

Background: Lipid A-specific antibodies are not effective in sepsis treatment; infection by Gram-negative bacteria can induce autoimmune disease.
Results: Antibody-combining sites orient lipid A to bury the LPS attachment point and can cross-react with ssDNA through terminal nucleotides.
Conclusion: Anti-lipid A antibodies cannot bind full-length LPS; phosphates play a crucial role in polyspecificity.
Significance: Antibody recognition of lipid A illuminates the genesis of autoimmunity.
Keywords: autoimmunity, lipid A, lipopolysaccharide (LPS), monoclonal antibody, x-ray crystallography
Abstract
Septic shock is a leading cause of death, and it results from an inflammatory cascade triggered by the presence of microbial products in the blood. Certain LPS from Gram-negative bacteria are very potent inducers and are responsible for a high percentage of septic shock cases. Despite decades of research, mAbs specific for lipid A (the endotoxic principle of LPS) have not been successfully developed into a clinical treatment for sepsis. To understand the molecular basis for the observed inability to translate in vitro specificity for lipid A into clinical potential, the structures of antigen-binding fragments of mAbs S1–15 and A6 have been determined both in complex with lipid A carbohydrate backbone and in the unliganded form. The two antibodies have separate germ line origins that generate two markedly different combining-site pockets that are complementary both in shape and charge to the antigen. mAb A6 binds lipid A through both variable light and heavy chain residues, whereas S1–15 utilizes exclusively the variable heavy chain. Both antibodies bind lipid A such that the GlcN-O6 attachment point for the core oligosaccharide is buried in the combining site, which explains the lack of LPS recognition. Longstanding reports of polyspecificity of anti-lipid A antibodies toward single-stranded DNA combined with observed homology of S1–15 and A6 and the reports of several single-stranded DNA-specific mAbs prompted the determination of the structure of S1–15 in complex with single-stranded DNA fragments, which may provide clues about the genesis of autoimmune diseases such as systemic lupus erythematosus, thyroiditis, and rheumatic autoimmune diseases.
Introduction
Bacterial Gram-positive and Gram-negative infections are a leading cause of septic shock, with over 750,000 cases annually in the United States and with a mortality rate between 28 and 50% (1–3). It is the third most common cause of death in Germany where it claims 60,000 lives a year (2). The inflammatory cascade at the onset of septic shock can be caused by the presence of bacterial lipopolysaccharide (LPS), which is shed from the outer membrane of Gram-negative bacteria (4).
The inflammatory cascade is initiated by the formation of a signaling complex of the lipid A moiety of LPS with Toll-like receptor 4 (TLR4) and co-receptor myeloid differentiation factor 2 (MD-2) (5–8). One heavily investigated therapeutic route has been focused on the potential of monoclonal antibodies (mAbs) to sequester LPS (9–11) to prevent formation of the LPS·MD-2·TLR4 signaling complex. Although antibodies specific for the lipid A, core, and O-polysaccharide components of LPS have been reported (10–19), the diversity of structures together with the rapid onset of septic shock have hindered the introduction of O-polysaccharide-specific antibodies into clinical use (4, 11, 20–22). In contrast to O-polysaccharide, there is strong structural conservation among the inner cores of relevant serovars (23); however, generation of broadly cross-reactive antibodies has proved challenging. To date, only mAb WN1 222-5 has been reported to be successful in neutralizing a wide range of Gram-negative bacteria, including Escherichia, Salmonella, Shigella, and Citrobacter (9, 10, 24).
Although lipid A structure is relatively conserved among pathogenic species, none of the numerous reported antibodies claimed to be specific for lipid A have led to successful clinical implementation (13–15, 25). Specific binding to lipid A was observed upon acid treatment of bacterial LPS, thereby liberating the lipid A fragment acting then as neoantigen, when embedded into erythrocytes or liposomes (25, 26). Of particular significance were mAbs A6 (IgG2b) isolated from mice immunized with heat-killed Escherichia coli J-5 cells (27) and S1–15, also referred to as S1 (25). Binding studies showed a preference of mAb A6 for the bisphosphorylated (i.e. native) lipid A, with weak binding to the monophosphorylated lipid A (25), although S1–15 displays high avidity for both the mono- and bisphosphorylated lipid A (25, 28). Neither antibody was observed to recognize intact LPS.
Our earlier work showed that such antibodies bind as well to the bisphosphorylated glucosamine (GlcN) disaccharide backbone (BBP)3 without the acyl chains (28), indicating that S1–15 and A6 recognize the carbohydrate backbone exclusively. Significantly, binding studies of the C6′ methyl-capped disaccharide showed that neither antibody bound lipid A lacking a free primary hydroxyl at C6 on the β-GlcN, which serves as the attachment point for inner core residues of the LPS (29).
Interestingly, a number of anti-lipid A antibodies have been reported to display significant polyspecificity to a range of antigens, including cardiolipin (30, 31), I antigen on B-lymphocytes (30), double-stranded DNA (dsDNA) (13), and single-stranded DNA (ssDNA) (14, 30), with reports of mice immunized with bacterial LPS resulting in mAbs cross-reactive to DNA (32, 33). Antibodies against ssDNA are noteworthy, as their induction has been implicated in autoimmune diseases such as lupus (34–37), thyroid disease (38), and rheumatic disorders (31, 39). Furthermore, studies have implicated a specific variable heavy chain family gene V(H)4–21 in dual recognition of lipid A and ssDNA, demonstrating a possible link between infection with Gram-negative bacteria and development of systemic lupus erythematosus and arthritis (31, 34).
To date there are no reports of crystal structures of any anti-lipid A mAb either unliganded or in complex. We now report binding data and crystal structures of unliganded and liganded antigen-binding fragments (Fabs) for two anti-lipid A mAbs, S1–15 and A6, to elucidate the reason these antibodies do not bind intact LPS. Furthermore, we present a structure of S1–15 in complex with ssDNA fragments that supports the polyspecific potential of anti-lipid A antibodies toward ssDNA.
Experimental Procedures
ELISA
ELISA against the immobilized neoglycoconjugates was performed as described earlier (29). For the binding assay, BBP and B4P were conjugated in a 5 m excess to BSA, which had been activated by divinylsulfone, were then immobilized at concentrations of 2.5, 5.0, 10, and 20 pmol of ligand per well, and reacted with mAbs A6 and S1–15, which were titrated 1:1 over 12 steps starting from 2 μg/ml concentration.
Germ Line Gene Usage Analysis
The nucleotide sequences of the variable regions were analyzed with the IMGT/V-quest and junctional analysis web applications (40, 41) to determine the murine germ line gene segments from which the lipid A-specific antibodies were derived.
Fab Preparation and Crystallization
Generation of mAbs S1–15 (IgG2b) (25) and A6 (IgG2b) (27) was described earlier. For large scale preparation, mAb S1–15 was isolated from ascites by affinity chromatography on BBP conjugated to AH-Sepharose (80 mg of ligand/2.5 ml of packed beads) followed by elution with 0.1 m glycine-HCl, pH 3.2 (29). Fractions were adjusted to pH 4 by addition of 1 m NaHCO3. Antibody A6 was isolated and purified in the same manner as S1–15.
Fab fragments of each antibody were prepared by digestion of the intact immunoglobulin with papain (Sigma). IgG was dialyzed into 25 mm HEPES (Sigma), pH 7.5, diluted to a concentration of 1.0–0.8 mg/ml, 2 mm EDTA (Sigma), and 5–6 mm DTT (Sigma). The digestion reaction was carried out at room temperature using a papain/IgG ratio of 1:200 molar eq for 2–3 h. The reaction was quenched by the addition of 10 mm iodoacetamide (Sigma) and dialyzed overnight into 25 mm HEPES buffer, pH 7.4. Fab fragment was purified by cation-exchange chromatography on a Shodex CM-825 column (Phenomenex, Torrance, CA) using a linear gradient of 0.0 to 0.5 m NaCl in 20 mm HEPES, pH 7.4. Purified Fab was concentrated to 10–12 mg/ml stock and stored at 4.0 °C.
The Fabs of A6 and S1–15 mAbs were mixed with 3 mm lipid A backbone bisphosphate: βGlcN4P(1→6)αGlcN1P, designated BBP (28), for liganded crystallization screening. Several crystals (0.25 × 0.35 × 0.30 mm3) were obtained for S1–15 in the presence of BBP after 2 months in a 16 °C room from hanging drop setup in PEG II suite crystal screen (Qiagen, Toronto, Canada), under condition 13 (0.1 m sodium chloride, 0.1 m bicine, pH 9.0, and 30% (w/v) PEG 550 methyl ether) using a 1:1 reservoir-to-protein ratio. Approximately cubic crystals (0.1 × 0.15 × 0.15 mm3) of A6 were grown initially at a hanging drop setup in a 16 °C room in the presence of BBP using Index crystal screen (Hampton Research, Aliso Viejo, CA) under condition 44 (0.1 m HEPES, pH 7.5, and 25% (w/v) PEG 3350) in 1:1 reservoir-to-protein ratio. Larger crystals (0.4 × 0.4 × 0.2 mm3) were obtained using a 1:1.5 reservoir-to-protein ratio.
Fresh papain digests of intact S1–15 and A6 were used by applying the protocol as above, for crystallization screening of unliganded Fabs and S1–15 Fab in the presence of oligonucleotides. Conditions were set using an Art Robbins Gryphon Xtallization Robot (San Jose, CA), with each 96-well sitting drop plate incubated in a 16 °C room. S1–15 unliganded crystals (0.4 × 0.3 × 0.25 mm3) were grown under condition 21 (25% (w/v) PEG 2000 MME) of the PEG II suite screen. Initial crystals of variable sizes for unliganded A6 Fab were grown under condition 16 (0.1 m Tris-HCl, pH 8.5, and 30% (w/v) PEG 1000) of PEG II suite crystal screen, and subsequently optimized (for diffraction quality) using 35% (w/v) PEG 1000. S1–15 Fab (12 mg/ml) was mixed with the deoxythymidine oligonucleotides of sequence 5′P-TTTTT-3′P (both ends phosphorylated) referred to as p5(dT)p and 5′-TTTTT-3′P (lacking the 5′-phosphate) referred to as 5(dT)p, in a 1:2 molar ratio. Crystals in presence of p5(dT)p oligonucleotides were initially obtained after a few days using the sitting drop method under condition 55 (0.075 m MgCl2, 0.1 m HEPES, pH 7.5, 28% (v/v) PEG 550 MME) of the index screen (Hampton Research). Larger crystals (0.35 × 0.3 × 0.3 mm3) were obtained via the hanging drop method and using 1:2–1:3 reservoir-to-protein ratios. A large crystal (0.7 × 0.4 × 0.4 mm3) was obtained in presence of 5(dT)p antigen, after a few days under condition 22 (0.1 m Tris-HCl, pH 8.5, 25% (v/v) PEG 550 MME) of the PEG suite crystal screen using sitting drop method.
Data Collection, Molecular Replacement, and Structure Refinement
Crystallization conditions for S1–15 liganded and unliganded, as well as A6 unliganded structures already contained levels of cryoprotectant suitable to form a glass upon freezing. Liganded A6 crystals were carefully dehydrated in a 16 °C room until the concentration of cryoprotectant (PEG 3350) reached appropriate levels. All crystals were flash-frozen to −160 °C using an Oxford Cryostream 700 crystal cooler (Oxford Cryosystems, Oxford, UK). The x-ray diffraction data sets were collected at the Canadian Macromolecular Crystallography Facility on beamline 08ID-1 of the Canadian Light Source (Saskatoon, Saskatchewan, Canada) at 0.979 Å wavelength, with a Marmosaic CCD300 detector, and processed using HKL2000 (HKL Research Inc., Charlottesville, VA).
The S1–15 structure in complex with lipid A was solved by molecular replacement using Phaser (42) with the constant domain and Fv fragments of unliganded Fab S55–3 (Protein Data Bank code 4ODS) as search models. The liganded structure of S1–15 was used subsequently as a search model for the unliganded S1–15 dataset and the ssDNA liganded datasets. The A6 liganded structure was also solved using the Fv and constant domains of the S1–15 liganded structure as search models. The liganded Fv and constant domains of A6 Fab were used as search models for the unliganded A6 structure.
Manual fitting of σ-A-weighted Fo − Fc and 2Fo − Fc electron density maps was carried out with Coot (43). Restrained refinement and translation, libration, and screw (TLS) refinement was carried out using REFMAC5 (44, 45). All stereo figures and r.m.s.d. calculations presented in this paper were made using SetoRibbon (available upon request from author S. V. E.). Electrostatic surface potential figures were made using Chimera molecular visualization software (46). Marvin Sketch Version 5.7.0 from ChemAxon was used for drawing chemical structures.
Results
Characterization of Lipid A-specific Antibodies through ELISA
The quantitative conjugate ELISA for mAbs S1–15 and A6 to BBP and B4P at concentrations of 2.5, 5, 10, and 20 pmol/well are shown in Fig. 1. Both antibodies bound to the immobilized BBP conjugate and much weaker to the B4P conjugate. Against BBP, the affinity of S1–15 was ∼4-fold weaker than A6. However, S1–15 showed stronger binding to the B4P-conjugate confirming the weaker dependence on the recognition of the phosphate at the anomeric position (25). Binding data against synthetic lipid A and derivatives thereof have been reported earlier (15, 25, 29).
FIGURE 1.

Quantitative conjugate ELISAs coated with graded concentrations of neoglycoconjugates corresponding to 2.5 (blue triangle), 5 (green triangle), 10 (red circle), and 20 (black square) pmol of ligand per well and reacted with mAbs (A) S1–15 and (B) A6 at concentrations indicated on the x axis are shown. Ligands used were bisphosphorylated lipid A backbone conjugated to BSA (BBP-divinyl sulfone BSA, left panel) and 4′-monophosphorylated lipid A backbone conjugated to BSA (right panel), and absorbance was measured at 405 nm. The chemical structure of the carbohydrate backbone of lipid A (BBP) used for crystallization trials (C). 1P and 4P refer to the position of the phosphates on the pyranose ring.
X-ray Diffraction Data Collection and Refinement
Data collection and refinement statistics for liganded and unliganded structures of the Fabs from lipid A-specific mAbs S1–15 (IgG2b) and A6 (IgG2b) are given in Table 1. Deacylated and purified lipid A (BBP) was used during crystallization trials (Fig. 1C).
TABLE 1.
Data collection and refinement statistics for liganded and unliganded structures of lipid A-specific antibodies, A6 and S1–15
| Crystal |
||||||
|---|---|---|---|---|---|---|
| S1–15 BBP | S1–15 unliganded | A6 BBP | A6 unliganded | S1–15 T5a | S1–15 T5 no 5′Pa | |
| PDB codes | 4ODT | 4ODU | 4ODV | 4ODW | 4Z8F | 4Z95 |
| Resolution (Å) | 25.0-1.95 (2.02-1.95) | 30.00-2.29 (2.34-2.29) | 25.0-2.15 (2.23-2.15) | 25.0-2.70 (2.80-2.70) | 25.0-1.75 (1.81-1.75) | 30.00-1.79 (1.85-1.79) |
| Space group | P43212 | P21 | P212121 | P1 | P43212 | P43212 |
| a (Å) | 77.5 | 75.8 | 37.6 | 42.1 | 77.6 | 77.8 |
| b (Å) | 77.5 | 77.6 | 64.3 | 69.2 | 77.6 | 77.8 |
| c (Å) | 156.2 | 72.0 | 154.2 | 69.1 | 156.3 | 156.5 |
| α (°) | 90.0 | 90.0 | 90.0 | 71.0 | 90.0 | 90.0 |
| β (°) | 90.0 | 96.8 | 90.0 | 72.4 | 90.0 | 90.0 |
| γ (°) | 90.0 | 90.0 | 90.0 | 88.3 | 90.0 | 90.0 |
| Volume Å3 | 937500 | 420390 | 372247 | 180558 | 940639 | 946473 |
| Wavelength (Å) | 0.979 | 0.979 | 0.979 | 0.979 | 0.979 | 0.979 |
| Mean B-factor (Å2) | 40.3 | 31.0 | 48.1 | 53.30 | 38.9 | 39.0 |
| Z | 1 | 2 | 1 | 2 | 1 | 1 |
| Unique reflections | 35,329 | 34,731 | 21,015 | 18,489 | 46,712 | 43,755 |
| Redundancy | 15.7 (15.8) | 5.0 (5.1) | 4.9 (4.5) | 2.0 (2.0) | 14.5 (14.5) | 9.9 (10.0) |
| 〈I/σ(I)〉 | 34.1 (4.91) | 24.4 (2.79) | 16.0 (4.13) | 18.2 (1.86) | 36. 0 (5.95) | 37.7 (3.7) |
| Rsym (%) | 8.8 (78.6) | 6.3 (61.5) | 8.5 (40.4) | 4.0 (47.2) | 5.90 (61.0) | 5.3 (74.0) |
| Completeness (%) | 100.0 (100.0) | 100.0 (100.0) | 99.7 (99.1) | 98.1 (98.3) | 99.9 (100.0) | 99.9 (100.0) |
| Protein atoms | 3375 | 6662 | 3338 | 6534 | 3375 | 3375 |
| Solvent atoms | 265 | 300 | 94 | 16 | 208 | 278 |
| Heterogenb atoms | 64 | 10 | 38 | 8 | 101 | 5 |
| Nucleic acids | 0 | 0 | 0 | 0 | 5 | 0 |
| Ramachandran outliers | 0 | 1 | 3 | 0 | 0 | 0 |
| Solvent content (%) | 49.0 | 44.9 | 36.8 | 37.3 | 48.4 | 49.9 |
| Refinement | ||||||
| Rwork (%) | 19.7 | 18.7 | 20.4 | 23.1 | 20.8 | 20.3 |
| Rfree (%) | 22.4 | 23.3 | 24.9 | 25.7 | 24.0 | 23.6 |
| r.m.s. bond lengths (Å) | 0.0091 | 0.010 | 0.011 | 0.011 | 0.0093 | 0.010 |
| r.m.s. bond angles (°) | 1.33 | 1.42 | 1.56 | 1.61 | 1.28 | 1.34 |
a Antigen T5 represents the DNA oligonucleotide 5′P-TTTTT-3′P.
b Heterogen atoms describe nonstandard residues, such as prosthetic groups, inhibitors, carbohydrates, solvents, and ions.
A6
Data were collected to 2.15 Å resolution from a crystal grown in the presence of BBP, which were consistent with orthorhombic space group P212121 with an Rsym of 0.085 and containing one molecule per asymmetric unit. Superb electron density was observed for the antigen (Fig. 2A) and for the remainder of the protein, with the exception of the solvent-exposed residues 128–136 and 157–161 on the H chain constant domain, which is often observed to be disordered in IgG2b Fab structures (47–50). Data were collected on crystals of the unliganded Fab of A6 to 2.75 Å resolution with Rsym of 0.041 in triclinic space group P1. The structure was solved with two molecules in the asymmetric unit, and it exhibited relatively higher thermal motion that manifested as poor electron density in solvent-exposed regions, particularly residues 127–134 and 157–161 on the constant domain of the H chain, some of which were excluded from the final model.
FIGURE 2.

Stereo diagram 2Fo − Fc electron density map (blue) contoured at 1. 0σ for the BBP lipid A analogue was observed in the combining sites of A6 (A) and S1–15 mAbs (B) and two single-stranded DNA fragments. p2(dT)p (top) and p3(dT)p (bottom) were observed in the combining site of S1–15 (C). To show the base stacking interaction, the trinucleotide (bottom) is bound to a symmetry-related Fab. Although the original antigen used was p5(dT)p, only two and three nucleotides could be modeled with confidence. Stereo diagram of S1–15 in complex with 5(dT)p ligand showing hydrogen bonds (orange dashed spheres) between residues of S1–15 and to the 3′P (D). The remainder of the antigen was disordered. Indirect hydrogen bonds mediated by waters (cyan spheres) are also shown. CDR loops of the light and heavy chain are colored white and gray, respectively. Color scheme is as follows: carbon, green; oxygen, red; nitrogen, blue; phosphorus, orange; water, cyan. Density (blue) for the phosphate is contoured at 1.0σ. Stereo view of A6 (E) and S1–15 (F) in complex with lipid A analogues BBP shows hydrogen bonds and water bridges between the antigen and S1–15. CDR loops of the light and heavy chain are colored white and gray, respectively. Strong hydrophobic contacts are shown as dashed lines (black). C6 hydroxyl group of second GlcN is the attachment point to inner core residues of LPS. Stereo diagram of S1–15 Fab in complex with two p5(dT)p ssDNA fragments is shown (G). Stereo diagram of S1–15 Fab in complex with two p5(dT)p ssDNA fragments is shown.
S1–15
Data of crystals of S1–15 grown in the presence of BBP collected to 1.95 Å resolution fit a tetragonal unit cell in Laue group 422 with an Rsym of 0.088 and with systematic absences corresponding to space groups P41212 and P43212. The structure was solved in space group P43212 by molecular replacement with one molecule in the asymmetric unit. Excellent electron density was observed for the antigen (Fig. 2B) and for the remainder of the protein with the exception of residues in the corresponding region as A6 structures.
Data for two additional liganded S1–15 Fab structures were collected as follows: one in complex with 5-stranded deoxythymidine (dT) oligonucleotide with both ends phosphorylated or p5(dT)p and a 5-oligothymidine nucleotide with no 5′-terminal phosphate called 5(dT)p. The data were collected to 1.75 and 1.79 Å resolution with Rsym of 0.059 and 0.053, respectively (Table 1). Both data sets were solved in the same space group and near-identical unit cells as the structure of S1–15 in complex with the lipid A analogue. The ssDNA ligand with both terminal phosphates showed density for two separate oligonucleotide fragments, one at each phosphate-binding site of lipid A. A dinucleotide with the 3′-terminal phosphate occupies the 1P lipid A recognition pocket, and on the 4P lipid A pocket, a trinucleotide could be confidently modeled, although the terminal 5′-thymine base was largely disordered. The two oligonucleotide fragments stabilized one another through stacking interactions via symmetry-related Fab molecules, showing electron density corresponding to a total of five oligonucleotides (Fig. 2C). In the second data set with 5(dT)p as ligand, density could only be observed for the 3′-phosphate group (Fig. 2D) as the rest of the oligonucleotide could not be modeled due to dynamic disorder.
Data were collected on the unliganded of S1–15 Fab to 2.29 Å resolution with Rsym of 0.063 and were solved in the monoclinic space group P21 containing two molecules in the asymmetric unit. As with the liganded structures, residues 128–132 on the H chain were disordered with some residues excluded from the final model.
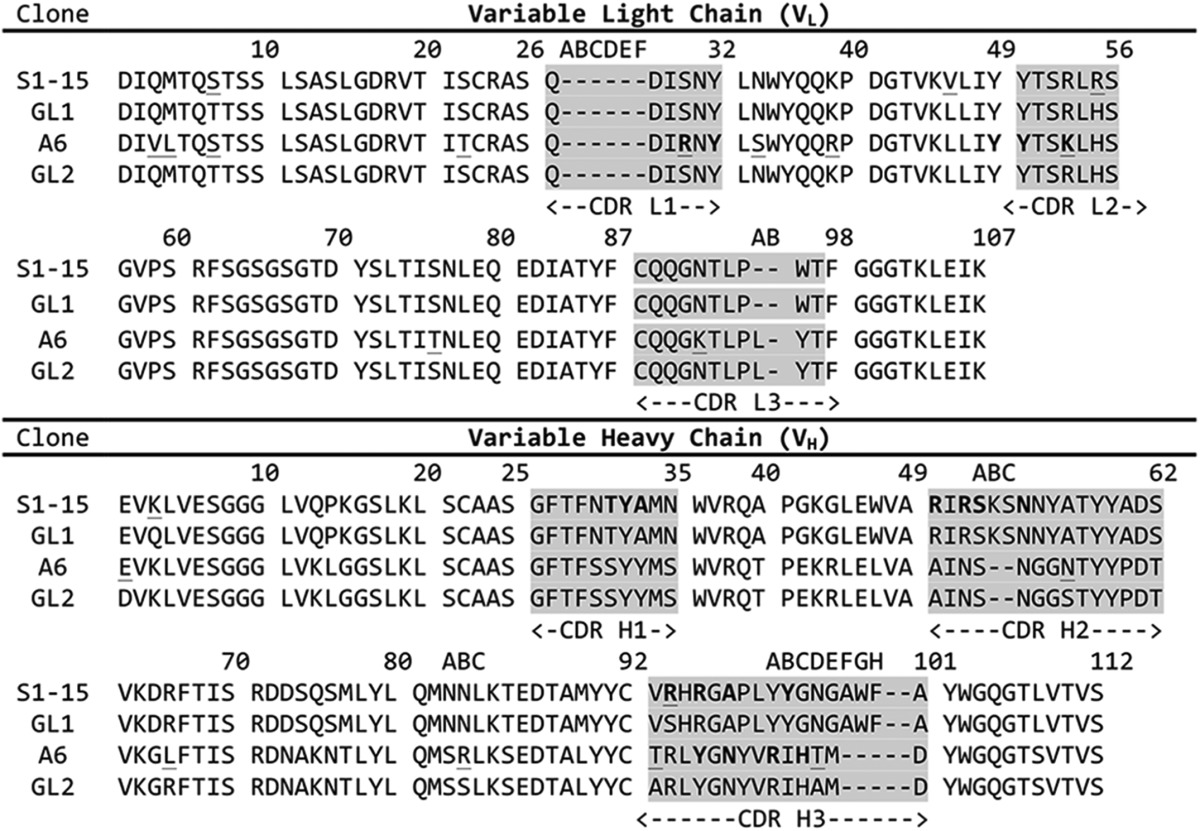
Germ Line Gene Usage and Sequence Comparison
The primary sequence comparisons for the variable loops of mAbs S1–15 and A6 are given in Table 2. IMGT germ line database analysis revealed that the nucleotide sequences of mAbs S1–15 and A6 arise from closely related V- and J-genes for the VL chain. The A6 VL chain was found to share 261/279 and 36/39 nucleotide identity with V-gene IGKV10–96*01 and J-gene IGKJ1*02, respectively (the * denotes the allele group). The S1–15 VH chain was found to share 292/294 nucleotide identity with the V-gene IGHV10–1*02, resulting in a single mutation of a Ser(H)-94 to Arg (residues are identified as H or L to denote the heavy chain and light chains, respectively) at the base of CDR H3. The D- and J-genes of S1–15 are identical in nucleotide sequence to their respective germ line genes IGHD2–1*01 and IGHJ3*01. The A6 VH chain V-, D-, and J-genes belong to IGHV5–6-2*01, IGHD2–1*01, and IGHJ4*01, respectively. The V-, D-, and J-genes of A6 share 280/288, 40/42, and 43/50 in nucleotide identity with their respective germ line genes. Overall, there are 16 amino acid mutations from the germ line gene in A6, six of which lie in the CDRs; there were only five amino acid mutations away from the germ line in S1–15 with only one in the combining site (Table 2).
TABLE 2.
Amino acid sequences of the CDR regions for S1–15 and A6 mAbs
The sequences of S1–15 and A6 likely originate from germ-line (GL) sequence 1 and 2, respectively. The VL chain germ line genes of A6 originate from identical genes as that of S1–15. Numbering is based on the Kabat scheme. Underlined amino acids indicate residues mutated from the germ line, and residues in boldface are contacting the lipid A antigen either directly or through water bridges.
A6 Recognition of Lipid A
There are 11 direct hydrogen bonds to the bisphosphorylated lipid A backbone BBP and two bridging water interactions from one water molecule (Fig. 2E and Table 3). There are three salt bridge interactions as follows: a bidentate salt bridge between 1P and Arg(H)-100A and a bifurcated salt bridge between 1P and Arg(L)-30. An additional contact is formed with the 1P through Tyr(L)-32. Tyr(L)-32 also shares a hydrogen bond with the ring oxygen of the first GlcN. The 4P forms two hydrogen bonds with Tyr(H)-96 and Tyr(L)-49 hydroxyl groups and a salt bridge with Lys(L)-53 residue. The final two hydrogen bonds stem from the terminal GlcN ring oxygen and C6 hydroxyl to Asn(H)-98 and His(H)-100C, respectively. Tyr(L)-50 forms multiple hydrophobic contacts (data not shown) with the hydrophobic face of the terminal GlcN, and an additional strong hydrophobic contact (3.45 Å) to the C6 of the first GlcN residue (Fig. 2E, dashed lines).
TABLE 3.
Hydrogen bond interactions between mAbs A6 and S1–15 to lipid A BBP and between S1–15 and oligonucleotide ligand
Lipid A numbering scheme is given in Fig. 2A.The distance cutoff for hydrogen bond assignment was 3.3 Å, except for charged residue interactions where the distance cutoff was 3.9 Å. Asterisk indicates charged residue interaction.
| Antibody and PDB code | Antigen |
Antibody |
CDR | Distance | ||
|---|---|---|---|---|---|---|
| Residue | Atom(s) | Residue | Atom | |||
| Å | ||||||
| A6 4ODV | GlcN4P | O6 | His(H)-100C | NE2 | H3 | 3.04 |
| O5 | Asn(H)-98 | ND2 | H3 | 3.16 | ||
| 4PO4 | Tyr(H)-96 | OH | H3 | 2.68 | ||
| Lys(L)-53 | NZ | L2 | 3.33* | |||
| Tyr(L)-49 | OH | L2 | 2.46 | |||
| GlcN1P | O5 | Tyr(H)-32 | OH | H1 | 3.15 | |
| 1PO4 | Tyr(H)-32 | OH | H1 | 2.24 | ||
| Arg(H)-100A | NE | H3 | 3.34* | |||
| Arg(H)-100A | NH2 | H3 | 2.87* | |||
| Arg(L)-30 | NH1 | L1 | 2.41* | |||
| Arg(L)-30 | NH2 | L1 | 2.57* | |||
| S1–15 4ODT | GlcN4P | O6 | Ala(H)-33 | N | H1 | 2.95 |
| Arg(H)-96 | O | H3 | 2.85 | |||
| 4PO4 | Arg(H)-52 | NE | H2 | 2.81* | ||
| Arg(H)-52 | NH2 | H2 | 2.70* | |||
| Arg(H)-96 | NH1 | H3 | 3.09* | |||
| GlcN1P | O5 | Ala(H)-98 | N | H3 | 3.06 | |
| 1PO4 | Tyr(H)-32 | OH | H1 | 2.62 | ||
| Tyr(H)-100B | OH | H3 | 2.52 | |||
| S1–15 4Z8F | dT chain E | 3′PO4 | Arg(H)-94 | NH1 | H3 | 2.96* |
| Arg(H)-94 | NH2 | H3 | 2.69* | |||
| Tyr(H)-100B | OH | H3 | 2.68 | |||
| Tyr(H)-102 | OH | H3 | 3.12 | |||
| Arg(L)-55 | NH2 | L2 | 2.71* | |||
| O2 | Thr(H)-28 | N | H1 | 2.85 | ||
| N3 | Thr(H)-28 | OG1 | H1 | 2.88 | ||
| dT chain A | 5′PO4 | Arg(H)-50 | NH1 | H3 | 2.39* | |
| Arg(H)-50 | NH1 | H3 | 2.91* | |||
| Arg(H)-52 | NE | H3 | 2.92* | |||
| Arg(H)-52 | NH2 | H3 | 2.87* | |||
| Arg(H)-96 | NH1 | H3 | 3.05* | |||
| Arg(H)-96 | NH2 | H3 | 2.93* | |||
| O5 | Asn(H)-53 | ND2 | H2 | 2.99 | ||
| O2 | Ser(H)-52 | OG | H2 | 3.06a | ||
a Poor electron density was observed for the terminal thymidine of chain A, and this hydrogen bond therefore cannot be assigned with confidence.
S1–15 Recognition of Lipid A
There are eight direct hydrogen bond contacts to the BBP and six bridging water interactions from four water molecules (Fig. 2F and Table 3). There are two salt bridge interactions as follows: a bidentate salt bridge between 4-phosphate (4P) and Arg(H)-52 and salt bridge between 4P and Arg(H)-96. There are also two additional weak electrostatic interactions as follows: one between 4P and Arg(H)-50 and another between 1P and Arg(H)-94 at a distance of 4.07 and 3.96 Å, respectively. The backbone amide of Ala(H)-33 and the backbone oxygen of Arg(H)-96 both participate in hydrogen bonds with the C6 hydroxyl of the β-GlcN (GlcN4P). A second backbone amide of Ala(H)-98 forms a hydrogen bond to the ring oxygen on the α-GlcN. The 1P group forms two hydrogen bonds with a side chain of Tyr(H)-32 and Tyr(H)-100B residues. Finally, there is a hydrophobic interaction (∼3.7 Å) between the Tyr(H)-32 phenyl ring and the C6 methyl group on the α-GlcN (Fig. 2F, dashed lines).
Comparison with DNA-specific Antibodies
The V-genes of numerous DNA-specific antibodies have been determined (35, 51–53), many of which belong to five subgroups of the J558 family as follows: VH238 (IGHV5-9-03 or 01*),4 133.16VH (IGHV1–82-01*), VH31 (IGHV1S19–01*), 6G6VH (IGHV7–3-04*), and pH3–6a (IGVH9–1-02*). Various antibodies utilizing these genes have been reported to be involved in polyspecificity to p-azophenylarsenate (54), cardiolipin (52), and other autoantigens (55). The V-genes of mAb A6 are closely related to several characterized anti-DNA mAbs (51, 52). The VL chain genes of A6 and S1–15 share over 90% nucleotide identity with several anti-DNA antibodies (52). One of the closest examples include an antibody isolated from hybridoma cell line 73.23 (52) with 96/108 amino acid sequence identity. Although the VH chain V-gene of A6 is not involved in the recognition of lipid A antigen, it is closely related to known anti-DNA V-gene known as VH238 (51), sharing 273/291 nucleotides and 85/98 amino acids.
S1–15 also shares VH genes with an ssDNA-specific autoantibody, BV04-01 (56), and is closely related to antibody Dna-1 (57). S1–15 shares 92% amino acid sequence identity with BV04-01 on the VH chain, although they do not share VL chain genes nor do their heavy chain V-genes match any of the five known anti-DNA V-gene subtypes. Conversely, S1–15 only shares 51% sequence identity with Dna-1 VH chain, but many of these differences are conservative substitutions. Only one of the critical residues contacting lipid A is not present in Dna-1, namely Arg(H)-96.
S1–15 Recognition of ssDNA
The crystal structure of S1–15 Fab in complex with p5(dT)p showed two different oligonucleotide fragments bound at each phosphate-binding site, with a total of 14 hydrogen bonds between ssDNA ligands (Table 3) and residues of the combining site. These include five salt bridges to the terminal phosphates, and 12 hydrogen bonds mediated by eight water molecules (Fig. 2G). There are three salt bridges to the 5′P, formed via Arg(H)-50, Arg(H)-52, and Arg(H)-96. The two latter residues form a bidentate salt bridge. There are also two additional electrostatic interactions to the 3′-terminal phosphate as follows: a bidentate salt bridge from Arg(H)-94 and salt bridge from Arg(L)-55. Tyr(H)-100B and Tyr(H)-102 residues are involved in the hydrogen bonding to the terminal 3′-phosphate. There are only two hydrogen bonds formed between thymidine bases of the ssDNA fragments and the antibody-combining site, both from Thr(H)-28 to the thymidine on the 3′-terminal end. One hydrogen bond is also formed between the ring oxygen of the deoxyribose moiety on the 5′-terminal end and Asn(H)-53. Finally, there are several hydrophobic interactions between Pro(H)-99 ring carbons and the 5′-terminal thymidine base (data not shown).
Crystallization of S1–15 with an ssDNA oligomer lacking the 5′-terminal phosphate resulted in the occupancy of only one phosphate-binding site. Density could only be observed for the terminal 3′P (Fig. 2D), with many of the same residues forming contacts to the 3′-phosphate as with the p5(dT)p complexed structure. Interestingly, the 3′P occupies a slightly different space, resulting in the loss of the hydrogen bond to Tyr(H)-102 and the formation of a hydrogen bond to Tyr(H)-32, rather than the water bridge observed in the p5(dT)p structure (Fig. 2D).
Least Square Superposition of Liganded and Unliganded Fv Structures
Least square alignments of the α-carbon backbones of the VL chains of the corresponding liganded and unliganded Fv structures of S1–15 and A6 are presented in Fig. 3, A and B. The alignment does not include the Fv structure of S1–15 in complex with oligonucleotides, because no significant changes in conformation were observed in comparison with the structure in complex with lipid A. The root mean square deviation (r.m.s.d.) between the liganded and unliganded S1–15 Fv structures are presented in the order of VL to VH, respectively: 0.12 and 0.31 Å for the entire Fv, including a maximum of 2.55 and 1.63 Å and a minimum of 0.02 and 0.06 Å. The only conformational change between the liganded and unliganded occurred for Ser(L)-7, which is responsible for the ∼2.55 Å shift (Fig. 3A). The alignment of A6 resulted in a mean r.m.s.d. of 0.16 and 0.52 Å, a maximum of 1.07 and 10.7 Å, and a minimum of 0.03 and 0.12 Å for the VL and VH, respectively. The three different Fv molecules of A6 showed a large conformational change around Lys(H)-64 on framework region 3 (FR3), resulting in 10.7 Å r.m.s.d. observed. A significant structural shift was also observed between liganded and the two unliganded Fab molecules on CDR H3 with an r.m.s.d. of 5.02 Å (Fig. 3B).
FIGURE 3.

Stereo view of Fv structure alignments between liganded and unliganded structures of S1–15 (A) and A6 (B) is shown. Alignments were carried out using the α-carbon trace of the liganded VL as the reference structure for each antibody. Displacement of CDR L1 and H3 is highlighted. Dark blue, liganded light chain. Cyan, unliganded light chains. Orange, unliganded heavy chains. Red, liganded heavy chain. Because of conformational differences, both unliganded molecules in the asymmetric unit were included in the alignment of A6. Stereo images of the electrostatic surface potentials for Fv structures of S1–15 (C) and A6 (D), bound to lipid A analogue. Blue regions represent relatively positive surface charge, and red represents negative surface charge. White represents neutral surface charge. α-Carbon alignment between BV04-01 Fv (blue) and S1–15 Fv (white) in complex with a trinucleotide (khaki) and lipid A (green), respectively (E), is shown. Transparent surface diagram showing S1–15 Fv and symmetry-related Fv molecule in complex with p5(dT)p (green) fragments (F) is shown. The VL and VH domains are colored blue and pink, respectively. Stereo images of the electrostatic surface potentials for S1–15 Fv in complex with p5(dT)p ssDNA fragments. The lipid A 4′P binding pocket of S1–15 is closely related to the 5′P-binding site of BV04-01. For clarity, only the α-carbon trace of the CDRs is shown for each antibody (G).
Discussion
Steric Hindrance Prevents S1–15 and A6 from Binding LPS
The liganded structures for mAbs S1–15 and A6 demonstrate the structural basis for the failure of these antibodies to recognize LPS-bound lipid A (29, 58), which is the steric hindrance surrounding the C6 hydroxyl of the second GlcN residue that forms the attachment point of the inner core. Furthermore, mAbs S1–15 and A6 possess a pocket specific to the OH-6 (Fig. 3, C and D) and form a hydrogen bond to this hydroxyl. Both factors are consistent with the observed loss of binding to lipid A upon the formation of the 6-O-methyl derivative (29).
S1–15 and A6 Lipid A Recognition Strategy
The binding mechanism of antibodies is often described as “lock and key” or “induced fit” based on the degree of dislocation of the CDRs when comparing bound and unbound states of the antibody (16, 59–64). Comparison between the unliganded and liganded structure of S1–15 showed that lock and key type, with its associated minimum entropic penalty upon binding (Fig. 3A). However, mAb A6 displayed a significant degree of induced fit, particularly in the FR3 of the VH where a maximum r.m.s.d. of 10.7 Å was observed (Fig. 3B).
An antibody-combining site can form a pocket or groove, or a combination of both depending on the nature of antigen recognition. Antibodies specific for proteins and larger structures often utilize grooves, although antibodies specific for oligosaccharides and haptens often utilize pockets. As expected, each of the lipid A-specific antibodies uses a pocket with high structural and charge complementarity to lipid A (Fig. 3, C and D). Carbohydrate-specific antibodies are usually observed to bind antigen along the VL/VH interface, which explains the difficulty that has been reported in attempts to use the phage display technique to isolate single domain antibodies (i.e. composed solely of a heavy or light chain variable domain) specific for carbohydrates larger than a monosaccharide (65, 66). Interestingly, the lipid A-specific antibody S1–15 Fab binds lipid A with no involvement of the light chain (Figs. 2F and 3C). Although antibodies have been observed to recognize monosaccharides in pockets formed exclusively by heavy CDRs (67), this is the only structural example to date of an antibody using a heavy chain binding pocket to bind a carbohydrate moiety larger than a monosaccharide.
Comparison between S1–15 and A6 Binding to Lipid A
Although there are similarities, the combining sites utilize strikingly different mechanisms to recognize the lipid A backbone. In contrast to S1–15, mAb A6 binds to the lipid A antigen with both VL and VH chain residues, despite sharing VL chain genes with S1–15. Most of the hydrogen bonds in the S1–15·lipid A complex are directed toward the 4P of the second GlcN (Fig. 2F), complementing ELISA data, where binding toward the 4P-monophosphorlyated lipid A antigen is largely retained (Fig. 1A). A6 forms multiple contacts through both VL and VH chain residues to both phosphates of the lipid A analogue BBP, with the majority of these contacts directed toward the 1P (Fig. 2E), and thus there is a significant loss of avidity to the 4P-monophosphorylated lipid A (Fig. 1B). One feature common to both antibodies is a strong hydrophobic contact between the C6 carbon of the first GlcN (Fig. 2, E and F, dashed lines) and Tyr residues His-32 and Leu-50.
From the conjugate ELISA, we determined the relative strength of binding, showing a 64-fold increase in avidity toward the bisphosphorylated lipid A backbone when comparing A6 to S1–15 at 2 pmol/well concentration of antigen (Fig. 1, A and B). This trend is consistent with the number of contacts and salt bridges formed between these antibodies and lipid A. A6 forms 11 hydrogen bonds, three of which are charged interactions. In contrast, S1–15 forms eight hydrogen bonds between lipid A and residues of S1–15, two of which are charged residue interactions.
Sequence Comparison to Anti-nucleotide Antibodies and the Basis for Polyspecificity
The structure of A6 and S1–15 in complex with lipid A carbohydrate backbone revealed distances of 11.9 and 12.1 Å, respectively, between the 1′- and 4′- phosphates, which approximate the distance (11.1 Å) observed in the trinucleotide in complex with antibody BV04-01 Fab.
Similarities in sequence had pointed toward potential polyspecificity of A6 and S1–15 toward ssDNA, as antibodies produced by immunization with lipid A carbohydrate backbone often share V-genes with DNA-specific antibodies. For example, the antibody belonging to the hybridoma cell line 73.23 possesses a related VL gene to A6 mAb, which not only binds ssDNA but dsDNA with lower avidity (52). Of the residues contacting lipid A for mAb A6, only Arg(L)-30 differs from anti-DNA antibody 73.23 (replaced by Ser(L)-30). Although the heavy chain V-region (coded by the V-gene) of A6 is not involved in recognition of lipid A, the regions corresponding to the D and J genes of CDR H3 play a crucial role. DNA-specific antibodies have been shown to have CDR H3 loops dominated by Arg and Tyr residues (51), which is a feature shared with the CDR H3 of A6 (Table 2).
Structural Basis for S1–15 ssDNA Polyspecificity
Crystals were observed growing in number of different conditions, and several structures determined where the oligonucleotide was highly disordered (data not shown). After considerable optimization over a large range of different conditions and ligands, a structure with interpretable density was obtained for S1–15 in complex with the 5(dT) oligonucleotide; however, the combining site was not spanned by a single molecular ssDNA, but it bound two separate oligonucleotides by their terminal 5′- and 3′-phosphate groups (Fig. 3E). This arrangement was stabilized by base stacking between symmetry-related oligonucleotides (Figs. 2C and 3F). The phosphate binding pocket on S1–15 that recognizes 1P of lipid A binds the terminal 3′-phosphate, although the 4P lipid A pocket binds the 5′-phosphate. Interestingly, higher temperature factors were observed for the 5′-terminal phosphate-binding site when refined at full occupancy, despite the fact that there are more interactions with the 5′P. This appears to be due to two reasons. First there are two base-specific hydrogen bonds out of the total 14, both occurring at the 3′-terminal end. Second, the 3′-terminal thymidine base formed stacking interactions with the neighboring thymidine nucleotide. Furthermore, in the S1–15 Fab structure in complex with the 5(dT)p (lacking 5′P), density is only observable for the 3′-terminal phosphate with the residual oligonucleotides being disordered. This suggests that the 5′P-binding site plays a crucial role in stabilizing the second oligonucleotide fragment via stacking interactions on the 3′P-binding site.
Finally, density for the thymidine base on the 5′ end was poor in the p5(dT)p liganded structure due to high thermal motion, consistent with the lack of stabilizing stacking interactions.
Structural Comparison with Single-stranded Nucleotide-specific Antibodies
Published anti-ssDNA antibody structures show that the backbone phosphates are usually oriented away from the combining site, with the CDRs interacting directly through stacking interactions with the hydrophobic bases.
Autoantibody BV04-01 does recognize the backbone phosphate groups in its complex with the trinucleotide p5(dT) (lacking the 3′P), with the central thymidine forming stacking interactions with a Tyr and a Trp residue on either side of the thymidine (56). Additionally, the 5′P and central phosphate groups of the trinucleotide contribute to binding through hydrogen bonds, and interestingly, the 5′P-binding site of BV04-01 contains many interactions that correspond to the 5′P (and the 4′P lipid A)-binding site of S1–15 (Fig. 3G), marking its importance for dual recognition of ssDNA and lipid A. Of the three phosphates of the trinucleotide bound to BV04-01, only one phosphate formed a salt bridge to the Fab.
Similarly the RNA-specific antibody Jel 103 Fab structure in complex with RNA nucleotide (Protein Data Bank code 1MRF) display base stacking interaction with a Tyr residue and two salt bridges to the phosphate moiety from Lys and Arg residues, respectively (47).
It is remarkable that S1–15 and A6 bind ssDNA at all, as both have undergone affinity maturation toward the lipid A carbohydrate antigen, and the corresponding germ line antibodies could exhibit alternative binding modes to ssDNA than observed here. Crucially, the higher avidity of decavalent IgM plays an important role in establishing polyspecificity toward single-stranded nucleic acids. Binding assays have shown significant binding to various ssDNA fragments for both antibodies (data not shown), and additional crystallization studies with these antigens are ongoing.
Conclusions
The structures of mAbs S1–15 and A6 provide a structural explanation of their inability to bind intact LPS.
The ability of antibodies to undergo somatic hypermutation implies that germ line antibodies in general must display a significant degree of cross-reactivity or polyspecificity to increase the number of antigens recognized. Although there has been significant progress toward structural characterization of antibody cross-reactivity, there is a dearth of structures demonstrating the polyspecific nature of antibodies. These structures show significant binding to terminal nucleotides and shorter ssDNA oligonucleotides via positively charged surface complementary to the terminal phosphates. Structural insights into the molecular basis for polyspecificity against ssDNA is of clinical interest and may provide crucial clues into how these antibodies arise as implicated in autoimmune diseases such as systemic lupus erythematosus, thyroiditis, and rheumatic autoimmune diseases.
Author Contributions
O. H. G. performed the experiments shown in Figs. 2 and 3, conceived some of the experiments, analyzed the data, and wrote a major part of the paper. S. M. L. performed the ELISA experiments shown in Fig. 1, conceived some of the experiments, analyzed the data, and contributed to the paper. T. R. performed some of the experiments. L. B. performed some of the experiments, analyzed the data, and contributed to the paper. P. K. provided reagents and contributed to the paper. H. B. provided reagents, conceived many of the experiments, and contributed to the paper. S. V. E. conceived many of the experiments and wrote a major part the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We gratefully acknowledge the technical assistance of Ute Agge, Veronika Susott, and Christine Schneider at the Research Center Borstel, Germany. Research described in this paper was performed at the Canadian Light Source, which is supported by the Canada Foundation for Innovation, Natural Sciences and Engineering Research Council of Canada, the University of Saskatchewan, the Government of Saskatchewan, Western Economic Diversification Canada, the National Research Council Canada, and the Canadian Institutes of Health Research.
This work was supported by the Natural Sciences and Engineering Research Council of Canada (to S. V. E.) and by Austrian Science Fund (Fonds zur Förderung der Wissenschaftlichen Forschung) Grant P22909 (to P. K.). The authors declare that they have no conflicts of interest with the contents of this article.
- BBP
- bisphosphorylated lipid A backbone
- CDR
- complementarity determining region
- Fab
- fragment antigen binding
- Fv
- fragment variable
- GlcN
- d-glucosamine
- VH
- variable heavy
- VL
- variable light
- B4P
- 4′-monophosphorylated lipid A backbone
- DVS
- divinyl sulfone
- ssDNA
- single-stranded DNA
- bicine
- N,N-bis(2-hydroxyethyl)glycine
- r.m.s.d.
- root mean square deviation.
REFERENCES
- 1. Angus D. C., Linde-Zwirble W. T., Lidicker J., Clermont G., Carcillo J., Pinsky M. R. (2001) Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Crit. Care Med. 29, 1303–1310 [DOI] [PubMed] [Google Scholar]
- 2. Engel C., Brunkhorst F. M., Bone H. G., Brunkhorst R., Gerlach H., Grond S., Gruendling M., Huhle G., Jaschinski U., John S., Mayer K., Oppert M., Olthoff D., Quintel M., Ragaller M., et al. (2007) Epidemiology of sepsis in Germany: results from a national prospective multicenter study. Intensive Care Med. 33, 606–618 [DOI] [PubMed] [Google Scholar]
- 3. Martin G. S. (2012) Sepsis, severe sepsis and septic shock: changes in incidence, pathogens and outcomes. Expert Rev. Anti.-Infect. Ther. 10, 701–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Buttenschoen K., Radermacher P., Bracht H. (2010) Endotoxin elimination in sepsis: physiology and therapeutic application. Langenbeck. Arch. Surg. 395, 597–605 [DOI] [PubMed] [Google Scholar]
- 5. Miller S. I., Ernst R. K., Bader M. W. (2005) LPS, TLR4 and infectious disease diversity. Nat. Rev. Microbiol. 3, 36–46 [DOI] [PubMed] [Google Scholar]
- 6. Kim H. M., Park B. S., Kim J. I., Kim S. E., Lee J., Oh S. C., Enkhbayar P., Matsushima N., Lee H., Yoo O. J., Lee J. O. (2007) Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist eritoran. Cell 130, 906–917 [DOI] [PubMed] [Google Scholar]
- 7. Miyake K. (2007) Innate immune sensing of pathogens and danger signals by cell surface Toll-like receptors. Semin. Immunol. 19, 3–10 [DOI] [PubMed] [Google Scholar]
- 8. Hold G., Bryant C. (2011) in Bacterial Lipopolysaccharides (Knirel Y., Valvano M., eds) pp. 371–387, Springer, Vienna [Google Scholar]
- 9. Di Padova F. E., Brade H., Barclay G. R., Poxton I. R., Liehl E., Schuetze E., Kocher H. P., Ramsay G., Schreier M. H., McClelland D. B. (1993) A broadly cross-protective monoclonal-antibody binding to Escherichia coli and Salmonella lipopolysaccharides. Infect. Immun. 61, 3863–3872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gomery K., Müller-Loennies S., Brooks C. L., Brade L., Kosma P., Di Padova F., Brade H., Evans S. V. (2012) Antibody WN1 222-5 mimics Toll-like receptor 4 binding in the recognition of LPS. Proc. Natl. Acad. Sci. U.S.A. 109, 20877–20882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Müller-Loennies S., Brade L., Brade H. (2007) Neutralizing and cross-reactive antibodies against enterobacterial lipopolysaccharide. Int. J. Med. Microbiol. 297, 321–340 [DOI] [PubMed] [Google Scholar]
- 12. Brade L., Brade H. (1985) Characterization of two different antibody specificities recognizing distinct antigenic determinants in free lipid A of Escherichia coli. Infect. Immun. 48, 776–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fujihara Y., Bogard W. C., Lei M. G., Daddona P. E., Morrison D. C. (1993) Monoclonal anti-lipid A IgM antibodies HA-1A and E-5 recognize distinct epitopes on lipopolysaccharide and lipid A. J. Infect. Dis. 168, 1429–1435 [DOI] [PubMed] [Google Scholar]
- 14. Helmerhorst E. J., Maaskant J. J., Appelmelk B. J. (1998) Anti-lipid A monoclonal antibody centoxin (HA-1A) binds to a wide variety of hydrophobic ligands. Infect. Immun. 66, 870–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kuhn H. M. (1993) Cross-reactivity of monoclonal antibodies and sera directed against lipid A and lipopolysaccharides. Infection 21, 179–186 [DOI] [PubMed] [Google Scholar]
- 16. Blackler R. J., Müller-Loennies S., Brade L., Kosma P., Brade H., Evans S. V. (2012) in Anticarbohydrate Antibodies: From Molecular Basis to Clinical Application (Kosma P., Müller-Loennies S., eds) pp. 75–120, Springer, Vienna [Google Scholar]
- 17. Rynkiewicz M. J., Lu Z., Hui J. H., Sharon J., Seaton B. A. (2012) Structural analysis of a protective epitope of the Francisella tularensis O-polysaccharide. Biochemistry 51, 5684–5694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vulliez-Le Normand B., Saul F. A., Phalipon A., Bélot F., Guerreiro C., Mulard L. A., Bentley G. A. (2008) Structures of synthetic O-antigen fragments from serotype 2a Shigella flexneri in complex with a protective monoclonal antibody. Proc. Natl. Acad. Sci. U.S.A. 105, 9976–9981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cygler M., Rose D. R., Bundle D. R. (1991) Recognition of a cell-surface oligosaccharide of pathogenic Salmonella by an antibody Fab fragment. Science 253, 442–445 [DOI] [PubMed] [Google Scholar]
- 20. Ianaro A., Tersigni M., D'Acquisto F. (2009) New insight in LPS antagonist. Mini-Rev. Med. Chem. 9, 306–317 [DOI] [PubMed] [Google Scholar]
- 21. Li J., Carr B., Goyal M., Gaieski D. F. (2011) Sepsis: the inflammatory foundation of pathophysiology and therapy. Hosp. Pract. 39, 99–112 [DOI] [PubMed] [Google Scholar]
- 22. Cross A. S. (2014) Anti-endotoxin vaccines: Back to the future. Virulence 5, 219–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Holst O. (2007) The structures of core regions from enterobacterial lipopolysaccharides–an update. FEMS Microbiol. Lett. 271, 3–11 [DOI] [PubMed] [Google Scholar]
- 24. Pollack M., Ohl C. A., Golenbock D. T., Di Padova F., Wahl L. M., Koles N. L., Guelde G., Monks B. G. (1997) Dual effects of LPS antibodies on cellular uptake of LPS and LPS-induced proinflammatory functions. J. Immunol. 159, 3519–3530 [PubMed] [Google Scholar]
- 25. Kuhn H. M., Brade L., Appelmelk B. J., Kusumoto S., Rietschel E. T., Brade H. (1992) Characterization of the epitope specificity of murine monoclonal antibodies directed against lipid A. Infect. Immun. 60, 2201–2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Galanos C., Lüderitz O., Westphal O. (1971) Preparation and properties of antisera against the lipid A component of bacterial lipopolysaccharides. Eur. J. Biochem. 24, 116–122 [DOI] [PubMed] [Google Scholar]
- 27. Appelmelk B. J., Verweij-van Vught A. M., Maaskant J. J., Schouten W. F., De Jonge A. J., Thijs L. G., Maclaren D. M. (1988) Production and characterisation of mouse monoclonal antibodies reacting with the lipopolysaccharide core region of Gram-negative bacilli. J. Med. Microbiol. 26, 107–114 [DOI] [PubMed] [Google Scholar]
- 28. Brade L., Holst O., Brade H. (1993) An artificial glycoconjugate containing the bisphosphorylated glucosamine disaccharide backbone of lipid-A binds lipid-A monoclonal antibodies. Infect. Immun. 61, 4514–4517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brade L., Engel R., Christ W. J., Rietschel E. T. (1997) A nonsubstituted primary hydroxyl group in position 6′ of free lipid A is required for binding of lipid A monoclonal antibodies. Infect. Immun. 65, 3961–3965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bhat N. M., Bieber M. M., Chapman C. J., Stevenson F. K., Teng N. N. (1993) Human antilipid A monoclonal antibodies bind to human B cells and the I antigen on cord red blood cells. J. Immunol. 151, 5011–5021 [PubMed] [Google Scholar]
- 31. Miller J. J., 3rd, Bieber M. M., Levinson J. E., Zhu S., Tsou E., Teng N. N. (1996) V(H)4–34(V(H)4.21) gene expression in the chronic arthritides of childhood: studies of associations with anti-lipid A antibodies, HLA antigens, and clinical features. J. Rheumatol. 23, 2132–2139 [PubMed] [Google Scholar]
- 32. Izui S., Kobayakawa T., Zryd M. J., Louis J., Lambert P. H. (1977) Mechanism for induction of anti-DNA antibodies by bacterial lipopolysaccharides in mice. II. Correlation between anti-DNA induction and polyclonal antibody formation by various polyclonal B lymphocyte activators. J. Immunol. 119, 2157–2162 [PubMed] [Google Scholar]
- 33. Sumazaki R., Fujita T., Kabashima T., Nishikaku F., Koyama A., Shibasaki M., Takita H. (1986) Monoclonal antibody against bacterial lipopolysaccharide cross-reacts with DNA-histone. Clin. Exp. Immunol. 66, 103–110 [PMC free article] [PubMed] [Google Scholar]
- 34. Spellerberg M., Chapman C., Hamblin T., Stevenson F. (1995) Dual recognition of lipid A and DNA by human antibodies encoded by the V(H)4–21 gene–a possible link between infection and lupus. Ann. NY. Acad. Sci. 764, 427–432 [DOI] [PubMed] [Google Scholar]
- 35. Tillman D. M., Jou N. T., Hill R. J., Marion T. N. (1992) Both IgM and IgG anti-DNA antibodies are the products of clonally selective B-cell stimulation in (Nzb × Nzw)F(1) mice. J. Exp. Med. 176, 761–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Robertson C. R., Pisetsky D. S. (1992) Specificity analysis of antibodies to single-stranded micrococcal DNA in the sera of normal human–subjects and patients with systemic lupus-erythematosus. Clin. Exp. Rheumatol. 10, 589–594 [PubMed] [Google Scholar]
- 37. Ravirajan C. T., Rowse L., MacGowan J. R., Isenberg D. A. (2001) An analysis of clinical disease activity and nephritis-associated serum autoantibody profiles in patients with systemic lupus erythematosus: a cross-sectional study. Rheumatology 40, 1405–1412 [DOI] [PubMed] [Google Scholar]
- 38. Pedro A. B., Romaldini J. H., Americo C., Takei K. (2006) Association of circulating antibodies against double-stranded and single-stranded DNA with thyroid autoantibodies in Graves' disease and Hashimoto's thyroiditis patients. Exp. Clin. Endocrinol. Diabetes 114, 35–38 [DOI] [PubMed] [Google Scholar]
- 39. Kaburaki J., Kuwana M., Ogasawara T., Takano M., Funatsu Y., Tojo T. (1992) Specificity of antibodies to single-stranded (ss)DNA in SLE patients with anti-phospholipid syndrome. Keio J. Med. 41, 10–15 [DOI] [PubMed] [Google Scholar]
- 40. Brochet X., Lefranc M. P., Giudicelli V. (2008) IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res. 36, W503–W508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Monod M. Y., Giudicelli V., Chaume D., Lefranc M. P. (2004) IMGT/Junction Analysis: the first tool for the analysis of the immunoglobulin and T cell receptor complex V-J and V-D-J junctions. Bioinformatics 20, 379–385 [DOI] [PubMed] [Google Scholar]
- 42. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 44. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 45. Winn M. D., Murshudov G. N., Papiz M. Z. (2003) Macromolecular TLS refinement in REFMAC at moderate resolutions. Method Enzymol. 374, 300–321 [DOI] [PubMed] [Google Scholar]
- 46. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 47. Pokkuluri P. R., Bouthillier F., Li Y., Kuderova A., Lee J., Cygler M. (1994) Preparation, characterization and crystallization of an antibody Fab fragment that recognizes RNA: Crystal structures of native Fab and 3 Fab-mononucleotide complexes. J. Mol. Biol. 243, 283–297 [DOI] [PubMed] [Google Scholar]
- 48. Tormo J., Stadler E., Skern T., Auer H., Kanzler O., Betzel C., Blaas D., Fita I. (1992) 3-Dimensional structure of the Fab fragment of a neutralizing antibody to human rhinovirus serotype-2. Protein Sci. 1, 1154–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mol C. D., Muir A. K., Cygler M., Lee J. S., Anderson W. F. (1994) Structure of an immunoglobulin Fab fragment specific for triple-stranded DNA. J. Biol. Chem. 269, 3615–3622 [PubMed] [Google Scholar]
- 50. Mol C. D., Muir A. K., Lee J. S., Anderson W. F. (1994) Structure of an immunoglobulin Fab fragment specific for poly(dG)·poly(dC). J. Biol. Chem. 269, 3605–3614 [PubMed] [Google Scholar]
- 51. Shlomchik M., Mascelli M., Shan H., Radic M. Z., Pisetsky D., Marshak-Rothstein A., Weigert M. (1990) Anti-DNA antibodies from autoimmune mice arise by clonal expansion and somatic mutation. J. Exp. Med. 171, 265–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ibrahim S. M., Weigert M., Basu C., Erikson J., Radic M. Z. (1995) Light-chain contribution to specificity in anti-DNA antibodies. J. Immunol. 155, 3223–3233 [PubMed] [Google Scholar]
- 53. Krishnan M. R., Jou N. T., Marion T. N. (1996) Correlation between the amino acid position of arginine in VH-CDR3 and specificity for native DNA among autoimmune antibodies. J. Immunol. 157, 2430–2439 [PubMed] [Google Scholar]
- 54. Rathbun G. A., Otani F., Milner E. C., Capra J. D., Tucker P. W. (1988) Molecular characterization of the A-J-J558 family of heavy-chain variable region gene segments. J. Mol. Biol. 202, 383–395 [DOI] [PubMed] [Google Scholar]
- 55. Monestier M., Bonin B., Migliorini P., Dang H., Datta S., Kuppers R., Rose N., Maurer P., Talal N., Bona C. (1987) Autoantibodies of various specificities encoded by genes from the Vh J558 family bind to foreign antigens and share idiotopes of antibodies specific for self and foreign antigens. J. Exp. Med. 166, 1109–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Herron J. N., He X. M., Ballard D. W., Blier P. R., Pace P. E., Bothwell A. L., Voss E. W., Jr., Edmundson A. B. (1991) An autoantibody to single-stranded-DNA- comparison of the 3-dimensional structures of the unliganded Fab and a deoxynucleotide Fab complex. Proteins 11, 159–175 [DOI] [PubMed] [Google Scholar]
- 57. Tanner J. J., Komissarov A. A., Deutscher S. L. (2001) Crystal structure of an antigen-binding fragment bound to single-stranded DNA. J. Mol. Biol. 314, 807–822 [DOI] [PubMed] [Google Scholar]
- 58. El-Samalouti V. T., Schletter J., Brade H., Brade L., Kusumoto S., Rietschel E. T., Flad H. D., Ulmer A. J. (1997) Detection of lipopolysaccharide(LPS)-binding membrane proteins by immuno-coprecipitation with LPS and anti-LPS antibodies. Eur. J. Biochem. 250, 418–424 [DOI] [PubMed] [Google Scholar]
- 59. Rini J. M., Schulze-Gahmen U., Wilson I. A. (1992) Structural evidence for induced fit as a mechanism for antibody-antigen recognition. Science 255, 959–965 [DOI] [PubMed] [Google Scholar]
- 60. Wilson I. A., Stanfield R. L. (1994) Antibody-antigen interactions: new structures and new conformational-changes. Curr. Opin. Struct. Biol. 4, 857–867 [DOI] [PubMed] [Google Scholar]
- 61. Schulze-Gahmen U., Rini J. M., Wilson I. A. (1993) Detailed analysis of the free and bound conformations of an antibody. X-ray structures of Fab 17/9 and 3 different Fab-peptide complexes. J. Mol. Biol. 234, 1098–1118 [DOI] [PubMed] [Google Scholar]
- 62. Blackler R. J., Müller-Loennies S., Brooks C. L., Evans D. W., Brade L., Kosma P., Brade H., Evans S. V. (2011) A common NH53K mutation in the combining site of antibodies raised against chlamydial LPS glycoconjugates significantly increases avidity. Biochemistry 50, 3357–3368 [DOI] [PubMed] [Google Scholar]
- 63. Brooks C. L., Müller-Loennies S., Brade L., Kosma P., Hirama T., MacKenzie C. R., Brade H., Evans S. V. (2008) Exploration of specificity in germline monoclonal antibody recognition of a range of natural and synthetic epitopes. J. Mol. Biol. 377, 450–468 [DOI] [PubMed] [Google Scholar]
- 64. Nguyen H. P., Seto N. O., MacKenzie C. R., Brade L., Kosma P., Brade H., Evans S. V. (2003) Germline antibody recognition of distinct carbohydrate epitopes. Nat. Struct. Biol. 10, 1019–1025 [DOI] [PubMed] [Google Scholar]
- 65. Vranken W., Tolkatchev D., Xu P., Tanha J., Chen Z., Narang S., Ni F. (2002) Solution structure of a llama single-domain antibody with hydrophobic residues typical of the VH/VL interface. Biochemistry 41, 8570–8579 [DOI] [PubMed] [Google Scholar]
- 66. Deng S. J., MacKenzie C. R., Sadowska J., Michniewicz J., Young N. M., Bundle D. R., Narang S. A. (1994) Selection of antibody single-chain variable fragments with improved carbohydrate-binding by phage display. J. Biol. Chem. 269, 9533–9538 [PubMed] [Google Scholar]
- 67. Kaminski M. J., MacKenzie C. R., Mooibroek M. J., Dahms T. E., Hirama T., Houghton A. N., Chapman P. B., Evans S. V. (1999) The role of homophilic binding in anti-tumor antibody R24 recognition of molecular surfaces. Demonstration of an intermolecular beta-sheet interaction between vh domains. J. Biol. Chem. 274, 5597–5604 [DOI] [PubMed] [Google Scholar]