

Background: UL97, the v-CDK encoded by HCMV, phosphorylates Rb.
Results: An LXCXE motif in UL97 helps activate E2F-dependent promoters independently of its stimulatory effect on Rb phosphorylation.
Conclusion: UL97 uses multiple ways to activate E2F-responsive transcription.
Significance: Identifying the presence of a novel way to activate E2F-mediated transcription increases our understanding of the cell cycle and oncogenesis.
Keywords: cancer; cyclin-dependent kinase (CDK); herpesvirus; oncogene; phosphorylation; retinoblastoma protein (pRb, RB); transcription; cytomegalovirus
Abstract
The retinoblastoma (Rb) tumor suppressor restricts cell cycle progression by repressing E2F-responsive transcription. Cellular cyclin-dependent kinase (CDK)-mediated Rb inactivation through phosphorylation disrupts Rb-E2F complexes, stimulating transcription. The human cytomegalovirus (HCMV) UL97 protein is a viral CDK (v-CDK) that phosphorylates Rb. Here we show that UL97 phosphorylates 11 of the 16 consensus CDK sites in Rb. A cleft within Rb accommodates peptides with the amino acid sequence LXCXE. UL97 contains three such motifs. We determined that the first LXCXE motif (L1) of UL97 and the Rb cleft enhance UL97-mediated Rb phosphorylation. A UL97 mutant with a non-functional L1 motif (UL97-L1m) displayed significantly reduced Rb phosphorylation at multiple sites. Curiously, however, it efficiently disrupted Rb-E2F complexes but failed to relieve Rb-mediated repression of E2F reporter constructs. The HCMV immediate early 1 protein cooperated with UL97-L1m to inactivate Rb in transfection assays, likely indicating that cells infected with a UL97-L1m mutant virus show no defects in growth or E2F-responsive gene expression because of redundant viral mechanisms to inactivate Rb. Our data suggest that UL97 possesses a mechanism to elicit E2F-dependent gene expression distinct from disruption of Rb-E2F complexes and dependent upon both the L1 motif of UL97 and the cleft region of Rb.
Introduction
The retinoblastoma (Rb)3 tumor suppressor protein is encoded by the RB1 gene, mutation of which was first associated with the development of retinoblastoma tumors in children (1). Rb is the master regulator of a pathway that controls cell cycle progression and that is aberrant in most human tumors (2). Additional pathways regulated by Rb and relevant to cancer include those controlling genome stability, senescence, apoptosis, differentiation, angiogenesis, and glutamine metabolism (3–7). Rb family members p107 and p130 serve similar functions and are regulated in an analogous manner. Many viruses modulate the Rb pathway, presumably to improve viral fitness (8–10). The mechanisms through which viruses inactivate the Rb pathway are of interest in terms of developing potential antiviral and anticancer therapeutics (11), designing oncolytic viruses as treatments for human cancers (12–14), and furthering our molecular understanding of cancer cell biology (15).
The 928-amino acid Rb protein (p105-Rb) is composed of an amino (N)-terminal domain (RbN) connected by an unstructured interdomain linker to the central pocket (divided into A and B domains) followed by an intrinsically disordered carboxyl (C)-terminal tail (RbC) (16–20). Sixteen putative cyclin-dependent kinase (CDK) phosphorylation sites within the Rb protein regulate its intramolecular structure and thus its intermolecular interactions with proteins that control cell cycle progression, most notably the members of the E2F transcription factor family.
E2F proteins (E2F1, -2, -3a, -3b, -4, -5, -6, -7, and -8) are defined by their conserved DNA binding domains (21, 22). By heterodimerizing with dimerization partner proteins, E2Fs associate with promoters containing E2F binding sites and modulate transcription of the downstream genes. Many of these genes encode proteins required for cell cycle progression and DNA replication. Hypophosphorylated Rb interacts with E2Fs and represses their ability to activate gene expression. Transcriptional repression is achieved by a masking of the transactivation domain of E2F through interaction with the A domain of the Rb pocket and the interface between the A and B pocket domains (18). In certain promoters, transcription is further suppressed by recruitment of histone deacetylases (HDACs) (23) through their LXCXE motifs that bind to the B pocket (19). Rb binding to the E2F1 protein in vivo and other E2F proteins in vitro also occurs through an interaction between the marked box domain of E2F and RbC (20).
Multiple CDKs phosphorylate Rb, resulting in disruption of Rb-E2F complexes, the activation of E2F-mediated gene expression, and concomitant cell cycle progression and DNA replication (4, 24). For many years, the accumulation of multiple phosphorylations, as opposed to phosphorylation at one or a few specific residues, was thought to be responsible for disrupting Rb-E2F complexes (24). However, recent evidence has pointed to specific phosphorylation events that disrupt individual contacts among HDACs, E2Fs, and Rb (17, 25). Phosphorylation at Thr821 and Thr826 stabilizes an intramolecular interaction between RbC and the pocket domain that blocks both the LXCXE-binding cleft as well as the region of RbC that contacts the marked box domain of E2F (20). Ser608 phosphorylation reorganizes a flexible loop within the Rb pocket domain, allowing it to interact intramolecularly with the A-B interface, thus directly blocking the site where the E2F transactivation domain binds (26). Furthermore, phosphorylation of Thr373 induces an intramolecular rearrangement allowing RbN to stably dock with the pocket domain, both blocking the LXCXE-binding cleft and altering the structure of the pocket to allosterically inhibit binding of the E2F transactivation domain (26).
Cyclin proteins bind to CDKs and promote their activation and substrate specificity (27, 28). All cyclins have a hydrophobic patch (HP) that interacts with RXL sequences such as those found in RbC (29). The D-type cyclins also contain an LXCXE motif. Binding through either one of these types of motifs facilitates CDK-mediated Rb phosphorylation (30, 31). However, it is presently unclear how or even whether LXCXE motifs or HPs control site selection for Rb phosphorylation.
The β-herpesvirus human cytomegalovirus (HCMV) is a significant pathogen that causes disease in neonates and adults with suppressed or impaired immune function (32). There is growing evidence implicating an association of the virus with human cancers, most notably glioblastoma multiforme brain tumors (33–36). HCMV inactivates Rb in multiple ways (8). The kinase encoded by the UL97 gene of HCMV phosphorylates Rb (37, 38). The pp71 tegument protein (39) is delivered to cells by infecting virions, migrates to the nucleus, and through its LXCXE-like motif binds to and induces the degradationof Rb (37, 40) in an uncommon proteasome-dependent, ubiquitin-independent manner (41). pp71 stimulates viral immediate early (IE) gene expression (42), and viral IE proteins, through transfection assays, have been implicated in the inactivation of Rb family members (8). Unlike pp71 and UL97, data linking the IE proteins to Rb inactivation during virus infection are absent.
UL97 and its homologs in the β- and γ-herpesviruses phosphorylate Rb and lamin A/C (thus disrupting the nuclear lamina) and mimic cellular CDK activity; thus they are considered viral CDKs (v-CDKs) (43). UL97 contains four putative Rb-binding motifs, three LXCXE-like motifs (37, 38), and an HP (8). Cells infected with a recombinant HCMV expressing a mutant UL97 (UL97-L1m) with the central cysteine of the first LXCXE motif (L1) changed to a glycine (C151G) showed lower steady state levels of total Rb protein and the Ser780 phosphorylated form (38) but displayed no growth defect (44).
Here we show that UL97-L1m is less capable of phosphorylating Rb than is wild-type UL97. Furthermore, we show that wild-type UL97 fails to fully phosphorylate an Rb cleft mutant that does not interact with the LXCXE-containing HDAC1 protein. Rb underphosphorylation in the absence of functional L1 or cleft sequences correlates with sustained Rb-mediated repression of E2F-responsive promoters. However, two independent lines of evidence demonstrate that underphosphorylation of Rb is not directly responsible for the inability of the UL97-L1m mutant to relieve Rb-mediated transcriptional repression. Our work reveals the molecular determinants of Rb and UL97 that facilitate phosphorylation but also implicates these same protein regions in an undefined mode for activation of E2F-responsive transcription.
Experimental Procedures
Cells
Saos-2 (containing truncated Rb) (45), U-2 OS, and primary human foreskin fibroblast (HFF) cells were grown and maintained at 37 °C in Dulbecco's modified Eagle's medium (DMEM; Invitrogen and Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich), 100 units/ml penicillin, 100 μg/ml streptomycin, and 0.292 mg/ml glutamine (Sigma-Aldrich). All infections were performed under serum-starved conditions. For serum starvation, HFF cells were incubated with DMEM with 0.1% FBS for 48 h. For serum stimulation, serum-starved HFF cells were incubated with 15% serum for 16 h. Saos-2 and U-2 OS cells were transfected using TransIT-2020 (Mirus).
Viruses
The wild-type HCMV strain was AD169. Viruses with UL97 deleted (Δ97), with kinase-inactive UL97 (in-frame deletion of codon 355, K355del; T3418), or with individual UL97 LXCXE motif disruptions (L1m, C151G; L2m, C428G; L3m, C693G) were described previously (38, 44, 46, 47). Viruses with all three UL97 LXCXE motifs disrupted (triple mutation (Tri); T3260), with the hydrophobic patch disrupted (W368A, HPm; T3727), or with all four motifs disrupted (quadruple mutation (Qd); T3719) were based on the bacterial artificial chromosome clone BA9 that expresses a secreted alkaline phosphatase reporter gene and in which UL97 is replaced by a bacterial galK gene and were generated as described previously (48). Growth curves and yield reduction assays for maribavir susceptibility were performed as described previously (49–51). For RNA analysis, maribavir (Acme Bioscience, A4028; 20 μm) was added at 2 h postinfection. Lentivirus infection was performed as described previously (52).
Plasmids
The following expression plasmids have been described: pCGN-HA UL97WT and pCGN-HA UL97KD (43), pSG5-HA UL97WT and pSG5-HA UL97KD (53), V5-tagged UL97 and kinase-dead derivative (54), pCMV-Rb (55), pSG5-HA-Rb (56), pCMV-Rb-N757F (57), pSG5-HA-Rb-ΔRXL (29), pE2F1-Luc(−242) and pE2F1-Luc(E2F−) (58), pHsOrc1-Luc(−1053) (59), pCycA-Luc (60), pEGFPhLA-WT (61), and pCMV-hRbΔCDK (62). The following expression plasmids were generated during this study by site-directed mutagenesis techniques: pCGN- and pSG5-based UL97 L1m (C151G), L2m (C428G), L3m (C693G), Tri (L1m + L2m + L3m), HPm (W368A), Qd (Tri + HPm); pCMV-Rb-3D (T373D/T821D/T826D), pCMV-Rb-3E (T373E/T821E/T826E), pCMV-Rb-3A (T356A/T821A/T826A), and pCMV-Rb-3Eb (T356E/T821E/T826E). pCMV-FLAG-Rb and pCMV-FLAG-RbΔCDK were constructed by inserting PCR products into HindIII and XbaI sites in p3xFLAG-CMV-10 (Sigma-Aldrich) using the In-Fusion HD cloning kit (Clontech). pSG5-IE1 was constructed by inserting a PCR product amplified from pCGN-IE1 (63) into the BamHI site of pSG5. All plasmids constructed by PCR were sequenced and confirmed to be correct. The sequences of primers for mutagenesis are available upon request.
Antibodies
Primary antibodies were purchased from Abcam (Rb-Thr(P)373, catalog number ab52975; Rb-Thr(P)826, ab4779 and ab133446), Abgent (Rb-Thr(P)356, AJ1682c; Rb-Ser(P)788, AP3238a), Affinity BioReagents (Rb-Ser(P)249/Thr(P)252, OPA1-03896), Ambion (GAPDH, AM4300), BIOSOURCE (Rb-Thr(P)821, 44-582G), Cell Signaling Technology (E2F1, 3742; Rb, 9309; Rb-Ser(P)608, 2181; Rb-Ser(P)780, 9307; Rb-Ser(P)795, 9301; Rb-Ser(P)807/Ser(P)811, 9308), Covance (HA, MMS-101P), Invitrogen (Rb-Ser(P)612, 44572G; V5, R960-25), Santa Cruz Biotechnology (E2F2, sc-633; E2F3, sc-878; E2F4, sc-866; p107, sc-318; p130, sc-317), Sigma-Aldrich (FLAG M2, F1804), Thermo Scientific (MCM7, MS-862), and Virusys (UL44, CA006-100). Antibodies against HCMV IE1 (1B12), pp71 (2H10-9), and UL97 have been described previously (63–65). Mouse TrueBlot Ultra (eBioscience, 18-8817) and rabbit TrueBlot (18-8816) were used as secondary antibodies for Western blotting of immunoprecipitation samples.
Treatment with siRNA
E2F4 siRNA (Hs_E2F4_1, catalog number SI00375417) and Allstars negative control siRNA (catalog number 1027281) were purchased from Qiagen. Saos-2 cells (2.5 × 105) were transfected with 80 pmol of each siRNA using Lipofectamine RNAiMAX (Invitrogen).
In Vivo Rb Phosphorylation Assay and Lamina Disruption Assay
To detect Rb phosphorylation, Saos-2 cells were transfected with wild-type and mutated Rb expression plasmid DNA together with wild-type and mutated HCMV UL97 expression plasmid DNA as described previously (43). At 48 h post-transfection, the transfected cells were harvested and subjected to Western blot analysis. A lamina disruption assay was performed in U-2 OS cells as described previously (43).
Luciferase Assays
Saos-2 cells (2.5 × 105/6-well) were transfected with each Rb expression vector (0.25 μg) together with pCGN vector carrying wild-type or mutated HCMV UL97 (1 μg) and pE2F1-Luc(−242), pE2F1-Luc(E2F−), pHsOrc1-Luc(−1053), or pCycA-Luc reporter plasmids (0.02 μg) using TransIT-2020. For cotransfection with IE1, Saos-2 cells were transfected with pSG5-IE1 (1.2 μg), pSG5-HA-Rb (0.6 μg), pSG5 vector carrying wild-type or mutated HCMV UL97 (1.2 μg), and pE2F1-Luc(−242) reporter plasmid (0.02 μg). Total DNA levels in transfections were balanced with pCGN vector. At 48 h post-transfection, luciferase activity was measured using a luciferase reporter assay system (Promega) and a Veritas microplate luminometer (Turner Biosystems) and normalized to the total protein concentration in the cell extract. Luciferase assays were conducted in biological triplicate and technical duplicate. Statistical analyses were determined using a two-tailed, unpaired Student's t test.
Immunoprecipitations
Saos-2 cells (1.6 × 106/100 mm) were transfected with pCMV-FLAG-Rb or pCMV-FLAG-RbΔCDK (1.8 μg) together with pCGN vector carrying wild-type/mutated HCMV UL97 (3.6 μg) using TransIT-2020. After 48 h, cells were suspended with modified CSK buffer (100 mm Pipes, pH 6.8, 500 mm NaCl, 300 mm sucrose, 1 mm EGTA, 1 mm MgCl2, 0.1% Triton X-100, 10 μg/ml pepstatin A, 25 μg/ml leupeptin, 1 mm PMSF, 25 mm NaF, 10 mm β-glycerophosphate), centrifuged, and diluted with an equal volume of ET gel buffer (50 mm Tris, pH 7.4, 0.1% Triton X-100, 1 mm EDTA). The extracts were immunoprecipitated with anti-FLAG or E2F1 antibodies (2 μg) and protein G-Sepharose beads (GE Healthcare). Then beads were washed three times with NET gel buffer (ET gel buffer with 150 mm NaCl) and eluted with SDS gel loading buffer.
Western Blotting
Rb-transfected Saos-2 cells or HCMV-infected fibroblasts were lysed in an SDS solution (1% SDS, 2% β-mercaptoethanol) by boiling for 10 min followed by vortexing as described previously (37). Equal amounts of proteins were separated by 7.5% SDS-PAGE and transferred onto nitrocellulose membranes. The phosphate affinity (Phos-tag, NARD Institute) electrophoresis was performed following the manufacturer's instructions. For experiments with λ-protein phosphatase, cells were suspended in lysis buffer A (66) without EDTA, NaF, β-glycerophosphate, and DTT. The obtained whole-cell lysates (120 μg/60 μl) were incubated in a 1× NEBuffer for protein metallophosphatases supplemented with 1 mm MgCl2 and 800 units of λ-protein phosphatase (New England Biolabs) for 20 min at 30 °C. Quantification of bands was performed in triplicate as described previously (67). Statistical analyses were determined using a two-tailed, unpaired Student's t test.
RNA Analysis
To quantify mRNA levels of E2F1 and Cdt1, total RNA was isolated as described previously (68). Equivalent amounts of DNase I-treated RNA were converted to cDNA using the SuperScript III First-Strand Synthesis Supermix for qRT-PCR (Invitrogen) according to the manufacturer's instructions. Quantitative PCR was performed as described previously (67) with gene-specific primer sets for human E2F1 (69), Cdt1 (70), and GAPDH (71). Data were analyzed by the comparative Ct method (72). Experiments were performed in biological triplicate, and statistical significance was determined using a two-tailed, unpaired Student's t test.
Results
v-CDK UL97 Phosphorylates Rb on 11 CDK Consensus Sites
Eleven phosphospecific antibodies that probe 13 of the 16 CDK consensus phosphorylation sites in the Rb protein are commercially available. We used these phosphospecific antibodies to demonstrate that, during HCMV infection, Rb becomes phosphorylated on at least 11 CDK consensus sites, all in a UL97-dependent manner (Fig. 1A and Table 1). Ser249 and Thr252 are not phosphorylated during HCMV infection (37), and phosphospecific antibodies to Thr5, Ser230, and Ser567 are unavailable. Phosphorylation was also detected by the different electrophoretic mobilities of Rb, which is either hypophosphorylated (faster migrating) or hyperphosphorylated (slower migrating) (Fig. 1A). An ectopically expressed Rb mutant protein (RbΔCDK) in which 14 of the 16 CDK phosphorylation sites were replaced with alanines (Ser230 and Ser567 remain) was not detectably phosphorylated by transfected UL97 as determined by electrophoretic mobility shift in conventional or phosphate affinity (Phos-tag) electrophoresis (Fig. 1B). We conclude that UL97 phosphorylates Rb on Thr356, Thr373, Ser608, Ser612, Ser780, Ser788, Ser795, Ser807, Ser811, Thr821, and Thr826 and that these are the major phosphorylation events on Rb during HCMV infection.
FIGURE 1.

UL97 phosphorylates Rb on 11 CDK consensus sites during HCMV infection. A, serum-starved HFFs were mock-infected (M) or infected with wild-type (WT) or UL97-null (Δ97) HCMV. At the indicated hour postinfection (hpi), protein lysates were harvested and analyzed by Western blotting with the indicated antibodies. B, Saos-2 cells were transfected with expression plasmids for FLAG-tagged alleles of wild-type Rb or Rb in which CDK consensus phosphorylation residues were replaced with alanines (RbΔCDK) together with either an empty vector (EV) or one expressing HA-tagged WT UL97 or a kinase-deficient (KD) UL97. Lysates harvested 48 h after transfection were separated by conventional (normal) or phosphate affinity (Phos-tag) electrophoresis and analyzed by Western blotting with the indicated antibodies. pS, phosphoserine; pT, phosphothreonine.
TABLE 1.
Rb phosphorylation during HCMV infection
| WTa | Δ97a | L1a | |
|---|---|---|---|
| Thr5 | NTb | NT | NT |
| Ser230 | NT | NT | NT |
| Ser249/Thr252 | −b | − | − |
| Thr356 | +b | − | − |
| Thr373 | + | − | + |
| Ser567 | NT | NT | NT |
| Ser608 | + | − | + |
| Ser612 | + | − | + |
| Ser780 | + | − | ±b |
| Ser788 | + | − | + |
| Ser795 | + | − | + |
| Ser807/Ser811 | + | − | ± |
| Thr821 | + | − | ± |
| Thr826 | + | − | − |
a WT, wild-type virus: Δ97, UL97-null virus; L1, L1 motif-mutated virus.
b NT, not tested: +, apparent; ±, marginal; −, absent.
The UL97 HP and Rb RXL Motifs Are Not Required for UL97-mediated Rb Phosphorylation
UL97 has four putative Rb-binding motifs, three LXCXE-like motifs and an HP-like sequence. To determine which of these were required for Rb phosphorylation and inactivation, we created UL97 mutant (m) alleles in which each LXCXE-like motif was individually disrupted by converting the central cysteine to a glycine residue (L1m, C151G; L2m, C428G; L3m, C693G), one in which all three were simultaneously disrupted (Tri), one in which the conserved tryptophane residue of the HP was converted to an alanine (HPm, W368A), and finally one where all four putative Rb-binding motifs were disrupted as described above (Qd). All mutant alleles expressed proteins to similar steady state levels (Fig. 2A). The L1m, L2m, L3m, and Tri mutants displayed wild-type patterns of autophosphorylation as judged by electrophoretic mobility shift (Fig. 2A), disrupted the nuclear lamina as well as the wild-type protein in transient assays (Fig. 2B), and in the context of recombinant viruses supported wild-type lytic replication kinetics in fibroblasts (Fig. 2C) (44). Thus we conclude that mutation of any or all of the LXCXE motifs does not detectably impair UL97 kinase activity or viral lytic replication. The HPm and Qd mutants, however, displayed reduced autophosphorylation (Fig. 2A), lamina disruption capability (Fig. 2B), and viral growth (Fig. 2C). Trp368 sits equidistant between two of the three residues of the catalytic triad (Lys355 and Glu380) essential for kinase activity. Mutation of this residue likely impairs general UL97 kinase activity based on reduced autophosphorylation (Fig. 2A) and substantial resistance to maribavir (Table 2). Therefore, results obtained with HPm mutant must be interpreted with caution.
FIGURE 2.
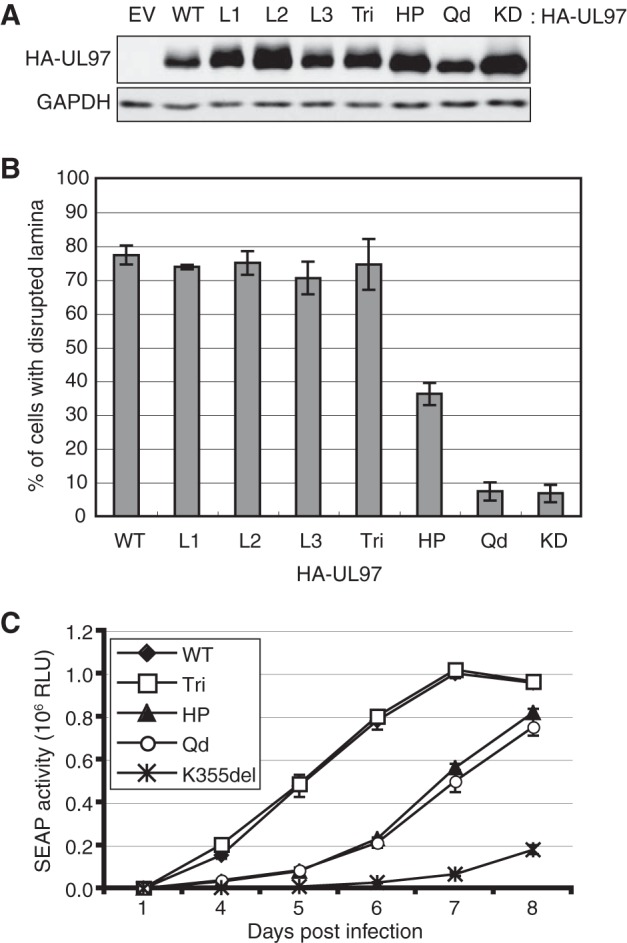
UL97 kinase activity is unaffected by mutation of the LXCXE motifs. A, Saos-2 cells were transfected with expression plasmids for Rb together with either an empty vector (EV) or HA-tagged alleles of wild-type UL97 or UL97 alleles with the following mutations: L1, C151G; L2, C428G; L3, C693G; Tri, C151G/C428G/C693G; HP, W368A; Qd, C151G/W368A/C428G/C693G; kinase-deficient (KD), K355Q. Lysates harvested 48 h after transfection were analyzed by Western blotting with the indicated antibodies. B, U-2 OS cells were transfected with an expression plasmid for a lamin A-GFP fusion protein together with an expression plasmid for the indicated HA-tagged UL97 wild-type or mutant allele. UL97-expressing cells (as determined by indirect immunofluorescence with the HA antibody) were scored for having an intact or disrupted nuclear lamina after visualizing GFP fluorescence. The percentage of UL97-positive cells displaying a disrupted lamina is shown. Error bars represent the S.D. from biological triplicate. C, low multiplicity growth curves in HFFs were quantitated by relative light units (RLU) of secreted alkaline phosphatase (SEAP) activity. See “Experimental Procedures” for details. Error bars represent the S.E. of 11–12 replicate growth curves set up over three separate dates.
TABLE 2.
Maribavir susceptibility phenotypes of UL97 mutant viruses
| Straina | UL97 genotypeb | Maribavir EC50c | Replicatesd |
|---|---|---|---|
| μm | |||
| T3261 | Wild type | 0.12 ± 0.03 | 22 |
| T3260 | C151G/C428G/C693G | 0.03 ± 0.01 | 13 |
| T3719 | C151G/W368A/C428G/C693G | 35 ± 7 | 12 |
| T3727 | W368A | 27 ± 7 | 12 |
a Serial number of recombinant CMV strain derived by transfection of bacterial artificial chromosome into fibroblasts.
b Mutation induced into bacterial artificial chromosome and recombinant virus.
c Mean drug concentration (μm) required to reduce secreted alkaline phosphatase growth by 50% at 6–7 days postinfection.
d Number of assays.
To circumvent the apparently attenuated kinase activity of the HPm and Qd mutants, we determined the ability of wild-type UL97 to phosphorylate Rb alleles lacking the RXL motifs with which the HP sequences interact. Rb has two RXL motifs within its C-terminal 99 amino acids. An allele truncated at codon 829 just prior to the first RXL motif is commonly used to express an RXL-deficient protein (29). Wild-type UL97 was equally able to phosphorylate full-length Rb (Rb1–928) and Rb-ΔRXL (Rb1–829) except for the Thr826 site (Fig. 3A and Table 3). We were unable to determine whether the inability to phosphorylate Thr826 of Rb-ΔRXL is a phosphorylation defect caused by the lack of RXL motifs or the proximity of the residue to the novel C terminus or whether phosphorylation still occurs but is undetectable because the truncation eliminates the full epitope recognized by the phosphospecific antibody. Rb-ΔRXL only minimally repressed an E2F1 reporter plasmid, but the observed inhibition was abrogated by UL97 (Fig. 3B), indicating that UL97 can functionally inactivate Rb-ΔRXL. We conclude that interactions between the hydrophobic patch of UL97 and the RXL motifs of Rb are not required for UL97-mediated Rb phosphorylation or inactivation.
FIGURE 3.

The RXL motif of Rb is dispensable for Rb phosphorylation by UL97. A, Saos-2 cells were transfected with expression plasmids for HA-tagged alleles of wild-type Rb or Rb in which the C-terminal 99 amino acids containing the RXL motifs were deleted (RxL) together with either an empty vector (EV) or one expressing V5-tagged WT UL97. Lysates harvested 48 h after transfection were analyzed by Western blotting with the indicated antibodies. B, Saos-2 cells were transfected with a luciferase reporter driven by the E2F1 promoter together with an expression plasmid for UL97 and either wild-type or RXL mutant Rb. Lysates harvested 48 h after transfection were analyzed for luciferase activity (top) and protein expression with the indicated antibodies (bottom). Luciferase activity was normalized to total protein concentration and is presented relative to the activity of the reporter without Rb or UL97 (set at 100%). Error bars denote the S.D. **, p < 0.01.
TABLE 3.
Rb phosphorylation in a transient transfection
| WT UL97a WT Rbb | UL97 L1a WT Rb | WT UL97 Rb CMb | WT UL97 Rb RXLb | |
|---|---|---|---|---|
| Thr5 | NTc | NT | NT | NT |
| Ser230 | NT | NT | NT | NT |
| Ser249/Thr252 | −c | − | − | − |
| Thr356 | +c | − | − | + |
| Thr373 | + | ±c | − | + |
| Ser567 | NT | NT | NT | NT |
| Ser608 | + | + | ± | + |
| Ser612 | + | + | ± | + |
| Ser780 | + | ± | ± | + |
| Ser788 | + | + | ± | + |
| Ser795 | + | + | − | + |
| Ser807/Ser811 | + | + | ± | + |
| Thr821 | + | ± | − | + |
| Thr826 | + | − | − | − |
a WT, wild-type UL97; L1, L1 motif-mutated UL97.
b WT, wild-type Rb; CM, Rb N757F mutant; RXL, Rb1–829 mutant.
c NT, not tested; +, apparent; ±, marginal; −, absent.
The First UL97 LXCXE Motif (L1) Is Required for Full Rb Phosphorylation
In transfection assays, LXCXE mutants L2m and L3m were as competent as wild-type UL97 at phosphorylating Rb on all residues analyzed (Fig. 4A). However, the L1 mutant (UL97-L1m) showed a defect in Rb phosphorylation as assayed both by electrophoretic mobility shift and phosphospecific antibody reactivity (Fig. 4, A and B). Specifically, UL97-L1m failed to phosphorylate Thr356 and Thr826 and showed modest reductions in phosphorylation of Thr373, Ser780, and Thr821 (Fig. 4B and Table 3). An N757F substitution creates a cleft mutant (CM) that renders Rb incapable of binding the HDAC1 protein that interacts with this Rb region through an LXCXE motif (57). Rb-CM was phosphorylated by UL97 less efficiently than was wild-type Rb as judged by electrophoretic mobility shift (Fig. 4C) and phosphatase treatment (Fig. 4D). Specifically, UL97 failed to phosphorylate Rb-CM on Thr356, Thr373, Ser795, Thr821, and Thr826 and showed reduced phosphorylation on Ser608, Ser612, Ser780, Ser788, and Ser807/Ser811 (Fig. 4C and Table 3). In fibroblasts infected with the UL97-L1m recombinant virus, Rb was significantly less phosphorylated on Thr356 and Thr826 and showed modestly reduced phosphorylation on Ser780, Ser807, Ser811, and Thr821 (Fig. 4, E and F, and Table 1). From these experiments, we conclude that the first LXCXE motif of UL97 and the LXCXE-binding cleft of Rb are required for full phosphorylation of Rb by UL97. Phosphorylation of residues Thr356, Thr821, and Thr826 appear to be the most dependent on this putative submolecular interaction.
FIGURE 4.

The first UL97 LXCXE motif and the Rb cleft mediate full phosphorylation of Rb by UL97. A, Saos-2 cells were transfected with an expression plasmid for Rb together with either an empty vector (EV) or one expressing HA-tagged WT UL97 or the indicated UL97 mutant allele. Lysates harvested 48 h after transfection were analyzed by Western blotting with the indicated antibodies. For detection of phospho-Ser249/Thr252, HFFs were serum-starved for 48 h and then stimulated with 15% serum. B, the graph represents the level of phosphorylated Rb normalized to the total Rb present from wild-type or L1m UL97-transfected cells shown in A. Values are presented relative to the value in wild-type UL97-transfected cells for each phosphorylation site (set at 1). Error bars denote the S.D. *, p < 0.05; **, p < 0.01; n.s., not significant. C, Saos-2 cells were transfected with expression plasmids for WT Rb or Rb with an N757F CM together with either an empty vector (EV) or one expressing V5-tagged wild-type UL97. Lysates harvested 48 h after transfection were analyzed by Western blotting with the indicated antibodies. D, Lysates prepared as in C were treated (+) or not (−) with λ-protein phosphatase (l-PPase) and analyzed as in C. E, serum-starved HFFs were mock-infected (M) or infected with WT HCMV or the indicated UL97 mutant virus at a multiplicity of infection of 1. At the indicated hour postinfection (hpi), protein lysates were harvested and analyzed by Western blotting with the indicated antibodies. F, the graph represents the level of phosphorylated Rb normalized to the total Rb present from wild-type or L1m virus-infected cells at 48 h postinfection shown in E except at a multiplicity of infection of 2. Values are presented relative to the value in wild-type virus-infected cells for each phosphorylation site (set at 1). Error bars denote the S.D. *, p < 0.05; **, p < 0.01; n.s., not significant. pS, phosphoserine; pT, phosphothreonine; KD, kinase-deficient.
The UL97 L1 Motif and the Rb Cleft Are Required for UL97-mediated Rb Inactivation during Reporter Assays
Phosphorylation inactivates the ability of Rb to suppress E2F-responsive gene expression. In a reporter assay, UL97 inhibited the ability of co-transfected Rb to repress the E2F1 promoter, and this depended upon the kinase activity of UL97 (Fig. 5A). Mutation of the CDK consensus phosphorylation sites in Rb did not prevent it from repressing the E2F1 reporter but did inhibit the ability of UL97 to counteract this repression (Fig. 5B). Furthermore, neither Rb nor UL97 affected reporter expression from a mutant E2F1 promoter lacking E2F binding sites (Fig. 5C). From this, we conclude that UL97 activates an E2F1 promoter-reporter by phosphorylating Rb and relieving Rb- and E2F-mediated repression. UL97 also relieved Rb repression of the Orc1 (Fig. 5D) and cyclin A (Fig. 5E) E2F-responsive promoters in reporter assays, indicating that UL97 can relieve Rb repression of multiple E2F-responsive promoters.
FIGURE 5.

Phosphorylation by UL97 relieves Rb-mediated repression of E2F-responsive promoters. A, Saos-2 cells were transfected with a luciferase reporter driven by the E2F1 promoter together with an empty vector (−) or an expression plasmid for Rb and the indicated allele of UL97. Lysates harvested 48 h after transfection were analyzed for luciferase activity (top) and protein expression with the indicated antibodies (bottom). Luciferase activity was normalized to total protein concentration and is presented relative to the activity of the reporter without Rb or UL97 (set at 100%). Error bars denote the S.D. *, p < 0.05; **, p < 0.01; n.s., not significant. B, luciferase and Western blot analyses were performed as in A except an Rb allele in which CDK consensus phosphorylation residues were replaced with alanines (RbΔCDK;Δ) was also included. C, luciferase and Western blot analyses were performed as in A except the reporter contained an E2F1 promoter in which the E2F binding sites were mutated. D, luciferase and Western blot analyses were performed as in A except the reporter contained the Orc1 promoter. E, luciferase and Western blot analyses were performed as in A except the reporter contained the cyclin A promoter. F, luciferase and Western blot analyses were performed as in A except an Rb allele with an N757F CM was also included. KD, kinase-deficient.
The UL97-L2m and -L3m proteins that fully phosphorylate Rb also counteracted Rb-mediated repression of the E2F1 (Fig. 5A), Orc1 (Fig. 5D), and cyclin A (Fig. 5E) reporters. However, the UL97-L1m protein that failed to fully phosphorylate Rb also failed to relieve Rb-mediated repression of each of these promoters (Fig. 5, A, D, and E). Rb-CM fails to bind HDAC1 but does bind E2F-1 and represses E2F-responsive promoters although not to wild-type levels (57). We found that Rb-CM was able to repress the E2F1 reporter, but this repression was not relieved by wild-type UL97 (Fig. 5F), which fails to fully phosphorylate this mutant Rb protein (Fig. 4C). From these experiments, we conclude that the first LXCXE motif of UL97 and the LXCXE-binding cleft of Rb are required for UL97 to inactivate the ability of Rb to repress E2F-responsive reporter constructs.
Although UL97-L1m Fails to Inactivate Rb during Reporter Assays, It Efficiently Disrupts Rb-E2F Complexes
The inefficiency with which UL97-L1m phosphorylates Rb at certain residues correlates with the inability of this mutant to inactivate Rb in reporter assays and the inability of wild-type UL97 to inactivate an Rb cleft mutant. These results implicate an L1-cleft interaction mediating full Rb phosphorylation and inactivation. To test this prediction, we generated Rb alleles encoding phosphomimetics at residues critical for disruption of Rb-E2F interactions and underphosphorylated by UL97-L1m. Critical residues for Rb phosphorylation-dependent inactivation include Thr373, Ser608, Thr821, and Thr826 (20, 26). Although Ser608 was phosphorylated well by UL97-L1m, the other residues were inefficiently phosphorylated (Fig. 4, A and B, and Table 3). Rb mutants in which Thr373, Thr821, and Thr826 were simultaneously converted to glutamate (3E) or aspartate (3D) were still able to repress an E2F1 promoter-reporter construct (Fig. 6A) and were inactivated by UL97 in a kinase-dependent manner (Fig. 6A). However, UL97-L1m still failed to inactivate Rb-3E or -3D even though residues inefficiently phosphorylated by this mutant kinase had phosphomimetic substitutions (Fig. 6A). Identical results were obtained with a similar construct in which the major L1-cleft-dependent phosphorylation sites Thr356, Thr821, and Thr826 were simultaneously changed to glutamate (3Eb) (Fig. 6B). Thus, inefficient Rb phosphorylation does not appear to be responsible for the inability of UL97-L1m to inactivate Rb in reporter assays.
FIGURE 6.

Underphosphorylation of Rb by UL97-L1m neither mediates sustained repression of E2F-responsive promoters by Rb nor affects disruption of Rb-E2F complexes. A, Saos-2 cells were transfected with a luciferase reporter driven by the E2F1 promoter together with an empty vector (−) or an expression plasmid for Rb alleles with specific phosphomimetic residue substitutions (3E, T373E/T821E/T826E; 3D, T373D/T821D/T826D) and the indicated allele of UL97. Lysates harvested 48 h after transfection were analyzed for luciferase activity (top) and protein expression with the indicated antibodies (bottom). Luciferase activity was normalized to total protein concentration and is presented relative to the activity of the reporter without Rb or UL97 (set at 100%). Error bars denote the S.D. **, p < 0.01. B, luciferase and Western blot analyses were performed as in A except WT Rb or Rb alleles with different phosphomimetic (3Eb, T356E/T821E/T826E) or non-phosphorylatable (3A, T356A/T821A/T826A) residue substitutions were also included. C, Saos-2 cells were transfected with expression plasmids for wild-type FLAG-tagged Rb and the indicated allele of UL97. Lysates harvested 48 h after transfection were subjected to immunoprecipitation (IP) with the FLAG antibody. Input lysates and immunoprecipitates were analyzed by Western blotting with the indicated antibodies. h.c., heavy chain. D, immunoprecipitation experiment as in C except an Rb allele in which CDK consensus phosphorylation residues were replaced with alanines (Δ) was transfected. E, immunoprecipitation experiment as in C except the E2F1 antibody was used for immunoprecipitation. KD, kinase-deficient.
If inefficient Rb phosphorylation is not the reason why UL97-L1m fails to rescue Rb-mediated repression in reporter assays of E2F-dependent transcription, then even the reduced level of Rb phosphorylation catalyzed by UL97-L1m should effectively disrupt Rb-E2F complexes. Activating E2F family members E2F1, E2F2, E2F3a, and E2F3b associate with transfected Rb in Saos-2 cells, and this association was equally disrupted by either wild-type or UL97-L1m (Fig. 6, C and E). Importantly, neither wild type nor UL97-L1m was able to disrupt these E2F complexes with the non-phosphorylatable RbΔCDK (Fig. 6D), indicating that UL97 disrupts Rb-E2F complexes through a phosphorylation-dependent process. This conclusion is supported by the inability of a kinase-deficient UL97 to disrupt E2F complexes with wild-type Rb (Fig. 6, C and E). Rb also interacts with the transcriptionally repressive E2F4 protein but is not known to interact with E2F5–8 (73, 74). Although we detected E2F4 expression in Saos-2 cells (Fig. 6, C and D), we did not detect Rb association with E2F4 and thus are unable to determine whether UL97-L1m disrupts such complexes. However, knockdown of E2F4 failed to rescue the ability of UL97-L1m to promote E2F1 reporter expression in the presence of Rb (Fig. 7A). As UL97-L1m disrupts Rb complexes with E2F1–3 and E2F4 is not required for Rb-mediated transcriptional repression in the presence of UL97-L1m, we conclude that the defect in the ability of UL97-L1m to inactivate Rb in a reporter assay does not result from inefficient disruption of Rb-E2F complexes.
FIGURE 7.

Knockdown of E2F4 or p107 and p130 does not allow UL97-L1m to relieve Rb-mediated suppression of E2F-responsive transcription. A, Saos-2 cells were transfected with an siRNA targeting E2F4 or a control siRNA for 24 h. Cells were then transfected with a luciferase reporter driven by the E2F1 promoter together with an empty vector (−) or an expression plasmid for Rb and the indicated allele of UL97. Lysates harvested 48 h after plasmid transfection were analyzed for luciferase activity (top) and protein expression with the indicated antibodies (bottom). Luciferase activity was normalized to total protein concentration and is presented relative to the activity of the reporter without Rb or UL97 (set at 100%). Error bars denote the S.D. *, p < 0.05; **, p < 0.01. B, luciferase and Western blot analyses were performed as in A except cells were transduced with lentiviruses expressing shRNAs targeting either GFP or p107 and p130. Luciferase activity is presented relative to the activity of the reporter without Rb or UL97 in each transduced cell (set at 100%). KD, kinase-deficient.
Knockdown of p107 or p130 Does Not Allow UL97-L1m to Relieve Rb-dependent Suppression of E2F-mediated Transcription
The ability of UL97-L1m to efficiently disrupt Rb complexes spurred us to investigate an alternative potential explanation for why this mutant failed to inactivate the ability of Rb to suppress E2F-dependent transcriptional activation. We considered that the other Rb family members, p107 and p130, may be suppressing E2F-dependent transcription in the presence of UL97-L1m. However, simultaneous knockdown of p107 and p130 failed to rescue the ability of UL97-L1m to promote E2F1 reporter expression in the presence of Rb (Fig. 7B). In total, our data suggest that UL97 possesses an LXCXE-dependent way to counteract Rb-mediated repression of E2F-responsive promoters distinct from disrupting complexes between the E2F proteins and the Rb family members.
The Inability of UL97-L1m to Promote E2F-dependent Gene Expression Is Complemented during HCMV Infection by IE1
Despite the inability of UL97-L1m to relieve Rb-mediated repression of E2F-responsive reporters, recombinant HCMV expressing this allele (44) and UL97 with disruption of all three LXCXE motifs (Fig. 2C) had no growth defect. L1m virus induced the accumulation of the E2F-responsive gene products E2F1 and MCM7 at the protein level as well as wild-type virus. The LXCXE triple mutant virus induced E2F1 protein accumulation as well as wild type and had only a small defect in MCM7 protein accumulation (Fig. 8, A and C). The E2F1 and MCM7 proteins were not induced during infection with a UL97-null HCMV (Fig. 8B). Wild type, UL97-L1m, and the triple mutant virus all induced similar steady state transcript levels of E2F1 and another E2F-responsive gene, Cdt1 (Fig. 8D). E2F1 and Cdt1 transcript levels in HCMV-infected cells were significantly reduced in the presence of the UL97 inhibitor maribavir (Fig. 8D). Thus, although UL97 kinase activity is absolutely required to induce these E2F-responsive genes during HCMV infection, its L1 motif is not (Fig. 8, A–D).
FIGURE 8.

The first UL97 LXCXE motif is dispensable for expression of the E2F-responsive genes E2F1, MCM7, and Cdt1 during HCMV infection likely because of complementation from the viral IE1 protein. A, serum-starved HFFs were mock-infected (M) or infected with WT HCMV or the indicated UL97 mutant virus at a multiplicity of infection of 1. At the indicated hour postinfection (hpi), protein lysates were harvested and analyzed by Western blotting with the indicated antibodies. B, Western blot analysis conducted as in A except during either WT or UL97-null (Δ97) HCMV infection. C, E2F1 and MCM7 protein levels at 48 h postinfection from the experiment shown in A were quantified and normalized to GAPDH protein, and values are presented relative to the value in wild-type HCMV-infected cells (set at 1). Error bars denote the S.D. **, p < 0.01; n.s., not significant. D, serum-starved HFFs were infected with each virus at a multiplicity of infection of 2 in the presence of maribavir (MBV) or DMSO (D). E2F1 and Cdt1 mRNA levels at 48 h postinfection were analyzed by quantitative PCR and normalized to GAPDH mRNA level. Values are presented relative to the value in mock infection (M) (set at 1). Error bars denote S.D. *, p < 0.05; **, p < 0.01; n.s., not significant. E, Saos-2 cells were transfected with a luciferase reporter driven by the E2F1 promoter together with an empty vector (−) or an expression plasmid for Rb, the indicated allele of UL97, or HCMV IE1. Lysates harvested 48 h after transfection were analyzed for luciferase activity (top) and protein expression with the indicated antibodies (bottom). Luciferase activity was normalized to total protein concentration and is presented relative to the activity of the reporter without Rb, UL97, or IE1 (set at 100%). P-Rb, hyperphosphorylated Rb; non-P-Rb, non-phosphorylated Rb. F, Saos-2 cells were transfected as in E. Lysates harvested 48 h after transfection were analyzed by Western blotting with the indicated antibodies. G, model for Rb inactivation by UL97. I, Rb represses E2F-responsive transcription. II, UL97 induces E2F-responsive transcription both by Rb phosphorylation-dependent disruption of Rb-E2F complexes, which is an LXCXE-independent event, and by an unknown L1 LXCXE-dependent function. III, a UL97 mutant with a non-functional L1 motif (UL97-L1m) induces partial phosphorylation of Rb and disruption of Rb-E2F complexes but fails to activate Rb-repressed E2F-responsive transcription. IV, the inability of UL97-L1m to activate Rb-repressed E2F-responsive transcription is complemented by HCMV IE1 through an unknown mechanism. KD, kinase-deficient.
HCMV encodes other proteins (pp71, IE1, and IE2) that inactivate Rb family members and/or induce E2F-reponsive gene expression (8), and one of these may cooperate with UL97-L1m during viral infection to activate E2F-dependent genes. Preliminary experiments failed to detect a synergistic effect between UL97-L1m and either pp71 or IE2, but the combination of UL97-L1m and IE1 activated the E2F1 promoter as well as wild-type UL97 (Fig. 8E). IE1 did not promote further phosphorylation of Rb (Fig. 8F); thus the observed stimulation must occur through some other mechanism. Our data indicate that an interaction between the first LXCXE motif of HCMV UL97 and the cleft of Rb is important for full Rb phosphorylation and the induction of E2F-responsive gene expression by UL97, but these defects can be negated by co-expression of the viral IE1 protein (Fig. 8G).
Discussion
Viral oncoproteins (19, 75, 76) and cellular chromatin modifiers (77) use LXCXE motifs to bind within the Rb cleft. The D-type cyclins also contain LXCXE motifs. Although these are dispensable for cyclins D1 and D2 to direct CDK-mediated Rb phosphorylation, the LXCXE motif of cyclin D2 (but not D1) is required for abrogating the growth inhibitory function of Rb (31). To our knowledge, the requirement for the cyclin D3 LXCXE motif in CDK-mediated Rb phosphorylation has not been examined. For the v-CDK UL97 of HCMV, two of the three LXCXE motifs within UL97 were completely dispensable for Rb phosphorylation, whereas ablation of the first motif, L1, diminished but did not abrogate Rb phosphorylation (Fig. 4, A, B, E, and F, and Tables 1 and 3). Likewise, a cleft mutant Rb protein was phosphorylated, albeit somewhat inefficiently, by wild-type UL97 (Fig. 4, C and D, and Table 3), so although the L1 LXCXE motif (presumably interacting with the Rb cleft) enhances Rb phosphorylation, it is not required for that reaction. The v-CDK encoded by Epstein-Barr virus, the BGLF4 protein, also phosphorylates Rb (43) but does not contain an LXCXE motif, again indicating that this motif is not an absolute requirement of CDK- or v-CDK-mediated Rb phosphorylation.
Despite the underphosphorylation of Rb catalyzed by UL97-L1m, this protein was still able to disrupt complexes between Rb and all the activating E2Fs (Fig. 6, C and E). Therefore, it is somewhat surprising that UL97-L1m was unable to relieve Rb-mediated repression of three different E2F-dependent promoter-reporters (Fig. 5, A, D, and E). Neither E2F4 nor p107/p130 seem to be responsible for this sustained transcriptional repression by Rb in the presence of UL97-L1m (Fig. 7). However, multiple potential phosphorylation targets of UL97 (but not UL97-L1m) may affect regulation of E2F-responsive transcription, including the E2F proteins, HDACs, or RNA polymerase II itself.
UL97 phosphorylates the C-terminal domain of RNA polymerase II on serine 5 (78), a modification that identifies polymerases in the initiation phase (79). Perhaps although UL97-L1m is able to displace Rb from an E2F-responsive promoter to allow for RNA polymerase II recruitment it may be unable to phosphorylate the C-terminal domain to facilitate initiation. UL97 has also been reported to phosphorylate HDAC1 and promote HCMV IE transcription (80). However, HDAC1 seems unlikely to be responsible for the transcriptional defect observed with UL97-L1m because the Rb cleft mutant (N757F) that does not associate with HDAC1 (57) still repressed the E2F-responsive promoter in the presence of UL97 (Fig. 5F).
Likely targets of the UL97 v-CDK are the E2F proteins themselves. Human E2F1 has 11 potential serine/threonine phosphorylation sites, E2F2 has two, and E2F3 has 10. We could find no published reports of CDK-mediated phosphorylation of E2F2 or E2F3. E2F1 is phosphorylated by CDK2-cyclin A, but all reports indicate this inhibits E2F-dependent transcription (81–84). CDK2-cyclin B phosphorylates E2F1 but does not detectably affect its activity (83), whereas neither CDK2-cyclin E nor CDK4/6-cyclin D phosphorylate E2F1 (83–85). Thus, the potential for CDK- (and thus v-CDK-) mediated regulation of E2F activity exists, but there is little precedence for this leading to transcriptional activation.
Unfortunately, only a few confirmed substrates for UL97 are known (86), although 290 have been proposed based on in vitro kinase reactions with protein arrays (87). As the average kinase phosphorylates only around 50 proteins (88), many of these are unlikely to be true substrates. A detailed comprehension of UL97 substrates should help us better understand this v-CDK.
A straightforward model predicts that UL97, through its LXCXE motifs, attaches to Rb, thus gaining access to substrates either associated with or adjacent to this tumor suppressor in the multiple complexes in which it exists (89). Mutation of the L1 motif would weaken this association, decreasing UL97 retention time in Rb complexes, resulting in decreased phosphorylation of Rb-affiliated proteins and therefore the maintenance of Rb-instituted transcriptional repression. However, we were unable to detect stable association of even wild-type UL97 with Rb (Fig. 6, C–E), making it impossible to determine whether UL97-L1m has a decreased affinity for the protein. The mechanism through which the L1 motif of UL97 relieves Rb-mediated transcription repression therefore requires more study.
Despite the defect of UL97-L1m in relieving Rb-mediated repression of E2F-responsive gene expression, a recombinant HCMV expressing this allele had no growth defect (Fig. 2C) (44) and showed wild-type levels of E2F-responsive gene expression (Fig. 8, A, C, and D). Our data indicate this is likely due to the viral IE1 protein (Fig. 8E). Interestingly, IE1 itself relieved Rb-mediated E2F-dependent transcriptional repression poorly (Fig. 8E) (90) and does not contain an LXCXE motif. However, IE1 has been implicated in promoting E2F protein phosphorylation (Ref. 91 and discussed above as a potential defect of the UL97-L1m protein). More work is needed to discover how IE1 and UL97-L1m cooperate to relieve Rb-mediated E2F-dependent transcriptional repression.
Finally, the role of Rb and its phosphorylation by UL97 during HCMV infection remains enigmatic. Through its ability to inhibit the transcription of E2F-responsive genes encoding nucleotide biosynthetic and DNA replication enzymes, Rb might have been considered as a potential inhibitor of HCMV viral DNA synthesis. However, we recently showed that Rb knockdown actually inhibits the accumulation of HCMV viral DNA, the late protein pp150, and progeny infectious virions (67) and that the known ability of UL97 to enhance viral DNA replication (92) appears to be independent of Rb phosphorylation.4 Perhaps identifying the Rb phosphorylation-independent positive effect of UL97 on E2F-dependent transcription observed here can illuminate not only how Rb and E2F control cell cycle progression but also how they impact HCMV infection.
Author Contributions
S. I. and R. F. K. designed the experiments. S. I. performed the experiments. M. H. and S. C. generated mutant viruses and performed growth curve analysis. S. I. and R. F. K. wrote the paper with comments from all authors.
Acknowledgments
We thank Adam Hume for generating mutant UL97 expression plasmids, Phil Balandyk and Ben Houser for expert technical assistance, Emily Albright for illustrations, and all the members of the laboratory for helpful discussions. We thank the following investigators for generous contributions of research materials: Peter D. Adams, Jim Alwine, Don Coen, Jim DeCapprio, David Gilbert, David Goodrich, Berthold Henglein, Maureen Murphy, Joe Nevins, Mark N. Prichard, Yongjun Yu, and Jean Y. J. Wang.
This work was supported, in whole or in part, by National Institutes of Health Grant AI080675 (to R. F. K.). This work was also supported by Department of Veterans Affairs Grant BX000925 (to S. C.). The authors declare that they have no conflicts of interest with the contents of this article.
C. V. Kuny and R. F. Kalejta, manuscript under revision.
- Rb
- retinoblastoma
- CDK
- cyclin-dependent kinase
- v-CDK
- viral CDK
- HCMV
- human cytomegalovirus
- IE
- immediate early
- HDAC
- histone deacetylase
- HP
- hydrophobic patch
- HFF
- human foreskin fibroblast
- Tri
- triple mutation
- Qd
- quadruple mutation
- m
- mutant
- CM
- cleft mutant.
REFERENCES
- 1. Friend S. H., Bernards R., Rogelj S., Weinberg R. A., Rapaport J. M., Albert D. M., Dryja T. P. (1986) A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 323, 643–646 [DOI] [PubMed] [Google Scholar]
- 2. Weinberg R. A. (1995) The retinoblastoma protein and cell cycle control. Cell 81, 323–330 [DOI] [PubMed] [Google Scholar]
- 3. Burkhart D. L., Sage J. (2008) Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 8, 671–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dick F. A., Rubin S. M. (2013) Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell Biol. 14, 297–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Di Fiore R., D'Anneo A., Tesoriere G., Vento R. (2013) RB1 in cancer: different mechanisms of RB1 inactivation and alterations of pRb pathway in tumorigenesis. J. Cell. Physiol. 228, 1676–1687 [DOI] [PubMed] [Google Scholar]
- 6. Indovina P., Marcelli E., Casini N., Rizzo V., Giordano A. (2013) Emerging roles of RB family: new defense mechanisms against tumor progression. J. Cell. Physiol. 228, 525–535 [DOI] [PubMed] [Google Scholar]
- 7. Nicolay B. N., Dyson N. J. (2013) The multiple connections between pRB and cell metabolism. Curr. Opin. Cell Biol. 25, 735–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hume A. J., Kalejta R. F. (2009) Regulation of the retinoblastoma proteins by the human herpesviruses. Cell Div. 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Helt A. M., Galloway D. A. (2003) Mechanisms by which DNA tumor virus oncoproteins target the Rb family of pocket proteins. Carcinogenesis 24, 159–169 [DOI] [PubMed] [Google Scholar]
- 10. Bellacchio E., Paggi M. G. (2013) Understanding the targeting of the RB family proteins by viral oncoproteins to defeat their oncogenic machinery. J. Cell. Physiol. 228, 285–291 [DOI] [PubMed] [Google Scholar]
- 11. Fera D., Schultz D. C., Hodawadekar S., Reichman M., Donover P. S., Melvin J., Troutman S., Kissil J. L., Huryn D. M., Marmorstein R. (2012) Identification and characterization of small molecule antagonists of pRb inactivation by viral oncoproteins. Chem. Biol. 19, 518–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jiang H., Gomez-Manzano C., Lang F. F., Alemany R., Fueyo J. (2009) Oncolytic adenovirus: preclinical and clinical studies in patients with human malignant gliomas. Curr. Gene Ther. 9, 422–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim M., Williamson C. T., Prudhomme J., Bebb D. G., Riabowol K., Lee P. W., Lees-Miller S. P., Mori Y., Rahman M. M., McFadden G., Johnston R. N. (2010) The viral tropism of two distinct oncolytic viruses, reovirus and myxoma virus, is modulated by cellular tumor suppressor gene status. Oncogene 29, 3990–3996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Russell S. J., Peng K. W., Bell J. C. (2012) Oncolytic virotherapy. Nat. Biotechnol. 30, 658–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moore P. S., Chang Y. (2010) Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 10, 878–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hassler M., Singh S., Yue W. W., Luczynski M., Lakbir R., Sanchez-Sanchez F., Bader T., Pearl L. H., Mittnacht S. (2007) Crystal structure of the retinoblastoma protein N domain provides insight into tumor suppression, ligand interaction, and holoprotein architecture. Mol. Cell 28, 371–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Heilmann A. M., Dyson N. J. (2012) Phosphorylation puts the pRb tumor suppressor into shape. Genes Dev. 26, 1128–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee C., Chang J. H., Lee H. S., Cho Y. (2002) Structural basis for the recognition of the E2F transactivation domain by the retinoblastoma tumor suppressor. Genes Dev. 16, 3199–3212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee J. O., Russo A. A., Pavletich N. P. (1998) Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature 391, 859–865 [DOI] [PubMed] [Google Scholar]
- 20. Rubin S. M., Gall A. L., Zheng N., Pavletich N. P. (2005) Structure of the Rb C-terminal domain bound to E2F1-DP1: a mechanism for phosphorylation-induced E2F release. Cell 123, 1093–1106 [DOI] [PubMed] [Google Scholar]
- 21. Chen D., Pacal M., Wenzel P., Knoepfler P. S., Leone G., Bremner R. (2009) Division and apoptosis of E2f-deficient retinal progenitors. Nature 462, 925–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Trimarchi J. M., Lees J. A. (2002) Sibling rivalry in the E2F family. Nat. Rev. Mol. Cell Biol. 3, 11–20 [DOI] [PubMed] [Google Scholar]
- 23. Brehm A., Miska E. A., McCance D. J., Reid J. L., Bannister A. J., Kouzarides T. (1998) Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 391, 597–601 [DOI] [PubMed] [Google Scholar]
- 24. Adams P. D. (2001) Regulation of the retinoblastoma tumor suppressor protein by cyclin/cdks. Biochim. Biophys. Acta 1471, M123–M133 [DOI] [PubMed] [Google Scholar]
- 25. Rubin S. M. (2013) Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem. Sci. 38, 12–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Burke J. R., Hura G. L., Rubin S. M. (2012) Structures of inactive retinoblastoma protein reveal multiple mechanisms for cell cycle control. Genes Dev. 26, 1156–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Malumbres M., Barbacid M. (2005) Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 30, 630–641 [DOI] [PubMed] [Google Scholar]
- 28. Satyanarayana A., Kaldis P. (2009) Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 28, 2925–2939 [DOI] [PubMed] [Google Scholar]
- 29. Adams P. D., Li X., Sellers W. R., Baker K. B., Leng X., Harper J. W., Taya Y., Kaelin W. G., Jr. (1999) Retinoblastoma protein contains a C-terminal motif that targets it for phosphorylation by cyclin-cdk complexes. Mol. Cell. Biol. 19, 1068–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schulman B. A., Lindstrom D. L., Harlow E. (1998) Substrate recruitment to cyclin-dependent kinase 2 by a multipurpose docking site on cyclin A. Proc. Natl. Acad. Sci. U.S.A. 95, 10453–10458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Baker G. L., Landis M. W., Hinds P. W. (2005) Multiple functions of D-type cyclins can antagonize pRb-mediated suppression of proliferation. Cell Cycle 4, 330–338 [PubMed] [Google Scholar]
- 32. Britt W. (2008) Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr. Top Microbiol. Immunol. 325, 417–470 [DOI] [PubMed] [Google Scholar]
- 33. Ranganathan P., Clark P. A., Kuo J. S., Salamat M. S., Kalejta R. F. (2012) Significant association of multiple human cytomegalovirus genomic loci with glioblastoma multiforme samples. J. Virol. 86, 854–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dziurzynski K., Chang S. M., Heimberger A. B., Kalejta R. F., McGregor Dallas S. R., Smit M., Soroceanu L., Cobbs C. S., and HCMV and Gliomas Symposium (2012) Consensus on the role of human cytomegalovirus in glioblastoma. Neuro Oncol. 14, 246–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Soroceanu L., Cobbs C. S. (2011) Is HCMV a tumor promoter? Virus. Res. 157, 193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mitchell D. A., Batich K. A., Gunn M. D., Huang M. N., Sanchez-Perez L., Nair S. K., Congdon K. L., Reap E. A., Archer G. E., Desjardins A., Friedman A. H., Friedman H. S., Herndon J. E., 2nd, Coan A., McLendon R. E., Reardon D. A., Vredenburgh J. J., Bigner D. D., Sampson J. H. (2015) Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 519, 366–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hume A. J., Finkel J. S., Kamil J. P., Coen D. M., Culbertson M. R., Kalejta R. F. (2008) Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science 320, 797–799 [DOI] [PubMed] [Google Scholar]
- 38. Prichard M. N., Sztul E., Daily S. L., Perry A. L., Frederick S. L., Gill R. B., Hartline C. B., Streblow D. N., Varnum S. M., Smith R. D., Kern E. R. (2008) Human cytomegalovirus UL97 kinase activity is required for the hyperphosphorylation of retinoblastoma protein and inhibits the formation of nuclear aggresomes. J. Virol. 82, 5054–5067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Penkert R. R., Kalejta R. F. (2012) Tale of a tegument transactivator: the past, present and future of human CMV pp71. Future Virol. 7, 855–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kalejta R. F., Bechtel J. T., Shenk T. (2003) Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol. Cell. Biol. 23, 1885–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kalejta R. F., Shenk T. (2003) Proteasome-dependent, ubiquitin-independent degradation of the Rb family of tumor suppressors by the human cytomegalovirus pp71 protein. Proc. Natl. Acad. Sci. U.S.A. 100, 3263–3268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Saffert R. T., Kalejta R. F. (2006) Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J. Virol. 80, 3863–3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kuny C. V., Chinchilla K., Culbertson M. R., Kalejta R. F. (2010) Cyclin-dependent kinase-like function is shared by the β- and γ-subset of the conserved herpesvirus protein kinases. PLoS Pathog. 6, e1001092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gill R. B., Frederick S. L., Hartline C. B., Chou S., Prichard M. N. (2009) Conserved retinoblastoma protein-binding motif in human cytomegalovirus UL97 kinase minimally impacts viral replication but affects susceptibility to maribavir. Virol J. 6, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shew J. Y., Lin B. T., Chen P. L., Tseng B. Y., Yang-Feng T. L., Lee W. H. (1990) C-terminal truncation of the retinoblastoma gene product leads to functional inactivation. Proc. Natl. Acad. Sci. U.S.A. 87, 6–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Prichard M. N., Gao N., Jairath S., Mulamba G., Krosky P., Coen D. M., Parker B. O., Pari G. S. (1999) A recombinant human cytomegalovirus with a large deletion in UL97 has a severe replication deficiency. J. Virol. 73, 5663–5670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chou S., Ercolani R. J., Marousek G., Bowlin T. L. (2013) Cytomegalovirus UL97 kinase catalytic domain mutations that confer multidrug resistance. Antimicrob. Agents Chemother. 57, 3375–3379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chou S. (2010) Recombinant phenotyping of cytomegalovirus UL97 kinase sequence variants for ganciclovir resistance. Antimicrob. Agents Chemother. 54, 2371–2378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chou S., Van Wechel L. C., Marousek G. I. (2006) Effect of cell culture conditions on the anticytomegalovirus activity of maribavir. Antimicrob. Agents Chemother. 50, 2557–2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chou S., Marousek G. I. (2008) Accelerated evolution of maribavir resistance in a cytomegalovirus exonuclease domain II mutant. J. Virol. 82, 246–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chou S. (2009) Diverse cytomegalovirus UL27 mutations adapt to loss of viral UL97 kinase activity under maribavir. Antimicrob. Agents Chemother. 53, 81–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yu Y., Alwine J. C. (2008) Interaction between simian virus 40 large T antigen and insulin receptor substrate 1 is disrupted by the K1 mutation, resulting in the loss of large T antigen-mediated phosphorylation of Akt. J. Virol. 82, 4521–4526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sun X., Bristol J. A., Iwahori S., Hagemeier S. R., Meng Q., Barlow E. A., Fingeroth J. D., Tarakanova V. L., Kalejta R. F., Kenney S. C. (2013) Hsp90 inhibitor 17-DMAG decreases expression of conserved herpesvirus protein kinases and reduces virus production in Epstein-Barr virus-infected cells. J. Virol. 87, 10126–10138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Prichard M. N., Britt W. J., Daily S. L., Hartline C. B., Kern E. R. (2005) Human cytomegalovirus UL97 Kinase is required for the normal intranuclear distribution of pp65 and virion morphogenesis. J. Virol. 79, 15494–15502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Qin X. Q., Chittenden T., Livingston D. M., Kaelin W. G., Jr. (1992) Identification of a growth suppression domain within the retinoblastoma gene product. Genes Dev. 6, 953–964 [DOI] [PubMed] [Google Scholar]
- 56. Sellers W. R., Novitch B. G., Miyake S., Heith A., Otterson G. A., Kaye F. J., Lassar A. B., Kaelin W. G., Jr. (1998) Stable binding to E2F is not required for the retinoblastoma protein to activate transcription, promote differentiation, and suppress tumor cell growth. Genes Dev. 12, 95–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen T. T., Wang J. Y. (2000) Establishment of irreversible growth arrest in myogenic differentiation requires the RB LXCXE-binding function. Mol. Cell. Biol. 20, 5571–5580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Johnson D. G., Ohtani K., Nevins J. R. (1994) Autoregulatory control of E2F1 expression in response to positive and negative regulators of cell cycle progression. Genes Dev. 8, 1514–1525 [DOI] [PubMed] [Google Scholar]
- 59. Ohtani K., DeGregori J., Leone G., Herendeen D. R., Kelly T. J., Nevins J. R. (1996) Expression of the HsOrc1 gene, a human ORC1 homolog, is regulated by cell proliferation via the E2F transcription factor. Mol. Cell. Biol. 16, 6977–6984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Henglein B., Chenivesse X., Wang J., Eick D., Bréchot C. (1994) Structure and cell cycle-regulated transcription of the human cyclin A gene. Proc. Natl. Acad. Sci. U.S.A. 91, 5490–5494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lee C. P., Huang Y. H., Lin S. F., Chang Y., Chang Y. H., Takada K., Chen M. R. (2008) Epstein-Barr virus BGLF4 kinase induces disassembly of the nuclear lamina to facilitate virion production. J. Virol. 82, 11913–11926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Leng X., Connell-Crowley L., Goodrich D., Harper J. W. (1997) S-phase entry upon ectopic expression of G1 cyclin-dependent kinases in the absence of retinoblastoma protein phosphorylation. Curr. Biol. 7, 709–712 [DOI] [PubMed] [Google Scholar]
- 63. Zhu H., Shen Y., Shenk T. (1995) Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J. Virol. 69, 7960–7970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. He Z., He Y. S., Kim Y., Chu L., Ohmstede C., Biron K. K., Coen D. M. (1997) The human cytomegalovirus UL97 protein is a protein kinase that autophosphorylates on serines and threonines. J. Virol. 71, 405–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nowak B., Sullivan C., Sarnow P., Thomas R., Bricout F., Nicolas J. C., Fleckenstein B., Levine A. J. (1984) Characterization of monoclonal antibodies and polyclonal immune sera directed against human cytomegalovirus virion proteins. Virology 132, 325–338 [DOI] [PubMed] [Google Scholar]
- 66. Iwahori S., Shirata N., Kawaguchi Y., Weller S. K., Sato Y., Kudoh A., Nakayama S., Isomura H., Tsurumi T. (2007) Enhanced phosphorylation of transcription factor Sp1 in response to herpes simplex virus type 1 infection is dependent on the ataxia telangiectasia-mutated protein. J. Virol. 81, 9653–9664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. VanDeusen H. R., Kalejta R. F. (2015) The retinoblastoma tumor suppressor promotes efficient human cytomegalovirus lytic replication. J. Virol. 89, 5012–5021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Albright E. R., Kalejta R. F. (2013) Myeloblastic cell lines mimic some but not all aspects of human cytomegalovirus experimental latency defined in primary CD34+ cell populations. J. Virol. 87, 9802–9812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fang F., Wang Y., Li R., Zhao Y., Guo Y., Jiang M., Sun J., Ma Y., Ren Z., Tian Z., Wei F., Yang D., Xiao W. (2010) Transcription factor E2F1 suppresses dendritic cell maturation. J. Immunol. 184, 6084–6091 [DOI] [PubMed] [Google Scholar]
- 70. Iwahori S., Kohmon D., Kobayashi J., Tani Y., Yugawa T., Komatsu K., Kiyono T., Sugimoto N., Fujita M. (2014) ATM regulates Cdt1 stability during the unperturbed S phase to prevent re-replication. Cell Cycle 13, 471–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Juckem L. K., Boehme K. W., Feire A. L., Compton T. (2008) Differential initiation of innate immune responses induced by human cytomegalovirus entry into fibroblast cells. J. Immunol. 180, 4965–4977 [DOI] [PubMed] [Google Scholar]
- 72. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔCT) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 73. Iaquinta P. J., Lees J. A. (2007) Life and death decisions by the E2F transcription factors. Curr. Opin. Cell Biol. 19, 649–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dimova D. K., Dyson N. J. (2005) The E2F transcriptional network: old acquaintances with new faces. Oncogene 24, 2810–2826 [DOI] [PubMed] [Google Scholar]
- 75. Lee C., Cho Y. (2002) Interactions of SV40 large T antigen and other viral proteins with retinoblastoma tumour suppressor. Rev. Med. Virol 12, 81–92 [DOI] [PubMed] [Google Scholar]
- 76. Kim H. Y., Ahn B. Y., Cho Y. (2001) Structural basis for the inactivation of retinoblastoma tumor suppressor by SV40 large T antigen. EMBO J. 20, 295–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dick F. A. (2007) Structure-function analysis of the retinoblastoma tumor suppressor protein—is the whole a sum of its parts? Cell Div. 2, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Baek M. C., Krosky P. M., Pearson A., Coen D. M. (2004) Phosphorylation of the RNA polymerase II carboxyl-terminal domain in human cytomegalovirus-infected cells and in vitro by the viral UL97 protein kinase. Virology 324, 184–193 [DOI] [PubMed] [Google Scholar]
- 79. Phatnani H. P., Greenleaf A. L. (2006) Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 20, 2922–2936 [DOI] [PubMed] [Google Scholar]
- 80. Bigley T. M., Reitsma J. M., Mirza S. P., Terhune S. S. (2013) Human cytomegalovirus pUL97 regulates the viral major immediate early promoter by phosphorylation-mediated disruption of histone deacetylase 1 binding. J. Virol. 87, 7393–7408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Peeper D. S., Keblusek P., Helin K., Toebes M., van der Eb A. J., Zantema A. (1995) Phosphorylation of a specific cdk site in E2F-1 affects its electrophoretic mobility and promotes pRB-binding in vitro. Oncogene 10, 39–48 [PubMed] [Google Scholar]
- 82. Xu M., Sheppard K. A., Peng C. Y., Yee A. S., Piwnica-Worms H. (1994) Cyclin A/CDK2 binds directly to E2F-1 and inhibits the DNA-binding activity of E2F-1/DP-1 by phosphorylation. Mol. Cell. Biol. 14, 8420–8431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Dynlacht B. D., Moberg K., Lees J. A., Harlow E., Zhu L. (1997) Specific regulation of E2F family members by cyclin-dependent kinases. Mol. Cell. Biol. 17, 3867–3875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kitagawa M., Higashi H., Suzuki-Takahashi I., Segawa K., Hanks S. K., Taya Y., Nishimura S., Okuyama A. (1995) Phosphorylation of E2F-1 by cyclin A-cdk2. Oncogene 10, 229–236 [PubMed] [Google Scholar]
- 85. Dynlacht B. D., Flores O., Lees J. A., Harlow E. (1994) Differential regulation of E2F transactivation by cyclin/cdk2 complexes. Genes Dev. 8, 1772–1786 [DOI] [PubMed] [Google Scholar]
- 86. Marschall M., Feichtinger S., Milbradt J. (2011) Regulatory roles of protein kinases in cytomegalovirus replication. Adv. Virus Res. 80, 69–101 [DOI] [PubMed] [Google Scholar]
- 87. Li R., Zhu J., Xie Z., Liao G., Liu J., Chen M. R., Hu S., Woodard C., Lin J., Taverna S. D., Desai P., Ambinder R. F., Hayward G. S., Qian J., Zhu H., Hayward S. D. (2011) Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell Host Microbe 10, 390–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ptacek J., Devgan G., Michaud G., Zhu H., Zhu X., Fasolo J., Guo H., Jona G., Breitkreutz A., Sopko R., McCartney R. R., Schmidt M. C., Rachidi N., Lee S. J., Mah A. S., Meng L., Stark M. J., Stern D. F., De Virgilio C., Tyers M., Andrews B., Gerstein M., Schweitzer B., Predki P. F., Snyder M. (2005) Global analysis of protein phosphorylation in yeast. Nature 438, 679–684 [DOI] [PubMed] [Google Scholar]
- 89. Morris E. J., Dyson N. J. (2001) Retinoblastoma protein partners. Adv. Cancer Res. 82, 1–54 [DOI] [PubMed] [Google Scholar]
- 90. Poma E. E., Kowalik T. F., Zhu L., Sinclair J. H., Huang E. S. (1996) The human cytomegalovirus IE1–72 protein interacts with the cellular p107 protein and relieves p107-mediated transcriptional repression of an E2F-responsive promoter. J. Virol. 70, 7867–7877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Pajovic S., Wong E. L., Black A. R., Azizkhan J. C. (1997) Identification of a viral kinase that phosphorylates specific E2Fs and pocket proteins. Mol. Cell. Biol. 17, 6459–6464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wolf D. G., Courcelle C. T., Prichard M. N., Mocarski E. S. (2001) Distinct and separate roles for herpesvirus-conserved UL97 kinase in cytomegalovirus DNA synthesis and encapsidation. Proc. Natl. Acad. Sci. U.S.A. 98, 1895–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]