystallographic symmetry (NCS) homodimer interface was analyzed using PDBePISA (20). Sequence conservation was mapped onto the surface of HsBBS91–407 using ConSurf (21) and a multiple sequence alignment (MSA) generated in Jalview (22) with the MUSCLE alignment algorithm. Sequences of HsBBS91–407 homologs were obtained via National Center for Biotechnology Information (NCBI) BLAST searches (23). The structure of HsBBS9 homologs was modeled using Phyre 2 one-to-one threading (24). Electrostatic potential was mapped onto the surface of HsBBS91–407 and the modeled homologs using the APBS plugin (25) in PyMOL (26). The G141R mutation was modeled into the wild type HsBBS91–407 structure using the mutagenesis tool in Coot.
Expression Test of HsBBS91–407 Constructs in Human Cell Culture
2 × 106 HEK 293T cells were grown in DMEM + 10% inactivated fetal bovine serum in 10-cm dishes. 24 h later, cells were transfected with 1 μg of DNA of either wild type HsBBS91–407 or HsBBS91–407 G141R cloned into a pRK5-FLAG expression plasmid, using PEI. 48 h after transfection, cells were harvested in Triton lysis buffer (40 mm Hepes, pH 7.4, 1% Triton X-100, 10 mm sodium pyrophosphate, 10 mm β-glycerophosphate, 2.5 mm MgCl2, and one Complete protease inhibitor tablet (Roche Diagnostics)). After a 20-min incubation, the soluble and insoluble fractions were separated by a 10-min centrifugation step at 21,000 × g. The soluble fractions were loaded onto an SDS-PAGE gel followed by Western blotting with anti-FLAG (Cell Signaling Technologies) and anti-Raptor (for loading control) antibodies.
Analytic 3C Proteolysis of His10-Arg8-ScSUMO-3C-HsBBS91–407 Point Mutants
2 μl of 1 mg/ml 3C protease was added to 48 μl of nickel eluate, and the reaction was allowed to proceed for 30 min at 37 °C. The reaction was quenched by the addition of 5× SDS-loading dye followed by boiling for 5 min. 10 μl of boiled sample was loaded onto an SDS-PAGE gel for analysis.
Size Exclusion Chromatography (SEC) Experiments
The HsBBS91–407 concentration-dependent shift experiment was performed in GF buffer 2. 100 μl of HsBBS91–407 was loaded onto a Superdex S75 10/300 size exclusion column at 10, 5, 2.5, or 1 mg/ml. The runs were performed in direct succession, and dilutions were performed immediately prior to the size exclusion run. HsBBS9405–887 concentration-dependent shift experiment was identically performed, except a Superdex S200 10/300 column was used. The initial HsBBS91–407, HsBBS9405–887binding test was performed in GF buffer 1. HsBBS91–407 and HsBBS9405–887 were mixed in an equimolar ratio and, after incubation at 4 °C, were loaded onto a Superdex S200 10/300 column equilibrated in GF buffer 1. For the low salt binding test, HsBBS91–407 and HsBBS9405–887 were mixed in an equimolar ratio in 10 mm Tris/HCl, pH 7.4, 100 mm NaCl, 10% glycerol, 1 mm DTT, and 0.1 mm EDTA prior to loading onto an S200 10/300 column equilibrated in the same buffer. For the high-salt binding test, equimolar amounts of HsBBS91–407 and HsBBS9405–887 were mixed in 10 mm Tris/HCl, pH 7.4, 500 mm NaCl, 1 mm DTT, and 0.1 mm EDTA prior to loading onto an appropriately equilibrated Superdex S200 10/300 column. The data were plotted in Prism (GraphPad Software).
Equilibrium Analytical Ultracentrifugation
Equilibrium analytical ultracentrifugation (AUC) on HsBBS91–407 and HsBBS9405–887 was performed, in GF buffer 2, in an Optima XL-1 analytical ultracentrifuge (Beckman Coulter) using an AN-50 titanium rotor (Beckman Coulter) with a six-channel, Epon rectangular centerpiece (1.2 cm, Beckman Coulter). A280 nm measurements were taken once the samples reached equilibrium, and buffer contribution to the absorbance was removed using a reference cell, which contained GF buffer 2. HsBBS91–407, at 0.37 mg/ml, was spun to 9,500, 12,000, and 18,000 rpm. Five absorbance scans were performed at each speed, with three replicates per scan. HsBBS9405–887 at 1.2 mg/ml, in duplicate, was spun to 9,500, 14,000, and 17,000 rpm. Three absorbance scans were performed at each speed, with three replicates per scan. The equilibrium absorbance data were analyzed using UltraScan II (27) and fit using nonlinear least squares fitting.
Results
Structure of the HsBBS91–407 N-terminal Domain
To structurally characterize H. sapiens BBS9, we recombinantly expressed the protein in E. coli. Full-length HsBBS91–887 suffered from low yield. To identify a construct suitable for crystallization, we performed a limited tryptic digest and identified a protease-resistant HsBBS9 fragment corresponding to the N-terminal domain, residues 1–374, via LC-MS/MS (Fig. 1A, data not shown). Subsequent expression, in bacteria, of HsBBS91–407 yielded sufficient material for structural analysis. We obtained crystals of the HsBBS9 N-terminal domain, which diffracted to 2.4 Å (Table 1). Despite knowing that HsBBS91–407 likely adopts a β-propeller fold, we found no molecular replacement solutions using various β-propeller templates. We also employed the Wide-Search Molecular Replacement (WSMR) protocol, available through the SBgrid software consortium (28), without success. The structure was ultimately solved using experimental phases that were obtained through single anomalous dispersion on SeMet-derivatized HsBBS91–407 crystals, which diffracted to 1.8 Å (Table 1). The model was refined against the SeMet dataset to a final Rwork of 16.9% and an Rfree of 20.0%. Overall the structure is well ordered, with an average protein B-factor of 35.4 Å2. Exceptions to this include the protein termini, as residues 1–2 and 370–407 are disordered, as well as several loop regions, where residues 113–117, 219–232, and 254–255 could not be built with confidence.
FIGURE 1.

The HsBBS91–407 N-terminal domain is a WD40 β-propeller. A, schematic diagram of the domain architecture of HsBBS9 with the relevant domain boundaries numbered. The N-terminal β-propeller domain, solved here, is colored white to blue from N to C terminus. Following the β-propeller is a γ-adaptin ear (GAE) β-sandwich domain, a mixed α/β platform domain, and a C-terminal α-helical domain (10). B, graphic representation of the HsBBS91–407 β-propeller viewed from the face and, as a result of a 90° rotation, the side. The structure is colored white to blue from N to C terminus as in A. β-Strands are depicted as arrows, helices are depicted as ribbons, and loops are depicted as lines. The individual blades are labeled. C, detailed schematic of HsBBS9 tertiary structure, colored as in A.
TABLE 1.
Data collection and refinement statistics
| Protein |
||
|---|---|---|
| BBS91–407 SeMet derivativea | BBS91–407 nativea | |
| Data collection | ||
| Space group | P21212 | P21212 |
| a, b, c (Å) | 136.42, 81.88, 85.65 | 136.14, 81.62, 85.49 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Wavelength (Å) | 0.9792 | 0.9792 |
| Resolution range (Å) | 44.70–1.80 (1.83–1.80) | 50.00–2.40 (2.44–2.40) |
| Total reflections | 327,946 | 120,972 |
| Unique reflections | 171,175 | 36,128 |
| Completeness (%) | 99.9 (99.9) | 95.2 (78.0) |
| Redundancy | 3.7 (3.7) | 3.3 (2.7) |
| Anomalous completeness (%) | 99.0 | |
| Rsym (%) | 8.6 (99.0) | 14.9 (57.0) |
| Rp.i.m. (%) | 5.1 (59.7) | 9.3 (40.1) |
| I/σ | 23.3 (1.1) | 8.8 (1.2) |
| CC1/2 (%) | 99.6 (58.3) | 99.1 (66.9) |
| Refinement | ||
| Resolution range (Å) | 36.94–1.80 | |
| Rwork (%) | 16.9 | |
| Rfree (%) | 20.0 | |
| Coordinate error (Å) | 0.2 | |
| Number of reflections | ||
| Total | 171,157 | |
| Rfree reflections | 3,941 | |
| Number of non-hydrogen atoms | 6,141 | |
| Protein atoms | 5,424 | |
| Water atoms | 717 | |
| RMS deviations | ||
| Bond lengths (Å) | 0.008 | |
| Bond angles (°) | 1.119 | |
| Average B factors (Å2) | ||
| Protein | 35.4 | |
| Water | 48.4 | |
| Ramachandran (%) | ||
| Favored (%) | 97.3 | |
| Allowed (%) | 2.7 | |
| Outlier (%) | 0.0 | |
| Clashscore | 5.6 | |
| MolProbity score | 1.52 | |
| MolProbity percentile | 93rd | |
| PDB ID | 4YD8 | |
a Organism: H. sapiens.
HsBBS91–407 comprises seven anti-parallel, four-stranded β-sheets that are radially arranged around a central, solvent-accessible pore (Fig. 1,B and C). HsBBS91–407 is ∼40 Å in diameter across its face and 20 Å in height when viewed side-on. HsBBS91–407 is a closed, seven-bladed β-propeller of the WD40 repeat family, which canonically is characterized by motifs 40 amino acids in length that end in tryptophan-aspartate motifs (29). Blades one through six (β1–β6), numbered from the N to the C terminus, each begin with the innermost strand (“A”) and trace to the outermost strand (“D”), which then connects to the A strand of the next blade. β7 is an exception to this topology because the N-terminal β-strand of the protein serves as the outermost D strand of this C-terminal, three-stranded β-sheet (A–C). This irregular connectivity, termed a “Velcro” closure, is common among β-propellers (30, 31). By physically tethering the N- and C-terminal blades together, this Velcro closure likely stabilizes the HsBBS91–407 fold by preventing opening of the propeller. HsBBS91–407 contains additional elaborations between the C and D strands of various blades, such as the helical insertion of β4 and the extended loops of β1 and β2.
Structural Analysis of the HsBBS91–407 β-Propeller
With the HsBBS91–407 structure in hand, we sought to gain insight into the function of the β-propeller through analogy to structurally similar proteins. We performed structure-based homology searches using the DALI server (32). In total, over 100 significant homologs were found (data not shown), as determined by Z scores above a predetermined threshold (Z > 10), and greater than half of the hits had an RMSD below 3.0 Å. The large number of hits obtained likely is due to the fact that HsBBS91–407 contains fairly few structural elaborations beyond the conserved, core scaffold that defines a propeller fold. Such elaborations would undoubtedly discriminate in the DALI search between various subclasses of seven-bladed propellers, reducing the number of significant solutions obtained. Despite this, three structural homologs, through their Z scores, stood out above the rest. The top scoring homolog was the recently solved C. reinhardtii (Cr) BBS1 N-terminal β-propeller (Protein Data Bank (PDB): 4v0m_B, RMSD 2.8 Å over 296 amino acids, SeqID 12%, Z = 31.5), which functions by binding the Arl6-GTPase that tethers the BBSome complex to membranes (12). The CrBBS1 N-terminal domain is also a fairly minimal propeller with few structured insertions. As the top solution, the CrBBS1 N-terminal domain and HsBBS91–407 are more similar to each other than they are to other propellers. This result likely extends to the other predicted, seven-bladed propellers within the BBSome, namely the BBS2 and BBS7 N-terminal domains. The other two high-scoring homologs were the WD40 domain of a Saccharomyces cerevisiae serine/threonine-protein kinase (Vps15, PDB: 3GRE_A, RMSD 2.4 Å over 291 amino acids, SeqID 11%, Z = 28.6), which binds the protein Gpa1 (33), and a WD40 protein of the Ski complex (Ski8, PDB: 1SQ9_A, RMSD 2.8 Å over 292 amino acids, SeqID 10%, Z = 27.4), which binds the proteins Ski2 and Ski3 (34). These hits illustrate that β-propellers are among the most common folds in biology, found in a wide range of proteins with diverse functions. Many β-propellers function as platforms for protein-protein interactions (31).
Given the propensity of β-propellers to form protein-protein interactions, we mapped the electrostatic potential onto the surface of HsBBS91–407 to see whether a charged binding site is apparent. This analysis reveals a large negative patch around the central pore on one face of the propeller (Fig. 2A). To determine whether this feature is conserved, we created an MSA sampling a diverse set of 17 BBS9 homologs and mapped sequence conservation onto the surface of HsBBS91–407 (Fig. 2B). This analysis revealed that the conservation of the charged residues responsible for the negative potential and those lining the pore opening, in general, is poor. Before dismissing this patch as an oddity of H. sapiens, we decided to create structural models of the 17 BBS9 β-propellers in the MSA, based on the H. sapiens structure, and map their surface electrostatic potential. This negative patch is notably present in BBS9 β-propellers across the MSA (Fig. 3). Although individual sequence positions are not conserved, these homologs have negatively charged residues at different positions and a relative lack of positive charge in the vicinity of the pore. Solvent-exposed carbonyl oxygens additionally contribute to the negative potential of this surface.
FIGURE 2.

A negative patch exists around the pore on one face of the HsBBS91–407 β-propeller. A, electrostatic potential mapped onto the solvent-accessible surface of HsBBS91–407. The electrostatic potential is colored from −5 kT/e (negative, red) to +5 kT/e (positive, blue). Both faces, rotated by 180° with respect to each other, are shown. B, sequence conservation mapped onto the solvent-accessible surface of HsBBS91–407. Conservation is colored from white (not conserved) to orange (conserved), and views are as in A.
FIGURE 3.

Electrostatic potential mapped onto the solvent-accessible surface of HsBBS91–407 and the modeled structures of the 17 homologs from the MSA, depicted in Fig. 8. Views and coloring are identical to Fig. 2A (left), with the electrostatic potential calculated from −5 kT/e to +5 kT/e.
Structural Analysis of HsBBS9 G141R, Which Causes BBS
Homozygosity mapping in a small, consanguineous family has revealed a SNP within the BBS9 N-terminal domain that causes BBS in humans (13). This SNP confers a missense mutation on residue 141, mutating the glycine at that position to an arginine. Gly141 is strictly conserved across the MSA with the exception of C. elegans BBS9, which has an Ile at this position and generally poor conservation in the region surrounding this residue (Fig. 4A). With the HsBBS91–407 crystal structure at hand, we can model the G141R mutation into the structure to assess why it is pathological. Gly141 is positioned within the tightly packed C terminus of the A strand of blade β3 (Fig. 4B), on the opposite face of the negatively charged patch described in Fig. 2. Immediately downstream of Gly141 is a highly curved loop that traces perpendicular to the A strand, packing Phe143 into the hydrophobic core of blade β3, before changing direction by 180° (Fig. 5A). Analyzing the Ramachandran plot reveals that Gly141 occupies a special position with dihedral angles that, although accessible to glycine, are forbidden to all other amino acids due to steric restraints imposed by the invariant presence of a β-carbon atom (Fig. 5B). Additionally, modeling all possible rotamers of arginine in position 141 of HsBBS91–407 reveals a steric clash between the β-carbon of the arginine side chain and the Cδ2 carbon of the Tyr186 aromatic ring (Fig. 5C). These carbon atoms are only 2.4 Å apart, which results in an overlap of 1 Å when hydrogen atoms are included. Based on these backbone and side chain restraints, we posit that there is an absolute structural requirement for a glycine at position 141.
FIGURE 4.

Location of Gly141 in HsBBS91–407. A, fragmentary MSA of BBS91–407 depicting the position and conservation of Gly141, labeled in red. Positions are colored white to blue, according to increasing sequence identity. B, graphic representation of HsBBS91–407 (gray) showing the position of Gly141 (green) and surrounding residues of interest as sticks. The boxed region is rotated by 45° for clarity.
FIGURE 5.

Detailed view of the position of glycine 141, which reveals steric restraints on the identity of this residue. A, view of main chain topology up- and downstream of glycine 141. The protein chain (green sticks) is shown for glycine 141 (left) or the G141R mutation (right) modeled as the most sterically permissive rotamer (fewest clashes). The final 2Fo − Fc electron density map is represented as a gray mesh. B, Ramachandran plots for glycine (left) or non-proline/glycine residues such as arginine (right). White dots show the distribution of φ and ψ angles for residues in the HsBBS91–407 structure. The position of glycine 141 and the G141R point mutant is shown (red dot). Favored regions (inner black line) and allowed regions (outer black line) are shown, and the plot is colored from white to black according to frequency of occurrence in the Top500 database of high-resolution protein structures. These plots were generated in PHENIX (18). C, view of the G141R-Tyr186 steric clash, rotated by 70° around the x axis relative to A. The arginine side chain is pointing out of the page. The atoms in close contact are labeled.
To test the effect of the G141R mutation in solution, we expressed the His10-Arg8-ScSUMO-3C-tagged HsBBS91–407 G141R point mutant in E. coli and compared the amount of purified protein with the level of wild type His10-Arg8-ScSUMO-3C-tagged HsBBS91–407 using a nickel affinity pulldown assay. When compared with wild type, the yield of HsBBS91–407 G141R after nickel affinity purification is marginal (Fig. 6A). This result is clearer after removal of the His10-Arg8-ScSUMO tag with 3C protease. Tagged HsBBS91–407 runs just under the 66-kDa marker in the same position as the E. coli GroEL chaperone, as determined by LC-MS/MS (data not shown), whereas untagged HsBBS91–407 runs unobstructed beneath the 3C protease band near the 43-kDa marker. This difference in protein level is reproducibly seen and is not due to clonal variation as the same result was obtained using five different clones of HsBBS91–407 G141R-producing E. coli (data not shown). To confirm that the observed difference is not an artifact of bacterial expression, we transiently transfected FLAG-tagged HsBBS91–407 and HsBBS91–407 G141R into the HEK 293T human cell line. Anti-FLAG Western blotting of the soluble fraction reveals that HsBBS91–407 G141R is barely visible, whereas the wild type protein is detected easily (Fig. 6B).
FIGURE 6.

Soluble protein levels of HsBBS91–407 G141R are substantially reduced when compared with wild type. A, Coomassie Brilliant Blue-stained SDS-PAGE gel of nickel affinity pulldown experiment, comparing purified protein levels of WT HsBBS91–407 (left) and HsBBS91–407 G141R (right) expressed in E. coli. Lane labels refer to samples taken throughout the nickel affinity purification: Total, crude lysate; Soluble, supernatant after lysate clarification; Insoluble, pellet after lysate clarification; Flow through, soluble sample that did not bind to nickel; Elution, soluble sample that bound nickel and was eluted with imidazole; 3C cleavage, eluate treated with 3C protease for 30 min at 37 °C for tag removal. In the elution fractions, the largest two bands (labeled with *) are E. coli contaminants. Other bands are labeled accordingly. B, anti-FLAG Western blot of soluble fraction from expression test of FLAG-tagged, wild type HsBBS91–407 (left) and FLAG-tagged HsBBS91–407 G141R (right) in HEK 293T cells. Two exposure times are shown, for clarity. Anti-Raptor antibody is used for the Raptor loading control. C, Coomassie Blue-stained SDS-PAGE gel from nickel affinity pulldown of HsBBS91–407 point mutants in E. coli. Shown are the 3C-treated, nickel elution fractions for two different clones of WT, disease mutant (G141R), and designed point mutants (labeled accordingly, above gel).
A prediction of our hypothesis, that a glycine is essential at position 141, is that any mutation regardless of identity will be equally deleterious. To test this prediction, we created a series of point mutants and tested their protein levels in E. coli again by analyzing the amount of protein isolated in a nickel pulldown on SDS-PAGE, after 3C cleavage of the affinity tag (Fig. 6C). Consistent with this prediction, mutating Gly141 to alanine results in the same virtual absence of the protein as observed with the G141R mutant. Interestingly, mutating the neighboring residue, Ser142, to a glycine fails to rescue protein levels in the context of both the arginine and the alanine mutant at position 141. Additionally, an attempt to remove the predicted side chain steric clash of Arg141 with Tyr186 by mutating the latter to alanine (Y186A) also failed to rescue purifiable protein amounts. In summary, the mutational analysis and the expression tests in two different hosts strongly suggest that the G141R mutation promotes a folding defect, effectively rendering HsBBS9 non-functional.
The HsBBS91–407 β-Propeller Forms a Homodimer in the Crystal Lattice
We analyzed packing contacts within our crystal to see whether potentially biologically relevant interactions are recapitulated within the lattice. The metric for relevance here is the predicted size or strength of a particular interaction. One such large contact occurs between two NCS mates where the β-propellers bind in a side-on fashion (Fig. 7A). The interface is symmetrical and primarily characterized by two domain swaps. In the first, each NCS mate contributes a fifth β-strand to blade β7 of the other HsBBS9 copy, forming a parallel β-sheet. This contributed strand emanates from the loop region between the C and D strands of blade β1 (Fig. 7A; inset). In the second domain swap, the C-terminal loop inserts into a surface pocket, between blades β6 and β7, on each NCS mate. In addition, the N-terminal loop packs against the loop-connecting strands A and B of blade β7. In total, this dimeric interface buries a surface area of 1,731 Å2 and comprises 24 hydrogen bonds and two salt bridges for a predicted free energy change of −23.8 kcal/mol upon binding (20).
FIGURE 7.

The HsBBS91–407 β-propeller forms a homodimer in the crystal lattice. A, overview of the HsBBS91–407 homodimer. Each NCS copy (one shown in blue, the other shown in gray) is depicted as a graphic representation. Inset left (solid line), close-up view of the β-strand domain swap. Inset right (dotted line), close-up view of the C-terminal loop domain swap, with interacting residues shown as sticks. Black dotted lines depict hydrogen bonds. The inset views are rotated relative to the overview for clarity. B, conservation mapped, from low (white) to high (orange), onto a solvent-accessible surface representation of HsBBS91–407 in the same orientation as the gray copy in A. The binding site of the symmetry mate is outlined (black). A 90° rotation of this starting orientation is shown, for clarity.
To validate this interaction, we examined sequence conservation on the HsBBS91–407 structure. This analysis reveals that the interface of the NCS dimer is moderately conserved, which makes it difficult to rule out this interaction as a crystallization artifact (Fig. 7B). By examining the MSA in detail, it becomes clear that two interface features are poorly conserved (Fig. 8). The first is the Asp8–His339 salt bridge, which can only be formed in a subset of species. Second, the β-strand domain swap is not conserved. The loop that contributes the fifth β-strand to blade β7 (residues 53–68 in H. sapiens) is of variable length in the MSA. Specifically, 11 out of 18 species in the alignment have a 4- or 5-amino acid deletion in this loop, likely making it too short to contribute this strand to the neighboring BBS9 β-propeller. Removing this interaction, however, still leaves the majority of Van der Waals contacts and 14 hydrogen bonds within the homodimer interface intact.
FIGURE 8.

MSA of the BBS9 N-terminal domain from 18 BBS9-containing eukaryotes. Residues are colored according to sequence identity, from white to blue. Numbered above the MSA, in red, are residues that participate in sequence-specific interactions within the HsBBS9 homodimer interface (hydrogen bonds and salt bridges). Residue ranges marked by black lines above the MSA denote amino acids buried by the interface (van der Waals interactions). Species were chosen for inclusion in the MSA based on Bork and co-workers and Dawson and co-workers (44, 45), to obtain a diverse sampling of eukaryotes and are ordered according to increasing distance from Homo sapiens. BBS9 is notably absent from land plants and fungi. The alignment comprises 13 metazoa (7 Chordata, 1 Arthropoda, 1 Nematoda, 1 Echinodermata, 1 Placozoa, 1 Cnidaria, and 1 Porifera), 1 Excavata, 1 protist (choanoflagellate), 1 plantae (green algae/Chlorophyta), and 2 protozoa (Chromalveolata).
Biophysical Characterization of HsBBS9 in Solution
To determine whether HsBBS91–407 dimerizes in solution, we performed SEC with purified HsBBS91–407, yielding a single peak at the approximate size of a monomer. Increasing the concentration of HsBBS91–407 had no effect on the elution volume, inconsistent with dimer formation (Fig. 9A). Due to the limited resolution of SEC, we set out to validate this result with equilibrium AUC. We tested a single protein concentration at three different speeds. The data for all three speeds fit globally to an ideal, non-interacting, one-component system with a molecular mass of 44,910 Da, indicative of a monomeric species (calculated molecular mass = 45,862 Da (35)) (Fig. 9B). These results demonstrate that the HsBBS91–407 homodimer does not form in solution.
FIGURE 9.

The HsBBS91–407 β-propeller does not form a homodimer in solution, unlike the HsBBS9405–887 C-terminal half. A, overlaid absorbance traces from SEC experiment on HsBBS91–407 at 1 mg/ml (light gray), 2.5 mg/ml (gray), 5 mg/ml (dark gray), and 10 mg/ml (black). Elution volume is indicated above each peak. A.U., absorbance units. B, top, absorbance data, at 280 nm, for HsBBS91–407 from equilibrium AUC experiment plotted against cell radius position in centimeters (see “Experimental Procedures” for details). Individual absorbance measures are depicted as blue dots, and non-linear least squares fits are shown (red lines). Data and fits are plotted for HsBBS91–407 at 9,500 rpm (top), 12,000 rpm (middle), and 18,000 rpm (bottom). B, bottom, residuals for model fit to data. The zero position is indicated by a black line. C, overlaid absorbance traces from SEC experiment on HsBBS9405–887 at 1 mg/ml (light gray), 2.5 mg/ml (gray), 5 mg/ml (dark gray), and 10 mg/ml (black). D, absorbance data, at 280 nm, for HsBBS9405–887 from equilibrium AUC experiment plotted as in B. Data and fits are plotted for HsBBS9405–887 at 9,500 rpm (top), 14,000 rpm (middle), and 17,000 rpm (bottom).
To determine whether other regions of HsBBS9 oligomerize, we performed SEC on the C-terminal half of the protein, HsBBS9405–887, which contains three folded domains according to structure prediction and can be recombinantly expressed and purified from E. coli. Unlike HsBBS91–407, the elution volume of HsBBS9405–887 decreases upon increasing protein concentration (Fig. 9C). The observed decrease in elution volume and broadening of the peak is consistent with dimer formation. To verify this result, we performed equilibrium AUC on HsBBS9405–887 at a single concentration, in duplicate, spun at three different speeds. The data fit well to a monomer-dimer equilibrium model, with a fitted molecular mass of 55,810 Da (monomer, calculated molecular mass = 54,419 Da (35)) and a dissociation constant of 33 μm (Fig. 9D). These data demonstrate that HsBBS9405–887 dimerizes in solution.
We went on to assess whether the N-terminal domain and C-terminal half of HsBBS9 bind to each other. HsBBS91–407 and HsBBS9405–887 were mixed in a 1:1 molar ratio prior to SEC in conditions of increasing ionic strength (100 mm NaCl, 10% v/v glycerol; 150 mm NaCl; 500 mm NaCl). Binding was not observed under any of these conditions (Fig. 10, A and B). This indicates that the HsBBS9 N-terminal β-propeller is flexibly tethered to the C-terminal half of the protein. Based on this series of experiments, we can conclude that HsBBS91–407 dimerization is a crystallization artifact.
FIGURE 10.
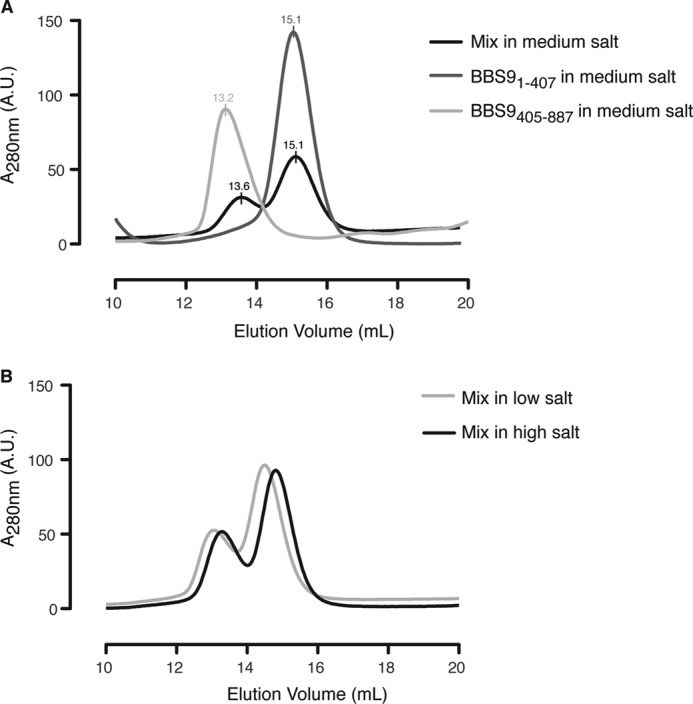
HsBBS91–407 and HsBBS9405–887 do not bind to each other on SEC. A, absorbance trace overlay from SEC on HsBBS91–407 (dark gray), HsBBS9405–887 (light gray), and the two mixed together in a 1:1 ratio (black) in medium salt buffer (150 mm NaCl; see “Experimental Procedures” for complete buffer composition). Peak elution volumes are noted above the curves and are approximately the same after mixing, indicating a lack of complex formation. A. U., arbitrary units. B, absorbance trace overlay from SEC on HsBBS1–407 and HsBBS405–887 mixed in low salt buffer (100 mm NaCl and 10% v/v glycerol, light gray) and high-salt buffer (500 mm NaCl, black). A net shift of both peaks occurs as a result of buffer differences. A third peak of smaller elution volume does not form, indicating that the proteins do not bind to each other.
Discussion
Here we analyzed the structural and biophysical properties of BBS9, a component of the octameric BBSome. We show that the N-terminal domain folds into a regular β-propeller. How does this information help in deducing the function of BBS9? The protein acts in the context of the octameric BBSome, which forms a membrane coat to select membrane-inserted cargo destined for the primary cilium. The cell has several such membrane coating systems, like COPI, COPII, clathrin and, more distantly related, the nuclear pore complex (36). The architecture of these assemblies is principally related, presumably because of a common evolutionary past (37, 38). Many of the architectural components within these coats are β-propellers, α-helical stacks, or a combination of both. However, the ways in which these elements come together to form the various coats seem to be quite diverse (39–41). Regarding BBS9, our data indicate that it is possibly involved in the higher-order assembly of a BBSome coat. We observed that the protein has a dimerization interface in its C-terminal half. In the context of building a BBSome coat, weak interactions, such as this, might work in an additive manner to build the polymerized assembly. Weak interactions could have evolved such that only small perturbations are required to shift the equilibrium between net polymerization and depolymerization.
In this context, the conserved negative patch on the BBS9 β-propeller domain is particularly intriguing. It is suggestive to propose that it forms a platform for a charge interaction with a peptide or protein of positive character. Other seven-bladed β-propellers are known to bind to positively charged peptides via their negatively charged pores. Examples include β′ COP, a COPI vesicle coat component, which binds endoplasmic reticulum retrieval motifs on cargo proteins (42), and the nucleosome-remodeling factor Nurf55, which binds to the positively charged N terminus of histone H3 via a negatively charged pore (43). Such an electrostatic interaction could be blocked easily by phosphorylation of the binding partner or selective interaction with phospholipids.
Our data indicate that the pathogenic G141R mutation in HsBBS9 likely causes misfolding of the β-propeller domain, which presumably abrogates any potential protein-protein interaction. Whether this interaction is within the BBSome octamer, or with a neighboring unit in the coat or even an external factor such as cargo, still needs to be understood.
Author Contributions
T. U. S. conceived the study. K. E. K. and T. U. S. designed the experiments, analyzed the data, and wrote the manuscript. K. E. K. performed the experiments.
Acknowledgments
We thank R. Saxton for performing the HEK 293T transient transfection and subsequent Western blot, N. Leksa for experimental advice, S. Bilokapic for help with crystal fishing, and E. Spooner for performing the LC-MS/MS. The x-ray crystallography work was conducted at the Advanced Photon Source (APS) NE-CAT beamlines, which are supported by award GM103403 from the NIGMS, National Institutes of Health. Use of the APS is supported by the Department of Energy, Office of Basic Energy Sciences, under Contract DE-AC02-06CH11357. The Biophysical Instrumentation Facility for the Study of Complex Macromolecular Systems (NSF-007031) is gratefully acknowledged.
This work was supported by National Institutes of Health Grant T32GM007287 (to K. E. K.) and the National Science Foundation Graduate Research Fellowship under Grant 1122374 (to K. E. K.). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 4YD8) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- BBSome
- Bardet-Biedl syndrome protein complex
- BBS9
- Bardet-Biedl syndrome 9 protein
- BBS
- Bardet-Biedl syndrome
- 3C
- human rhinovirus 3C protease
- SeMet
- selenomethionine
- NCS
- non-crystallographic symmetry
- MSA
- multiple sequence alignment
- SEC
- size exclusion chromatography
- AUC
- analytic ultracentrifugation
- RMSD
- root mean square deviation
- HsBBS9
- H. sapiens BBS9
- CrBBS1
- C. reinhardtii BBS1
- ScSUMO
- S. cerevisiae small ubiquitin-like modifier.
REFERENCES
- 1. Nachury M. V., Seeley E. S., Jin H. (2010) Trafficking to the ciliary membrane: how to get across the periciliary diffusion barrier? Annu. Rev. Cell Dev. Biol. 26, 59–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Singla V., Reiter J. F. (2006) The primary cilium as the cell's antenna: signaling at a sensory organelle. Science 313, 629–633 [DOI] [PubMed] [Google Scholar]
- 3. Corbit K. C., Aanstad P., Singla V., Norman A. R., Stainier D. Y. R., Reiter J. F. (2005) Vertebrate Smoothened functions at the primary cilium. Nature 437, 1018–1021 [DOI] [PubMed] [Google Scholar]
- 4. Hu Q., Milenkovic L., Jin H., Scott M. P., Nachury M. V., Spiliotis E. T., Nelson W. J. (2010) A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science 329, 436–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chih B., Liu P., Chinn Y., Chalouni C., Komuves L. G., Hass P. E., Sandoval W., Peterson A. S. (2012) A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat. Cell Biol. 14, 61–72 [DOI] [PubMed] [Google Scholar]
- 6. Nachury M. V., Loktev A. V., Zhang Q., Westlake C. J., Peränen J., Merdes A., Slusarski D. C., Scheller R. H., Bazan J. F., Sheffield V. C., Jackson P. K. (2007) A core complex of bbs proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 129, 1201–1213 [DOI] [PubMed] [Google Scholar]
- 7. Loktev A. V., Zhang Q., Beck J. S., Searby C. C., Scheetz T. E., Bazan J. F., Slusarski D. C., Sheffield V. C., Jackson P. K., Nachury M. V. (2008) A BBSome subunit links ciliogenesis, microtubule stability, and acetylation. Dev. Cell 15, 854–865 [DOI] [PubMed] [Google Scholar]
- 8. Tobin J. L., Beales P. L. (2009) The nonmotile ciliopathies. Genet Med. 11, 386–402 [DOI] [PubMed] [Google Scholar]
- 9. Berbari N. F., Lewis J. S., Bishop G. A., Askwith C. C., Mykytyn K. (2008) Bardet-Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proc. Natl. Acad. Sci. U.S.A. 105, 4242–4246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jin H., White S. R., Shida T., Schulz S., Aguiar M., Gygi S. P., Bazan J. F., Nachury M. V. (2010) The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell 141, 1208–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wei Q., Zhang Y., Li Y., Zhang Q., Ling K., Hu J. (2012) The BBSome controls IFT assembly and turnaround in cilia. Nat. Cell Biol. 14, 950–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mourão A., Nager A. R., Nachury M. V., Lorentzen E. (2014) Structural basis for membrane targeting of the BBSome by ARL6. Nat. Struct. Mol. Biol. 21, 1035–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nishimura D. Y., Swiderski R. E., Searby C. C., Berg E. M., Ferguson A. L., Hennekam R., Merin S., Weleber R. G., Biesecker L. G., Stone E. M., Sheffield V. C. (2005) Comparative genomics and gene expression analysis identifies BBS9, a new Bardet-Biedl syndrome gene. Am. J. Hum. Genet. 77, 1021–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Veleri S., Bishop K., Dalle Nogare D. E., English M. A., Foskett T. J., Chitnis A., Sood R., Liu P., Swaroop A. (2012) Knockdown of Bardet-Biedl syndrome gene BBS9/PTHB1 leads to cilia defects. PLoS ONE 7, e34389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Andersen K. R., Leksa N. C., Schwartz T. U. (2013) Optimized E. coli expression strain LOBSTR eliminates common contaminants from His-tag purification. Proteins 81, 1857–1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brohawn S. G., Leksa N. C., Spear E. D., Rajashankar K. R., Schwartz T. U. (2008) Structural evidence for common ancestry of the nuclear pore complex and vesicle coats. Science 322, 1369–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Method Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 18. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 21. Ashkenazy H., Erez E., Martz E., Pupko T., Ben-Tal N. (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38, W529–W533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Waterhouse A. M., Procter J. B., Martin D. M. A., Clamp M., Barton G. J. (2009) Jalview Version 2: a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- 24. Kelley L. A., Sternberg M. J. E. (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371 [DOI] [PubMed] [Google Scholar]
- 25. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.3r1, Schrödinger, LLC, New York [Google Scholar]
- 27. Demeler B. (2005) UltraScan: a comprehensive data analysis software package for analytical ultracentrifugation experiments. in Modern Analytical Ultracentrifugation: Techniques and Methods, pp. 210–229, Royal Society of Chemistry, Royal Society of Chemistry, Cambridge, UK, 10.1039/9781847552617-00210 [DOI] [Google Scholar]
- 28. Morin A., Eisenbraun B., Key J., Sanschagrin P. C., Timony M. A., Ottaviano M., Sliz P. (2013) Collaboration gets the most out of software. eLife 2, e01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fong H. K., Hurley J. B., Hopkins R. S., Miake-Lye R., Johnson M. S., Doolittle R. F., Simon M. I. (1986) Repetitive segmental structure of the transducin β subunit: homology with the CDC4 gene and identification of related mRNAs. Proc. Natl. Acad. Sci. U.S.A. 83, 2162–2166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wall M. A., Coleman D. E., Lee E., Iñiguez-Lluhi J. A., Posner B. A., Gilman A. G., Sprang S. R. (1995) The structure of the G protein heterotrimer Giα1β1γ2. Cell 83, 1047–1058 [DOI] [PubMed] [Google Scholar]
- 31. Xu C., Min J. (2011) Structure and function of WD40 domain proteins. Protein Cell 2, 202–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Heenan E. J., Vanhooke J. L., Temple B. R., Betts L., Sondek J. E., Dohlman H. G. (2009) Structure and function of Vps15 in the endosomal G protein signaling pathway. Biochemistry 48, 6390–6401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brown J. T., Bai X., Johnson A. W. (2000) The yeast antiviral proteins Ski2p, Ski3p, and Ski8p exist as a complex in vivo. RNA 6, 449–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wilkins M. R., Gasteiger E., Bairoch A., Sanchez J. C., Williams K. L., Appel R. D., Hochstrasser D. F. (1999) Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 112, 531–552 [DOI] [PubMed] [Google Scholar]
- 36. Brohawn S. G., Partridge J. R., Whittle J. R. R., Schwartz T. U. (2009) The nuclear pore complex has entered the atomic age. Structure 17, 1156–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Devos D., Dokudovskaya S., Alber F., Williams R., Chait B. T., Sali A., Rout M. P. (2004) Components of coated vesicles and nuclear pore complexes share a common molecular architecture. Plos Biol. 2, e380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mans B. J., Anantharaman V., Aravind L., Koonin E. V. (2004) Comparative genomics, evolution and origins of the nuclear envelope and nuclear pore complex. Cell Cycle 3, 1612–1637 [DOI] [PubMed] [Google Scholar]
- 39. Fotin A., Cheng Y., Sliz P., Grigorieff N., Harrison S. C., Kirchhausen T., Walz T. (2004) Molecular model for a complete clathrin lattice from electron cryomicroscopy. Nature 432, 573–579 [DOI] [PubMed] [Google Scholar]
- 40. Fath S., Mancias J. D., Bi X., Goldberg J. (2007) Structure and organization of coat proteins in the COPII cage. Cell 129, 1325–1336 [DOI] [PubMed] [Google Scholar]
- 41. Lee C., Goldberg J. (2010) Structure of coatomer cage proteins and the relationship among COPI, COPII, and clathrin vesicle coats. Cell 142, 123–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ma W., Goldberg J. (2013) Rules for the recognition of dilysine retrieval motifs by coatomer. EMBO J. 32, 926–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schmitges F. W., Prusty A. B., Faty M., Stützer A., Lingaraju G. M., Aiwazian J., Sack R., Hess D., Li L., Zhou S., Bunker R. D., Wirth U., Bouwmeester T., Bauer A., Ly-Hartig N., Zhao K., Chan H., Gu J., Gut H., Fischle W., Müller J., Thomä N. H. (2011) Histone methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell 42, 330–341 [DOI] [PubMed] [Google Scholar]
- 44. Ciccarelli F. D., Doerks T., von Mering C., Creevey C. J., Snel B., Bork P. (2006) Toward automatic reconstruction of a highly resolved tree of life. Science 311, 1283–1287 [DOI] [PubMed] [Google Scholar]
- 45. Fritz-Laylin L. K., Prochnik S. E., Ginger M. L., Dacks J. B., Carpenter M. L., Field M. C., Kuo A., Paredez A., Chapman J., Pham J., Shu S., Neupane R., Cipriano M., Mancuso J., Tu H., Salamov A., Lindquist E., Shapiro H., Lucas S., Grigoriev I. V., Cande W. Z., Fulton C., Rokhsar D. S., Dawson S. C. (2010) The genome of Naegleria gruberi illuminates early eukaryotic versatility. Cell 140, 631–642 [DOI] [PubMed] [Google Scholar]