

Background: The JAK-STAT3 signaling pathway is one of the critical pathways regulating cell proliferation and differentiation.
Results: Knockdown of endogenous STAT3 enhances VSMC contractile phenotype by promoting the association of the myocardin-SRF-CArG complex.
Conclusion: The JAK-STAT3 signaling pathway is a central regulator of the phenotypic switch of VSMCs.
Significance: The phenotypic switch of VSMCs can be controlled by modulation of JAK-STAT3 signaling.
Keywords: cardiovascular, cardiovascular disease, cell signaling, transcription factor, transcription regulation
Abstract
The JAK-STAT3 signaling pathway is one of the critical pathways regulating cell proliferation, differentiation, and apoptosis. Myocardin is regarded as a key mediator for the change of smooth muscle phenotypes. However, the relationship between STAT3 and myocardin in the vascular smooth muscle cell (VSMC) phenotypic switch has not been investigated. The goal of this study was to investigate the molecular mechanism by which STAT3 affects the myocardin-regulated VSMC phenotypic switch. Data presented in this study demonstrated that STAT3 was rapidly up-regulated after stimulation with VEGF. Inhibition of the STAT3 activation process impaired VSMC proliferation and enhanced the expression of VSMC contractile genes by increasing serum-response factor binding to the CArG-containing regions of VSMC-specific contractile genes. In contrast, the interaction between serum-response factor and its co-activator myocardin was reduced by overexpression of STAT3. In addition, treated VEGF inhibited the transcription activity of myocardin, and overexpression of STAT3 inhibited myocardin-induced up-regulation of VSMC contractile phenotype-specific genes. Although myocardin and STAT3 are negatively correlated, interestingly, both of them can enhance the expression of VEGF, suggesting a feedback loop to regulate the VSMC phenotypic switch. Taken together, these results indicate that the JAK-STAT3 signaling pathway plays a key role in controlling the phenotypic switch of VSMCs through the interactions between STAT3 and myocardin by various coordinated gene regulation pathways and feedback loops.
Introduction
Vascular smooth muscle cells (VSMCs)3 are characterized in part by the ability to modulate their phenotypes in response to the environmental stimuli through a process characterized by decreased gene expression of VSMC contractile markers, increased proliferation, migration, and matrix synthesis (1). Phenotypic switch is one of the major cellular events underlying many VSMC-related pathological conditions, such as atherosclerosis, postangioplasty restenosis, hypertension, and tumor angiogenesis (1). Unraveling the mechanisms involved in VSMC phenotypic switch is an important step toward a better understanding of the pathology of these diseases as well as in designing therapeutic agents for their treatment and prevention.
The switch between the contractile and synthetic VSMC phenotypes is tightly controlled through a synergistic and coordinated molecular regulatory network (2–4). Nearly all of the VSMC-specific contractile protein genes and many other genes, which contain CArG boxes within their promoters, are involved in cellular migration, proliferation, and extracellular matrix production during the process of VSMC phenotypic switch (1, 5, 6). Myocardin is regarded as a key mediator in smooth muscle phenotype change (4). The cellular mechanisms that drive SMC plasticity (e.g. the switch between contractile and synthetic phenotypes) are partially dependent on the interactions between myocardin and SRF (7). The myocardin-SRF complex, through its interaction with the promoters containing multiple CArG boxes (CC(A/T)6GG), can directly activate a battery of contractile smooth muscle genes, including h1 calponin (CNN1), transgelin (SM22 or TAGLN), α-smooth muscle actin (ACTA2), and smooth muscle myosin heavy chain (MYH11) (11–13). In addition, myocardin/SRF decreases SMC proliferation by increasing the expression of the cell cycle inhibiting gene cyclin-dependent kinase inhibitor 1A (CDKN1A) (14) and inhibiting NFκB-dependent cell cycle progression (15), thus indirectly promoting the contractile phenotype. Although myocardin is considered as a critical component of the contractile “switch” in SMC, the signals that regulate myocardin are only partially understood. Extracellular signals can mediate this molecular switch through alterations in effector pathways, and indeed myocardin-mediated gene activation is regulated by ligands such as TGFβ1 and angiotensin II (16), as well as intracellular signal transduction mediators such as YAP1, glycogen synthase kinase 3 (GSK3), and extracellular signal-regulated kinase (ERK) and the C terminus of Hsc70-interacting protein (ChIP) (17–20). Although the regulation of myocardin gene expression has been widely explored, there are few studies that address the cellular signal pathways underlying the regulation of myocardin-mediated SMC phenotypic changes.
Activation of the signal transducer and activator of transcription-3 (STAT3) by the Janus kinase-2 (JAK2) affects various biological processes, such as proliferation, cell survival, and inflammation (21). JAK2-dependent phosphorylation of STAT3 is induced in SMCs in response to various growth factors and cytokines, including platelet-derived growth factor (PDGF)-BB, angiotensin II, or interleukin (IL)-6 or vascular endothelial growth factor (VEGF) (22–24). STAT3 has been implicated as a central regulator of tumor progression through its transcriptional up-regulation of VEGF, MCL-1, and survivin, etc. (25). It has been suggested that STAT3 is critically involved in vascular injury, but the details of how STAT3-dependent genes are regulated as well as the functional significance of suppressing this pathway during the development of vascular proliferative diseases remain elusive. Previous studies, both in vitro and in vivo (in neointimal cells), have shown that phosphorylation of STAT3 induces trans-activation of cyclin D1 and survivin in SMCs, which therefore promotes proliferation and migration of SMCs as well as reducing apoptotic cell death (26). More recent evidence also suggest that STAT3 could be involved in VEGF-induced endothelial cell (EC) signaling and activation (27, 28). STAT3 is activated upon VEGF stimulation in EC by a VEGFR2-dependent and SRC-dependent mechanism, and this activation also mediates the induction of BCL2 by VEGF (29).
Although the JAK-STAT pathway has received extensive study, its role in the VSMC phenotypic switch is still poorly understood, In particular, the links between STAT3 and myocardin, as well as their interaction and coordination on how to control VSMC phenotypic switch under the situation of VEGF stimulation, have not been developed. Here, we reveal for the first time the role of STAT3 and myocardin in the VSMC phenotypic switch. We found that VEGF activates STAT3 but inhibits the transcription activity of myocardin during the VSMC phenotypic switch. Activated STAT3 not only led to enhanced proliferation of VSMCs but also suppressed the expression of VSMC-specific contractile protein genes. Furthermore, STAT3 affects VSMC phenotypic switch via interference with the myocardin-SRF-CArG box tertiary complex formation. Importantly, both myocardin and STAT3 can enhance the expression of VEGF, which leads to a feedback loop to regulate the VSMC phenotypic switch. Our study provides new insights into the mechanisms controlling the phenotypic switch of VSMC and identifies a potential therapeutic target for ameliorating VSMC-related diseases, and it might facilitate the development of novel approaches in vascular engineering.
Experimental Procedures
Cell Lines
T/G HA-VSMC cell lines (ATCC) were grown in Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum at 37 °C in a 5% CO2 incubator.
Plasmids
The vector pcDNA3.1 (Promega) alone was used as a negative control. The promoter regions of ACTA-2 (−3270/+295) and SM22α (−234/+246) were amplified by PCR followed by cloning into pGL3 luciferase reporter vector, respectively. The primers used to create ACTA2-luc and SM22α-luc were as follows: ACTA-2, F-5′ GGGGGTACCGTCTGTCTCACCTCTAGAACTAACT 3′ and R-5′ CCCCTCGAGCATACCAAACTACATATTAGCCCTG 3′; SM22α, F-5′ CCGGTACCTAAAAGGCTTTTCCCGGGCC 3′ and R-5′ CCCTCGAGGACTGAGAGGGTGGGTTTCC 3′; the vector pGL-3(Promega) was used as a control.
VSMC Proliferation Assays
Cell proliferation was determined by the growth curves of VSMCs; cell counting was performed as described previously (30). Briefly, VSMCs treated with VEGF or LIF or AG490 were seeded at a density of 6 × 105 cells/ml, respectively. Cell numbers were counted with a hemocytometer for the indicated periods. Each count was an average of three repeats, and each data point in the curve was the average of four experiments.
Enzyme-linked Immunosorbent Assay (ELISA)
The concentration of VEGF in the conditioned media derived from treated and untreated cells was measured using human VEGF Quantikine ELISA kit (R&D Systems) according to the manufacturer's instructions. The experiment was repeated in triplicate.
siRNA Gene Silencing
For functional studies of myocardin and STAT3, an siRNA-based silencing approach was used. Customized siRNA myocardin (sense, ACACCGAUUCAGCUACCUAdTdT, and antisense, dTdTUGUGGCUAAGUCGAUGGAU) and siRNA-STAT3 (sense, AGUCAGGUUGCUGGUCAAAdTdT, and antisense, dTdTUCAGUCCAACGACCAGUUU) were purchased from RIBOBIO. HA-VSMCs were transfected using Lipofectamine® 2000 reagent (Life Technologies, Inc.) according to the manufacturer's instructions. Transfected cells were preincubated for 48 h before further use. The efficiency of the siRNA-mediated myocardin and STAT3 knockdown was verified by Western blot.
Reverse Transcription-PCR (RT-PCR) and Quantitative Real Time RT-PCR
RT-PCR and quantitative RT-PCR analyses were carried out as described previously (31). Briefly, total cellular RNA was extracted from cultured cells with TRIzol reagent (Invitrogen), and 2 μg of total RNA was reverse-transcribed using Moloney murine leukemia virus reverse transcriptase (Promega) according to the manufacturer's instructions. The thermal cycle profile was as follows: denaturation for 30 s at 95 °C, annealing for 45 s at 52–58 °C depending on the primers used, and extension for 45 s at 72 °C. Each PCR was carried out for 28–32 cycles. PCR products were visualized on 2% agarose gels stained with ethidium bromide under UV trans-illumination. Quantitative RT-PCR was performed in Applied Biosystems StepOneTM real time PCR system. Fast SYBR® Green master mix was obtained from Applied Biosystems. Data were presented as the relative level normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
The PCR primer sequences are as follows: GAPDH, F-5′ ATTCAACGGCACAGTCAAGG 3′, and R-5′GCAGAAGGGGCGGAGATGA 3′; cyclin D1, F-5′ GCGAGGAACAGAAGTGC 3′, and R-5′ GAGTTGTCGGTGTAGATGC 3′; osteopontin (OPN), F-5′ CTGCCAGCAACCGAAGTTT 3′, and R-5′ ACCATTCAACTCCTCGCTTTC 3′; PCNA, F-5′ ATGAAATGAAGTTGATGGAT 3′, and R-5′ TTGAAGAGAGTGGAGTGGCT 3′; ACTA2, F-5′ TTGAGAAGAGTTACGAGTTG 3′, and R-5′ AGGACATTGTTAGCATAGAG 3′; SM22α, F-5′ ACCCACCCTCCATGGTCTTC 3′, and R-5′ CTTATGCTCCTGCGCTTTCT 3′; SMMHC, F-5′ AAGAAACACAGACCAGGCGT 3′, and R-5′ GGGTGAGTATCCAGCGGAAA 3′; VEGF, F-5′ GAGGGCAGAATCATCACGAA 3′, and R-5′ GGCTCCAGGGCATTAGACA 3′; myocardin, F-5′ GAACCAAATGAACAGATGGT 3′, and R-5′ GGTGTGACAGGAAAAGTGAC 3′; and STAT3, F-5′ GTCAGATGCCAAATGC 3′, and R-5′ ACCAGGTCCCAAGAGT 3′.
Western Blotting
Western blotting was performed as described previously (31). The total proteins of treated and mock-treated cells were immunoblotted and hybridized with specific antibodies overnight at 4 °C and then incubated with IR dye-conjugated secondary antibodies for 30 min at room temperature. The membranes were visualized by OdysseyTM infrared imaging system (Gene Co.). β-Actin expression was used as an internal control to show equal loading of the protein samples. The following antibodies were used: rabbit STAT3 (Abcam); rabbit p-STAT3(705) (Abcam); rabbit ACTA2 (Abcam); rabbit SM22α (Abcam); rabbit SMMHC (Abcam); rabbit cyclin D1 (Abcam); mouse PCNA (Abcam); mouse myocardin (Sigma); goat OPN (Santa Cruz Biotechnology); mouse ERK1/2 (Santa Cruz Biotechnology); mouse p-ERK1/2 (Santa Cruz Biotechnology); mouse SRF (Santa Cruz Biotechnology); and mouse β-actin (Santa Cruz Biotechnology). The following secondary antibodies were used: DyeTM-800-conjugated anti-rabbit antibodies; IR DyeTM-680-conjugated anti-mouse antibodies; and IR DyeTM-680-conjugated anti-goat antibodies (Li-COR).
Immunofluoresence Staining
Immunocytochemistry assays were performed as described previously (31). The cells after treatment were fixed in 4% paraformaldehyde for 15 min, blocked with normal goat serum for 20 min at room temperature, and then incubated with specific antibodies in a humid chamber overnight. The following antibodies used: rabbit ACTA2 (Abcam): rabbit SM22α (Abcam); rabbit FLAG (Proteintech); and mouse Myc (Proteintech). After incubating and washing with PBS twice, the cells were incubated with appropriate secondary antibodies (fluorescein isothiocyanate (FITC) goat anti-rabbit, FITC goat anti-mouse, or TRITC goat anti-rabbit) for 30 min at 37 °C followed by washing with PBS. The samples were then visualized using laser scanning confocal microscope (Olympus).
Luciferase Reporter Assays
Luciferase assays were performed as described previously (32). 24 hours after transfection, luciferase activity was measured with SynergyTM 4 (Biotek). Transfection efficiencies were normalized to the total protein concentrations in each luciferase assay. Data were collected based on three or more independent experiments, presented as the mean ± S.D. of triplicates from representative repeats.
ChIP Assay
ChIP analysis was performed using a commercially available kit, enzymatic ChIP (magnetic beads) (Cell Signaling Technology). Proteins bound to DNA were cross-linked using formaldehyde at a final concentration of 1% for 20 min at room temperature. Protein-DNA complexes were immunoprecipitated using primary antibodies for Myc-myocardin (Proteintech). Myocardin and ACTA-2 or myocardin and SM22α promoter complexes were measured by PCR. The primers used for the amplification of the human ACTA2 promoter region between −3237 and −3122 bp were as follows: ACTA2-GArG, F-5′ ATGGAGCATCCGACAAT 3′, and R-5′ TATCACAAGGTGGCAAT 3′; human SM22a promoter region between −132 and +13 bp were as follows: SM22α-GArG, F-5′ TGCTCCAACTTGGTGTCTTTC 3′, and R-5′ TGCCGCCGTGGTGATGT 3′. The samples were electrophoresed using a 2% agarose gel and visualized by ethidium bromide staining.
Co-immunoprecipitation
For the co-immunoprecipitation assay, Myc-myocardin/FLAG-STAT3 or Myc-myocardin/FLAG-STAT3/SRF (Addgene) were co-transfected into HEK293 cells with pcDNA3.1 vehicle. The co-immunoprecipitation assay was performed as described previously (33). Briefly, cells were lysed in lysis buffer (50 mm Tris-HCl, pH 7.8, 137 mm NaCl, 1 mm EDTA, 0.1% Triton X-100) with protease inhibitor (Roche Applied Science). The supernatants were collected after centrifugation and precleared with protein G-agarose for 1 h at 4 °C and then incubated with antibodies as indicated overnight at 4 °C. Normal IgG was used as a negative control. Immunoreactivity and band density were visualized by the Odyssey system (LI-COR Biosciences, Lincoln, NE), according to the manufacturer's instructions.
Statistical Analysis
Data were expressed as mean ± S.E. accompanied by the number of independent experiments performed and analyzed by t test. Differences at p < 0.05 were considered statistically significant.
Results
Activated STAT3 Correlates with VSMC Synthetic Phenotype
Previous studies have shown that VEGF is a key regulator of physiological and pathological angiogenesis, which inhibits VSMC apoptosis and promotes VSMC proliferation by suppressing the expression of VSMC marker genes (34, 35). Our data revealed that although the STAT3 mRNA level in HA-VSMCs was not significantly changed after incubating with 50 ng/ml VEGF for different time periods (12, 24, and 48 h), the phosphorylated (p)-STAT3 protein expression was significantly elevated to 3–6-fold compared with control levels (Fig. 1, A and C). The activated STAT3 correlates with the down-regulation of VSMC-specific genes (including myocardin, SMA, SMMHC, and SM22α) in either the mRNA or protein levels. It also enhance the expression of pro-proliferation genes, such as cyclin D1, OPN, and PCNA, in their mRNA or protein levels (Fig. 1, A–D). These observations are consistent with the effects of VEGF on the VSMC phenotypic switch and proliferation (34), suggesting that activated STAT3 signaling pathway is positively correlated with the synthetic VSMC phenotype.
FIGURE 1.

STAT3 is activated during VSMC phenotypic switch induced by VEGF. T/G HA-VSMCs after serum starvation (DMEM + 0.5% FBS) for 48 h were stimulated with 50 ng/ml VEGF for the indicated periods (12, 24, and 48 h). A and B, expression of STAT3, myocardin, markers of VSMC proliferation, and contractile phenotypes were analyzed by quantitative RT-PCR. GAPDH served as an internal control. n = 3. Data are expressed as mean ± S.D. (#, p > 0.05; *, p < 0.05; **, p < 0.01.. C and D, representative Western blot.
Activated STAT3 Promotes SMC Proliferation
It has been shown that activated STAT3 can enhance cell proliferation in other systems (36). We then tested whether inhibiting STAT3 is also able to impair VSMC proliferation. After inhibiting STAT3 with AG490 in HA-VSMCs cells, cell proliferation was evidently reduced compared with controls as shown in Fig. 2A. However, activating STAT3 with LIF enhanced cell proliferation compared with controls (Fig. 2A). Consistent with impaired HA-VSMC proliferation, the mRNA levels of the proliferative markers cyclin D1, OPN, and PCNA were all decreased in HA-VSMCs after treatment with AG490 and increased after treatment with LIF and VEGF (Fig. 2B). Similarly, the protein level of cyclin D1, OPN, and PCNA was down-regulated when treated with AG490 and up-regulated when treated with LIF and VEGF (Fig. 2C). Collectively, these data indicate that the expression of STAT3 can control the proliferation of HA-VSMCs.
FIGURE 2.

STAT3 regulated VSMC proliferation. T/G HA-VSMCs were treated with 50 ng/ml VEGF for 48 h or 2 ng/ml LIF for 30 min or 50 μmol/ml AG490 for 24 h. A, cell proliferation was measured by counting cell numbers as described under “Experimental Procedures.” n = 4. Data are expressed as mean ± S.D. (#, p > 0.05; *, p < 0.05; **, p < 0.01.) B, expression of VSMC proliferation markers was analyzed by quantitative RT-PCR. GAPDH served as internal control. n = 3. Data are expressed as mean ± S.D. (#, p > 0.05; *, p < 0.05; **, p < 0.01.) C, representative Western blot analysis. β-Actin served as internal control.
STAT3 Suppresses the Expression of VSMC-specific Contractile Protein Genes
To investigate the effect of STAT3 on VSMC contractile phenotype-specific gene expression, HA-VSMCs were incubated in growth medium with or without AG490 and LIF. SMC gene expressions were measured by quantitative RT-PCR or Western blot (Fig. 3, A and C). After treatment with AG490 in HA-VSMCs, STAT3-mediated inhibition of the ACTA2 and SM22α promoters was attenuated based on luciferase assay (Fig. 3G). These results indicate that both VSMC-specific genes associated with a contractile phenotype (ACTA2 and SM22α) were significantly up-regulated after inhibiting STAT3 (Fig. 3, A and C). To further confirm the results, myocardin and myocardin/STAT3 were overexpressed in HA-VSMCs cells for 24 h, and then ACTA2 and SM22α were examined by quantitative PCR, Western blotting, and immunofluorescence staining (Fig. 3, B, D and E). Our data showed that myocardin remarkably increased the mRNA and protein level of VSMC contractile phenotype-specific genes (Fig. 3, B and D), whereas overexpression of STAT3 abrogated the effects of myocardin on both its up-regulation (Fig. 3, B and D) and activation of the downstream genes ACTA2 and SM22α (Fig. 3H). The above data were further confirmed by the siRNA approach, and the protein levels of VSMC-specific contractile genes (SM22α and ACTA2) were markedly decreased in HA-VSMCs when transfected with si-myocardin. Overexpression of STAT3 further decreased the expression of the VSMC-specific contractile genes in myocardin-knockdown HA-VSMCs (Fig. 3I). Similarly, the protein levels of the pro-proliferation genes (cyclin D1, OPN, and PCNA) in HA-VSMCs were decreased after transfection with si-STAT3 and further decreased with overexpression of myocardin (Fig. 3J). Taken together, these data demonstrated that activated STAT3 is a repressor of the CArG-dependent VSMC contractile phenotype-specific genes by antagonizing myocardin-mediated activation.
FIGURE 3.

STAT3 regulated contractile phenotype-specific gene expression in VSMCs. A, T/G HA-VSMCs were treated with 50 ng/ml VEGF for 48 h or 2 ng/ml LIF for 30 min or 50 μmol/ml AG490 for 24 h, respectively. The expression of VSMC contractile phenotype genes was analyzed by quantitative RT-PCR. GAPDH served as internal control. n = 3. Data are expressed as mean ± S.D. (*, p < 0.05; **, p < 0.01.) B, T/G HA-VSMCs were co-transfected with myocardin and/or STAT3 expression plasmids for 24 h and then using quantitative RT-PCR to detect gene expression as indicated. C, T/G HA-VSMCs were treated the same as described in A. The expression of VSMC contractile phenotype genes was analyzed by Western blotting. β-Actin served as internal control. D, T/G HA-VSMCs were co-transfected with myocardin and/or STAT3 expression plasmids and then using Western blotting to detect gene expression as indicated. E and F, representative image showing ACTA2 or SM22α expression in T/G HA-VSMCs cells co-transfected with myocardin and/or STAT3 expression plasmids for 24 h. The left panels (green) show anti-ACTA2 or SM22α antibody reactivity to demonstrate gross morphology. The middle panels (blue) show the DAPI staining for nuclei. The right panels show double immunostaining for ACTA2 or SM22α and nuclei. Scale, 50 μm. G, luciferase reporter for the ACTA2 and SM22α promoter was transfected into T/G HA-VSMCs for 24 h. Cells were treated the same as described in A. The luciferase activity was measured as described under “Experimental Procedures.” The basal activity of DMSO on ACTA2 and SM22α promoter activity was normalized to 1. Data are expressed as mean ± S.D. (*, p < 0.05.) n = 3. H, luciferase reporter for the ACTA2 and SM22α promoter was co-transfected with myocardin and/or STAT3 expression plasmids in T/G HA-VSMCs, and the luciferase activity was measured 48 h after transfection as described under “Experimental Procedures.” The basal activity of pcDNA3.1 empty vector on ACTA2 and SM22α promoter activity was normalized to 1. Data are expressed as mean ± S.D. (**, p < 0.01.) n = 3. I, T/G HA-VSMCs were transfected with si-STAT3 for 48 h or overexpression of myocardin in STAT3 knockdown HA-VSMCs for 24 h, respectively. The expression of the pro-proliferation genes was analyzed by Western blotting. β-Actin served as internal control. J, T/G HA-VSMCs were transfected with si-myocardin 48 h or overexpression of STAT3 in myocardin knockdown HA-VSMCs for 24 h, respectively. The expression of the VSMC contractile phenotype genes was analyzed by Western blotting. β-Actin served as internal control.
STAT3 Modulates SMC Phenotype through Interaction with Myocardin-SRF Complex
To address the underlying mechanism on how STAT3 interacts with myocardin to modulate SMC phenotype, a series of co-immunoprecipitation assays were performed. Our results showed that STAT3 interacted specifically with myocardin as evidenced in Fig. 4A. Immunoprecipitation of myocardin with anti-Myc pulls down STAT3 as determined by Western blot with anti-FLAG in HA-VSMCs transiently transfected with plasmids expressing Myc-myocardin and FLAG-STAT3. In addition, overexpression of STAT3 attenuated the interaction between myocardin and SRF in a dose-dependent manner (Fig. 4B) as evidenced by the reduced SRF in the complexes that were pulled down with anti-Myc. These results demonstrated a direct interaction between myocardin and STAT3. We not only determined the interaction between myocardin and STAT3 by the co-immunoprecipitation assays, but immunostaining was also performed and demonstrated that STAT3 was co-localized with myocardin in the nucleus of HA-VSMCs (Fig. 4C). These results strongly suggest that myocardin and STAT3 are physically interacted in vivo and form a complex in HA-VSMCs.
FIGURE 4.

Interaction between STAT3 and myocardin in vivo. A, HEK293 cells were transfected with pcDNA3.1-myocardin-Myc and/or pcDNA3.1-STAT3-FLAG expression plasmids. The specificity of binding between STAT3 and myocardin was confirmed by precipitation with Myc antibody followed by immunoblotting (IB) with anti-FLAG antibody. IP, immunoprecipitation. B, HEK293 cells were transfected with pcDNA-myocardin-Myc and SRF. pcDNA3.1-STAT3-FLAG expression plasmid was introduced with increased dosage. Total cell lysates were precipitated with Myc antibody followed by immunoblotting with anti-SRF antibodies, FLAG, and Myc as indicated. IB, immunoblot. C, after co-transfection with Myc-myocardin and FLAG-STAT3 to T/G HA-VSMCs for 24 h, immunostaining was used to identify the interaction of myocardin and STAT3. The 1st panel (green) shows STAT3; the 2nd panel (red) shows myocardin, and the last panel shows double immunostaining for myocardin and STAT3. Nuclei were stained with DAPI (blue). Scale, 50 μm.
To address the functional relevance of these interactions, ChIP assays were performed in HA-VSMCs transfected with myocardin, myocardin/STAT3, or vector. Cross-linked chromatin was immunoprecipitated with specific anti-myocardin antibody or mock-treated control (no antibody). The precipitated chromatin DNA was then purified and amplified by PCR with specific primers targeting CArG boxes in ACTA2 and SM22α promoters. As shown in Fig. 5, A and B, compared with the zero PCR signal in IgG control and the mock-treated controls (No Ab), there were significant PCR signals from the myocardin precipitation, suggesting that myocardin binds to the CArG boxes of the ACTA2 and SM22α promoter. More importantly, the ChIP assays showed that overexpression of STAT3 in HA-VSMCs attenuated the myocardin-driven activation of the ACTA2 promoter and SM22α promoter (Fig. 5, A and B). Taken together, these data indicate that STAT3 interacts with myocardin to mediate the down-regulation of contractile phenotype-specific genes in VSMC and promotes proliferation.
FIGURE 5.

STAT3 attenuates the SRF-dependent transcriptional activation of CArG box containing contractile VSMC-specific genes. A and B, T/G HA-VSMCs were transiently transfected with myocardin, myocardin/STAT3, or a control vector (pcDNA3.1) for 24 h, and ChIP assays were performed by PCR with primers targeting ACTA2 and SM22α as described under “Experimental Procedures.” Sheared DNA-protein complexes were immunoprecipitated by using an anti-Myc-myocardin antibody followed by PCR to detect the endogenous CArG regions. The amount of DNA in each sample (2% input) is shown in the 2nd lane. Immunoprecipitations without primary antibody (No Ab) serve as a mock-treated control and IgG as a negative control.
STAT3 and Myocardin Can Regulate the Expression of VEGF
It is a well known fact that VEGF is a crucial regulatory molecule during angiogenesis (37, 38), which regulates the key steps of the angiogenic process, particularly cell proliferation and migration (39, 40). Previous studies demonstrated that VEGF-mediated STAT3 activation inhibits retinal vascularization by down-regulating local erythropoietin expression (24). STAT3 is activated upon VEGF stimulation of endothelial cells (EC) in vitro and in vivo by a VEGFR2 and Src-dependent mechanism, and this activation mediates BCL2 induction (29).
Recent studies reported that VEGF is directly regulated by constitutive STAT3 activity, which is associated with meningioma differentiation (41). STAT3 has an important role during the occurrence and development of human meningioma by regulating VEGF expression (41). In addition, STAT3 has been implicated as being a central regulator of tumor progression through its transcriptional up-regulation of VEGF, MCL-1, and survivin, among others (25). TNF-activated STAT3 was involved in regulating MSC production of VEGF, and ablation of the STAT3 gene partly blocked TNF-stimulated release of VEGF (42). Our results also show that VEGF production in HA-VSMCs is mediated by STAT3 (Fig. 6E), and overexpression of STAT3 can promote the transcriptional activation and expression of VEGF (Fig. 6, C and D). So VEGF-STAT3-VEGF forms a positive feedback regulation to regulate smooth muscle phenotype conversion.
FIGURE 6.

STAT3 and myocardin regulated the expression and activity of VEGF. A, luciferase reporter for the ACTA2 and SM22α promoter and myocardin was transfected into T/G HA-VSMCs for 24 h. Cells were treated with 50 ng/ml VEGF 48 h, and the luciferase activity was measured as described under “Experimental Procedures.” The basal activity of DMSO on ACTA2 and SM22α promoter activity was normalized to 1. Data are expressed as mean ± S.D. (*, p < 0.05.) n = 3. B, T/G HA-VSMCs were treated with different concentrations (25, 50, and 100 ng/ml) of VEGF for 48 h and then used Western blotting to detect ERK1/2 and the phosphorylation of ERK1/2. C, T/G HA-VSMCs were transfected with different concentrations of myocardin (100, 200, 300, 400, and 500 ng) and STAT3 (100, 200, 300, 400, and 500 ng) for 24 h. The expression of VEGF was analyzed by quantitative RT-PCR. GAPDH served as internal control. n = 3. Data are expressed as mean ± S.D. (*, p < 0.05.) D, luciferase reporter for the VEGF promoter and myocardin or STAT3 was transfected into T/G HA-VSMCs for 24 h, and the luciferase activity was measured as described under “Experimental Procedures.” The basal activity of DMSO on VEGF promoter activity was normalized to 1. Data are expressed as mean ± S.D. (*, p < 0.05; **, p < 0.01.) n = 3. E, T/G HA-VSMCs were transfected with different concentrations of myocardin (100, 200, 300, 400, and 500 ng) and STAT3 (100, 200, 300, 400, and 500 ng) for 24 h. The concentration of VEGF was measured using human VEGF Quantikine ELISA k it (R&D Systems) according to the manufacturer's instructions. (#, p > 0.05; *, p < 0.05.)
In this study, we found that VEGF can weaken myocardin-driven activation of the ACTA2 promoter and SM22α promoter in HA-VSMCs (Fig. 6A). VEGF can also increase phospho-ERK and stimulate the ERK1/2 signaling pathway (43, 44). A recent study reported that a single treatment of VEGF activated ERK1/2 and AKT signaling pathways in the adult rat hippocampus and in cultured hippocampal neuronal progenitor cells (45). Consistent with these observations, our results showed that VEGF could phosphorylate ERK1/2 to activate it in HA-VSMCs (Fig. 6B). It has been shown that phosphorylation of myocardin by ERK1/2 reduced its induction of the transcription of smooth muscle genes (19). Therefore, the negative regulation of myocardin by VEGF appears to be mediated by ERK1/2. Interestingly, results in this study also showed that myocardin promoted the transcription activation and expression of VEGF in a dose-dependent manner in HA-VSMCs (Fig. 6, C–E). Thus, VEGF-ERK1/2-myocardin-VEGF forms a negative feedback loop regulating smooth muscle phenotype conversion.
In summary, the above studies support a functional positive role of the JAK-STAT3 pathway in coordinating a VSMC proliferative phenotype switch upon VEGF activation, which is through direct interaction with myocardin. Most importantly, this study reveals feedback loops between VEGF and myocardin or STAT3 (Fig. 7).
FIGURE 7.
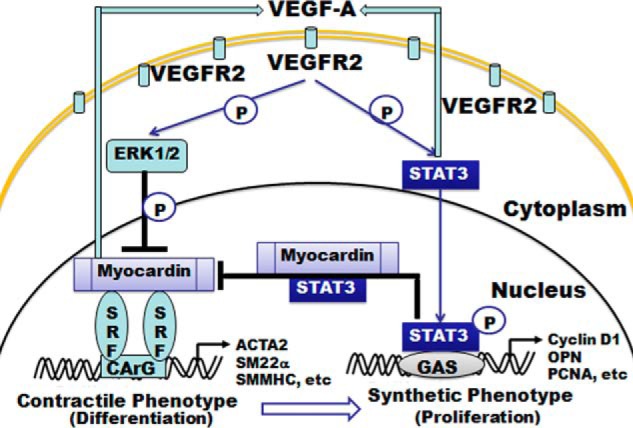
Proposed model on how STAT3 and myocardin regulate VSMC phenotypic switch stimulated by VEGF. VEGF activates STAT3 via phosphorylation. The activated STAT3 then suppresses myocardin activity, which results in reduced expression of contractile genes through an as yet undefined mechanism and increased proliferation markers in the VSMCs. Both myocardin and STAT3 can enhance the expression of VEGF, thus creating a feedback loop to regulate VSMC phenotypic switch.
Discussion
Previous studies demonstrated that the Janus kinase (JAK)/STAT signaling pathway plays a significant role in various physiological processes, including immune function, cell growth, differentiation, and hematopoiesis (46). Besides being activated by a cytokine or interferon, JAK/STAT signaling can be triggered by hypoxia and reactive oxygen species. Downstream effects include regulation of angiogenic genes such as VEGF and hypoxia inducible factor-1 (47). Recent studies reported that the downstream targets of STAT3 are important in preventing apoptosis and enhancing invasion (BCLxl and cyclin D1) as well as promoting metastasis and angiogenesis (VEGF) (48). Previous studies showed that VEGF could activate VEGFR2 in cultured EC, which rapidly induce the associated molecules SRC and STAT3. STAT3 is phosphorylated by a VEGFR2 and SRC-dependent mechanism. Therefore, VEGF is a primary activator of endothelial STAT3 (29). It was also reported that VEGF induced STAT3 phosphorylation and nuclear localization in MS1, human umbilical vein endothelial cells, and rat Müller cells (24, 29). In contrast, VEGF could also be regulated by constitutive STAT3 activity, which was observed during the development and differentiation of human meningioma (41). VEGF released in MSCs under normoxia was associated with constitutive STAT3 activity as well, where STAT3 deficiency led to the decreased production of VEGF in MSCs (42). Recently, it was reported that activated STAT3 mediates VEGF's induction of EC BCL2 and contributes to the protection of EC from apoptosis (29). Phospho-STAT3 induces overexpression of VEGF, which is decreased during angiogenesis by inhibition with antagonists of VEGF-VEGFR signaling (29). Our results also show that VEGF production in HA-VSMCs is mediated by STAT3 (Fig. 6E), and overexpression of STAT3 can promote the transcriptional activation and expression of VEGF (Fig. 6, C and D). So VEGF-STAT3-VEGF forms a positive feedback loop to regulate smooth muscle phenotype conversion.
VEGF is the key regulator of physiological and pathological angiogenesis (49). VEGF activates signaling in ECs after binding cognate receptors on the cell surface. Its two best characterized receptors are the tyrosine kinases, VEGF receptor 1 (VEGFR1) and VEGF receptor 2 (VEGFR2) (29). VEGFR2 signaling activates a variety of downstream mediators in ECs, including SRC, RAS, and members of the PI3K-AKT and RAF MEK-ERK pathways (46) and is responsible for many of the characteristic effects of VEGF on EC, including cell proliferation, survival, chemotaxis, and increased vascular permeability (50–53). In vitro cultured ECs revealed a role for SRF in VEGF-mediated angiogenesis, actin polymerization, and EC migration (54). The effects of VEGF on SRF activity have been suggested to require both MAPK/ERK and RhoA signaling. In vivo EC SRF loss-of-function studies have only been performed during embryogenesis so far (55). The genes encoding VEGF-R2 (Kdr; also known as Vegfr2 or Flk1) and VE-cadherin (Cdh5), among others, were identified as SRF target genes that could be stimulated upon VEGF signaling (55). VEGF-induced myocardin-related transcription factor translocation from the cytoplasm to the nucleus activates the expression of SRF target genes with cytoskeletal functions in ECs, which is essential for motile activities of tip cells to ensure appropriate vascularization of the postnatal retina (56).
The cellular mechanisms that drive SMC plasticity (e.g. the switch between contractile and synthetic phenotypes) are partially dependent on myocardin interaction with SRF (7). Myocardin binds CArG boxes to regulate growth-responsive genes such as c-fos and multiple SMC marker genes (57). Differentiated SMCs express a number of proteins that are part of the cytoskeleton and/or are believed to be involved in the regulation of contraction such as CNN1, SM22/TAGLN, SMA/ACTA2, and MYH11 (8–13). It is known that the phenotypic switch is caused by coordinated repression/activation of SMC-specific genes, such as SMA, SM22α, and SM-MHC. However, the mechanisms that regulate this process are not well understood. We speculate that myocardin-mediated induction of SMC genes plays an important role in switching vascular SM precursor cells from a “synthetic” to a “contractile” state during vascular injury and repair.
Myocardin activity is also regulated by post-translational modifications, such as phosphorylating myocardin at four sites (Ser-812, Ser-859, Ser-866, and Thr-893) in its transcriptional activation domain. Phosphorylation of myocardin by ERK1/2 reduces its induction of SM gene transcription (19). Myocardin, when phosphorylated by GSK-3β, can be ubiquitinated by the C terminus of ChIP, a cytosolic E3 ligase, resulting in proteasomal degradation and reduction of myocardin transcriptional activity (20). In addition, ERK1/2 signaling is activated via VEGF and plays a role in cell death by oxygen glucose deprivation (44). A single treatment of VEGF activated ERK1/2 and AKT signaling pathways in the adult rat hippocampus and in cultured hippocampal neuronal progenitor cells (45). VEGF increases phospho-ERK and phospho-AKT in phrenic motor neurons. VEGFA-165 increased phosphorylation of signaling molecules downstream from VEGFR-2 within the phrenic motor nucleus, including ERK (43). ERK can phosphorylate and activate HIF-1α thereby increasing VEGF expression (58). Consistent with the above findings, our results showed that VEGF could phosphorylate ERK1/2 to activate ERK1/2 in HA-VSMCs (Fig. 6B). By ERK1/2 phosphorylation that attenuates myocardin transcriptional activity, VEGF weakens the myocardin-driven activation of the ACTA2 promoter and SM22α promoter in HA-VSMCs. Interestingly, in this study we revealed that myocardin also promotes the transcriptional activation, and expression of VEGF and VEGF-ERK1/2-myocardin-VEGF forms a negative feedback loop regulating smooth muscle phenotype conversion.
In summary, VSMC phenotypic switch from contractile to synthetic upon VEGF stimuli is related to STAT3's controlling of myocardin activity, which regulates VSMC contractile gene expression and, through an as yet undefined mechanism, increased proliferation markers. The interaction between myocardin and STAT3 is likely to be a primary and essential component to regulate VSMC proliferation by facilitating the phenotypic switch from contractile to synthetic phenotypes. Collectively, the present findings provide new insight into the molecular mechanism of smooth muscle phenotype conversion. These data suggest that STAT3 and myocardin might be novel targets for both the diagnosis and treatment of human vascular diseases.
Author Contributions
X.-H. L. and N. W. conceived and coordinated the study and wrote the manuscript. D.-W. Z. and D.-L. Z. performed and analyzed the experiments shown in Figs. 1 and 2. L. Z. and W.J. X. designed, performed, and analyzed the experiments shown in Fig. 6. L.-Y. B. provided technical assistance and contributed to the preparation of the figures. W.-J. M. edited and reviewed the manuscript. T.-C. Z. and J. D. designed the manuscript. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by National Natural Science Foundation of China Grants 30970615, 31071126, 31000343, 31171303, 31171297, and 31200955, Program for Changjiang Scholars and Innovative Research Team in University of Ministry of Education of China Grant IRT1166, Research Fund for the Doctoral Program of Higher Education of China Grant 20111208110001, and Key Project of Chinese Ministry of Education Grant 212010. The authors declare that they have no conflicts of interest with the contents of this article.
- VSMC
- vascular smooth muscle cell
- SMC
- smooth muscle cell
- EC
- endothelial cell
- SRF
- serum-response factor
- F
- forward
- R
- reverse
- OPN
- osteopontin
- PCNA
- proliferating cell nuclear antigen
- TRITC
- tetramethylrhodamine isothiocyanate
- VEGFR
- VEGF receptor
- MSC
- mesenchymal stem cell.
REFERENCES
- 1. Owens G. K., Kumar M. S., Wamhoff B. R. (2004) Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 84, 767–801 [DOI] [PubMed] [Google Scholar]
- 2. Majesky M. W. (2007) Developmental basis of vascular smooth muscle diversity. Arterioscler. Thromb. Vasc. Biol. 27, 1248–1258 [DOI] [PubMed] [Google Scholar]
- 3. Xie C., Ritchie R. P., Huang H., Zhang J., Chen Y. E. (2011) Smooth muscle cell differentiation in vitro: models and underlying molecular mechanisms. Arterioscler. Thromb. Vasc. Biol. 31, 1485–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang Z., Wang D. Z., Hockemeyer D., McAnally J., Nordheim A., Olson E. N. (2004) Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature 428, 185–189 [DOI] [PubMed] [Google Scholar]
- 5. Miano J. M. (2003) Serum response factor: toggling between disparate programs of gene expression. J. Mol. Cell. Cardiol. 35, 577–593 [DOI] [PubMed] [Google Scholar]
- 6. Sun Q., Chen G., Streb J. W., Long X., Yang Y., Stoeckert C. J., Jr., Miano J. M. (2006) Defining the mammalian CArGome. Genome Res. 16, 197–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang D., Chang P. S., Wang Z., Sutherland L., Richardson J. A., Small E., Krieg P. A., Olson E. N. (2001) Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 105, 851–862 [DOI] [PubMed] [Google Scholar]
- 8. Chen J., Kitchen C. M., Streb J. W., Miano J. M. (2002) Myocardin: a component of a molecular switch for smooth muscle differentiation. J. Mol. Cell. Cardiol. 34, 1345–1356 [DOI] [PubMed] [Google Scholar]
- 9. Du K. L., Ip H. S., Li J., Chen M., Dandre F., Yu W., Lu M. M., Owens G. K., Parmacek M. S. (2003) Myocardin is a critical serum response factor cofactor in the transcriptional program regulating smooth muscle cell differentiation. Mol. Cell. Biol. 23, 2425–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang Z., Wang D. Z., Pipes G. C., Olson E. N. (2003) Myocardin is a master regulator of smooth muscle gene expression. Proc. Natl. Acad. Sci. U.S.A. 100, 7129–7134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yoshida T., Sinha S., Dandré F., Wamhoff B. R., Hoofnagle M. H., Kremer B. E., Wang D. Z., Olson E. N., Owens G. K. (2003) Myocardin is a key regulator of CArG-dependent transcription of multiple smooth muscle marker genes. Circ. Res. 92, 856–864 [DOI] [PubMed] [Google Scholar]
- 12. Yoshida T., Kawai-Kowase K., Owens G. K. (2004) Forced expression of myocardin is not sufficient for induction of smooth muscle differentiation in multipotential embryonic cells. Arterioscler. Thromb. Vasc. Biol. 24, 1596–1601 [DOI] [PubMed] [Google Scholar]
- 13. Long X., Bell R. D., Gerthoffer W. T., Zlokovic B. V., Miano J. M. (2008) Myocardin is sufficient for a smooth muscle-like contractile phenotype. Arterioscler. Thromb. Vasc. Biol. 28, 1505–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kimura Y., Morita T., Hayashi K., Miki T., Sobue K. (2010) Myocardin functions as an effective inducer of growth arrest and differentiation in human uterine leiomyosarcoma cells. Cancer Res. 70, 501–511 [DOI] [PubMed] [Google Scholar]
- 15. Tang R. H., Zheng X. L., Callis T. E., Stansfield W. E., He J., Baldwin A. S., Wang D. Z., Selzman C. H. (2008) Myocardin inhibits cellular proliferation by inhibiting NF-κB(p65)-dependent cell cycle progression. Proc. Natl. Acad. Sci. U.S.A. 105, 3362–3367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chiu C. Z., Wang B. W., Chung T. H., Shyu K. G. (2010) Angiotensin II and the ERK pathway mediate the induction of myocardin by hypoxia in cultured rat neonatal cardiomyocytes. Clin. Sci. 119, 273–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xie C., Guo Y., Zhu T., Zhang J., Ma P. X., Chen Y. E. (2012) Yap1 protein regulates vascular smooth muscle cell phenotypic switch by interaction with myocardin. J. Biol. Chem. 287, 14598–14605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Badorff C., Seeger F. H., Zeiher A. M., Dimmeler S. (2005) Glycogen synthase kinase 3β inhibits myocardin-dependent transcription and hypertrophy induction through site-specific phosphorylation. Circ. Res. 97, 645–654 [DOI] [PubMed] [Google Scholar]
- 19. Taurin S., Sandbo N., Yau D. M., Sethakorn N., Kach J., Dulin N. O. (2009) Phosphorylation of myocardin by extracellular signal-regulated kinase. J. Biol. Chem. 284, 33789–33794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xie P., Fan Y., Zhang H., Zhang Y., She M., Gu D., Patterson C., Li H. (2009) ChIP represses myocardin-induced smooth muscle cell differentiation via ubiquitin-mediated proteasomal degradation. Mol. Cell. Biol. 29, 2398–2408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grote K., Luchtefeld M., Schieffer B. (2005) JANUS under stress-role of JAK/STAT signaling pathway in vascular diseases. Vascul. Pharmacol. 43, 357–363 [DOI] [PubMed] [Google Scholar]
- 22. Neeli I., Liu Z., Dronadula N., Ma Z. A., Rao G. N. (2004) An essential role of the Jak-2/STAT-3/cytosolic phospholipase A(2) axis in platelet-derived growth factor BB-induced vascular smooth muscle cell motility. J. Biol. Chem. 279, 46122–46128 [DOI] [PubMed] [Google Scholar]
- 23. Wang D., Liu Z., Li Q., Karpurapu M., Kundumani-Sridharan V., Cao H., Dronadula N., Rizvi F., Bajpai A. K., Zhang C., Müller-Newen G., Harris K. W., Rao G. N. (2007) An essential role for gp130 in neointima formation following arterial injury. Circ. Res. 100, 807–816 [DOI] [PubMed] [Google Scholar]
- 24. Wang H., Byfield G., Jiang Y., Smith G. W., McCloskey M., Hartnett M. E. (2012) VEGF-mediated STAT3 activation inhibits retinal vascularization by down-regulating local erythropoietin expression. Am. J. Pathol. 180, 1243–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Devarajan E., Huang S. (2009) STAT3 as a central regulator of tumor metastases. Curr. Mol. Med. 9, 626–633 [DOI] [PubMed] [Google Scholar]
- 26. Daniel J. M., Dutzmann J., Bielenberg W., Widmer-Teske R., Gündüz D., Hamm C. W., Sedding D. G. (2012) Inhibition of STAT3 signaling prevents vascular smooth muscle cell proliferation and neointima formation. Basic Res. Cardiol. 107, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bartoli M., Platt D., Lemtalsi T., Gu X., Brooks S. E., Marrero M. B., Caldwell R. B. (2003) VEGF differentially activates STAT3 in microvascular endothelial cells. FASEB J. 17, 1562–1564 [DOI] [PubMed] [Google Scholar]
- 28. Yahata Y., Shirakata Y., Tokumaru S., Yamasaki K., Sayama K., Hanakawa Y., Detmar M., Hashimoto K. (2003) Nuclear translocation of phosphorylated STAT3 is essential for vascular endothelial growth factor-induced human dermal microvascular endothelial cell migration and tube formation. J. Biol. Chem. 278, 40026–40031 [DOI] [PubMed] [Google Scholar]
- 29. Chen S. H., Murphy D. A., Lassoued W., Thurston G., Feldman M. D., Lee W. M. (2008) Activated STAT3 is a mediator and biomarker of VEGF endothelial activation. Cancer Biol. Ther. 7, 1994–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen K. H., Guo X., Ma D., Guo Y., Li Q., Yang D., Li P., Qiu X., Wen S., Xiao R. P., Tang J. (2004) Dysregulation of HSG triggers vascular proliferative disorders. Nat. Cell Biol. 6, 872–883 [DOI] [PubMed] [Google Scholar]
- 31. Liao X. H., Wang N., Liu Q. X., Qin T., Cao B., Cao D. S., Zhang T. C. (2011) Myocardin-related transcription factor-A induces cardiomyocyte hypertrophy. IUBMB Life 63, 54–61 [DOI] [PubMed] [Google Scholar]
- 32. Liao X. H., Li Y. Q., Wang N., Zheng L., Xing W. J., Zhao D. W., Yan T. B., Wang Y., Liu L. Y., Sun X. G., Hu P., Zhou H., Zhang T. C. (2014) Re-expression and epigenetic modification of maspin induced apoptosis in MCF-7 cells mediated by myocardin. Cell. Signal. 26, 1335–1346 [DOI] [PubMed] [Google Scholar]
- 33. Liao X. H., Wang N., Liu L. Y., Zheng L., Xing W. J., Zhao D. W., Sun X. G., Hu P., Dong J., Zhang T. C. (2014) MRTF-A and STAT3 synergistically promote breast cancer cell migration. Cell. Signal. 26, 2370–2380 [DOI] [PubMed] [Google Scholar]
- 34. Shi X., Guo L. W., Seedial S. M., Si Y., Wang B., Takayama T., Suwanabol P. A., Ghosh S., DiRenzo D., Liu B., Kent K. C. (2014) TGF-β/Smad3 inhibit vascular smooth muscle cell apoptosis through an autocrine signaling mechanism involving VEGF-A. Cell Death Dis. 5, e1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Demyanets S., Kaun C., Rychli K., Pfaffenberger S., Kastl S. P., Hohensinner P. J., Rega G., Katsaros K. M., Afonyushkin T., Bochkov V. N., Paireder M., Huk I., Maurer G., Huber K., Wojta J. (2011) Oncostatin M-enhanced vascular endothelial growth factor expression in human vascular smooth muscle cells involvesPI3K-, p38 MAPK-, Erk1/2- and STAT1/STAT3-dependent pathways and is attenuated by interferon-γ. Basic Res. Cardiol. 106, 217–231 [DOI] [PubMed] [Google Scholar]
- 36. Fukada T., Ohtani T., Yoshida Y., Shirogane T., Nishida K., Nakajima K., Hibi M., Hirano T. (1998) STAT3 orchestrates contradictory signals in cytokine-induced G1 to S cell-cycle transition. EMBO J. 17, 6670–6677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Carmeliet P., Jain R. K. (2000) Angiogenesis in cancer and other diseases. Nature 407, 249–257 [DOI] [PubMed] [Google Scholar]
- 38. Ferrara N. (2009) Vascular endothelial growth factor. Arterioscler. Thromb. Vasc. Biol. 29, 789–791 [DOI] [PubMed] [Google Scholar]
- 39. Ferrara N. (2009) VEGF-A: a critical regulator of blood vessel growth. Eur. Cytokine Netw. 20, 158–163 [DOI] [PubMed] [Google Scholar]
- 40. Lamalice L., Houle F., Huot J. (2006) Phosphorylation of Tyr1214 within VEGFR-2 triggers the recruitment of Nck and activation of Fyn leading to SAPK2/p38 activation and endothelial cell migration in response to VEGF. J. Biol. Chem. 281, 34009–34020 [DOI] [PubMed] [Google Scholar]
- 41. Zhang M. X., Zhao X., Wang Z. G., Zhao W. M., Wang Y. S. (2010) Constitutive activation of signal transducer and activator of transcription 3 regulates expression of vascular endothelial growth factor in human meningioma differentiation. J. Cancer Res. Clin. Oncol. 136, 981–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang M., Zhang W., Crisostomo P., Markel T., Meldrum K. K., Fu X. Y., Meldrum D. R. (2007) STAT3 mediates bone marrow mesenchymal stem cell VEGF production. J. Mol. Cell. Cardiol. 42, 1009–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dale-Nagle E. A., Satriotomo I., Mitchell G. S. (2011) Spinal vascular endothelial growth factor induces phrenic motor facilitation via extracellular signal-regulated kinase and Akt signaling. J. Neurosci. 31, 7682–7690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Narasimhan P., Liu J., Song Y. S., Massengale J. L., Chan P. H. (2009) VEGF stimulates the ERK1/2 signaling pathway and apoptosis in cerebral endothelial cells after ischemic conditions. Stroke 40, 1467–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fournier N. M., Lee B., Banasr M., Elsayed M., Duman R. S. (2012) Vascular endothelial growth factor regulates adult hippocampal cell proliferation through MEK/ERK- and PI3K/Akt-dependent signaling. Neuropharmacology 63, 642–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Niwa Y., Kanda H., Shikauchi Y., Saiura A., Matsubara K., Kitagawa T., Yamamoto J., Kubo T., Yoshikawa H. (2005) Methylation silencing of SOCS-3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene 24, 6406–6417 [DOI] [PubMed] [Google Scholar]
- 47. Xu Q., Briggs J., Park S., Niu G., Kortylewski M., Zhang S., Gritsko T., Turkson J., Kay H., Semenza G. L., Cheng J. Q., Jove R., Yu H. (2005) Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 24, 5552–5560 [DOI] [PubMed] [Google Scholar]
- 48. Haridas V., Nishimura G., Xu Z. X., Connolly F., Hanausek M., Walaszek Z., Zoltaszek R., Gutterman J. U. (2009) Avicin D: a protein reactive plant isoprenoid dephosphorylates Stat 3 by regulating both kinase and phosphatase activities. PLoS One 4, e5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Belgore F., Blann A., Neil D., Ahmed A. S., Lip G. Y. (2004) Localisation of members of the vascular endothelial growth factor (VEGF) family and their receptors in human atherosclerotic arteries. J. Clin. Pathol. 57, 266–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jin J., Yuan F., Shen M. Q., Feng Y. F., He Q. L. (2013) Vascular endothelial growth factor regulates primate choroid-retinal endothelial cell proliferation and tube formation through PI3K/Akt and MEK/ERK dependent signaling. Mol. Cell. Biochem. 381, 267–272 [DOI] [PubMed] [Google Scholar]
- 51. Tallquist M. D., Soriano P., Klinghoffer R. A. (1999) Growth factor signaling pathways in vascular development. Oncogene 18, 7917–7932 [DOI] [PubMed] [Google Scholar]
- 52. Watanabe Y., Dvorak H. F. (1997) Vascular permeability factor/vascular endothelial growth factor inhibits anchorage-disruption-induced apoptosis in microvessel endothelial cells by inducing scaffold formation. Exp. Cell Res. 233, 340–349 [DOI] [PubMed] [Google Scholar]
- 53. Alon T., Hemo I., Itin A., Pe'er J., Stone J., Keshet E. (1995) Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat. Med. 1, 1024–1028 [DOI] [PubMed] [Google Scholar]
- 54. Chai J., Jones M. K., Tarnawski A. S. (2004) Serum response factor is a critical requirement for VEGF signaling in endothelial cells and VEGF-induced angiogenesis. FASEB J. 18, 1264–1266 [DOI] [PubMed] [Google Scholar]
- 55. Franco C. A., Mericskay M., Parlakian A., Gary-Bobo G., Gao-Li J., Paulin D., Gustafsson E., Li Z. (2008) Serum response factor is required for sprouting angiogenesis and vascular integrity. Dev. Cell 15, 448–461 [DOI] [PubMed] [Google Scholar]
- 56. Weinl C., Riehle H., Park D., Stritt C., Beck S., Huber G., Wolburg H., Olson E. N., Seeliger M. W., Adams R. H., Nordheim A. (2013) Endothelial SRF/MRTF ablation causes vascular disease phenotypes in murine retinae. J. Clin. Invest. 123, 2193–2206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mack C. P., Thompson M. M., Lawrenz-Smith S., Owens G. K. (2000) Smooth muscle α-actin CArG elements coordinate formation of a smooth muscle cell-selective, serum response factor-containing activation complex. Circ. Res. 86, 221–232 [DOI] [PubMed] [Google Scholar]
- 58. Richard D. E., Berra E., Gothié E., Roux D., Pouysségur J. (1999) p42/p44 mitogen activated protein kinases phosphorylate hypoxia-inducible factor 1α (HIF-1α) and enhance the transcriptional activity of HIF-1. J. Biol. Chem. 274, 32631–32637 [DOI] [PubMed] [Google Scholar]