

Background: PDE12 degrades 2′,5′-oligoadenylate, a second messenger involved in the antiviral action of interferon.
Results: Inactivation of the PDE12 gene and novel inhibitors of the enzyme render cells resistant to more than one virus.
Conclusion: PDE12 negatively regulates the innate immune response, and inhibitors of PDE12 have antiviral activity.
Significance: PDE12 inhibitors have the potential to be broadly acting antiviral medicines.
Keywords: antiviral agent, gene knockout, innate immunity, interferon, protein structure
Abstract
2′,5′-Oligoadenylate synthetase (OAS) enzymes and RNase-L constitute a major effector arm of interferon (IFN)-mediated antiviral defense. OAS produces a unique oligonucleotide second messenger, 2′,5′-oligoadenylate (2–5A), that binds and activates RNase-L. This pathway is down-regulated by virus- and host-encoded enzymes that degrade 2–5A. Phosphodiesterase 12 (PDE12) was the first cellular 2–5A- degrading enzyme to be purified and described at a molecular level. Inhibition of PDE12 may up-regulate the OAS/RNase-L pathway in response to viral infection resulting in increased resistance to a variety of viral pathogens. We generated a PDE12-null cell line, HeLaΔPDE12, using transcription activator-like effector nuclease-mediated gene inactivation. This cell line has increased 2–5A levels in response to IFN and poly(I-C), a double-stranded RNA mimic compared with the parental cell line. Moreover, HeLaΔPDE12 cells were resistant to viral pathogens, including encephalomyocarditis virus, human rhinovirus, and respiratory syncytial virus. Based on these results, we used DNA-encoded chemical library screening to identify starting points for inhibitor lead optimization. Compounds derived from this effort raise 2–5A levels and exhibit antiviral activity comparable with the effects observed with PDE12 gene inactivation. The crystal structure of PDE12 complexed with an inhibitor was solved providing insights into the structure-activity relationships of inhibitor potency and selectivity.
Introduction
Conventional antiviral drugs target essential viral proteins such as the RNA or DNA polymerase or protease. Such agents have provided highly effective medicines for pathogens such as human immunodeficiency virus and hepatitis C. Drawbacks to these types of direct acting antiviral agents can be the emergence of resistance or efficacy against a narrow range of strains within a divergent family. Alternative approaches that augment host innate defense or target essential host pathways required by the virus may provide benefit by increasing the barrier to resistance or by acting against a broad array of viral pathogens. The discovery of agents with activity against multiple viral pathogens is particularly appealing to help combat the threat of emerging diseases like pandemic influenza, severe acute respiratory syndrome, Middle East respiratory syndrome, and Ebola (1).
The first line of defense against viral infections occurs within the epithelial cell layer invaded by the pathogen. This function is regulated in mammalian cells through the biological activity of type 1 interferon (IFN). The enzymes oligoadenylate synthetase (OAS)2 and RNase-L constitute one of the major effector pathways of IFN action (2–4). IFN induces the expression of certain OAS enzymes. The activity of OAS is tightly regulated at the post-translational level by the binding of pathogen-associated double-stranded RNA intermediates. Once activated, OAS polymerizes ATP in a template-independent fashion to a length of 4–10 nucleotides. Interestingly, OAS produces 2′,5′-linked adenylate polymers (2–5A) as opposed to the 3′,5′-linkage found in typical nucleic acids like mRNA and DNA. RNase-L normally exists in an inactive monomeric state. 2–5A binds to an ankyrin repeat regulatory domain that triggers the dimerization and activation of the RNase. RNase-L promotes an antiviral state within the cell through direct cleavage of viral genomes and intermediates as well as through the regulation of specific host mRNAs. High levels of prolonged RNase-L activity lead to cell death (5), so it is important that the enzyme be tightly regulated. In addition to regulating the synthesis of 2–5A through OAS, host cells down-regulate the ligand through cleavage by 2′,5′-specific phosphodiesterase (2′5′-PDE) (6, 7).
Viral pathogens have evolved several mechanisms to overcome the OAS/RNA-L pathway (4, 8). For example, a conserved region in the RNA of poliovirus binds directly to RNase-L and inhibits substrate cleavage in a competitive manner (9). RNase-L is also inhibited by virus-encoded proteins such as the L* protein of Theiler's virus (10). OAS activation is prevented by the influenza A NS1 protein that binds to virus double-stranded RNA blocking the activation of 2–5A synthesis (11). Members of the coronavirus and rotavirus families have been shown to encode their own 2′5′-PDEs (12, 13). Thus, inhibition of RNase-L activity and inhibition of RNase-L activation through regulation of 2–5A appear to be major mechanisms for pathogen survival. Consequently, overcoming pathogen inhibition of this pathway may provide broadly acting antiviral therapies.
Although some viruses encode their own 2′5′-PDEs (14), others may be dependent upon host cell enzymes to down-regulate RNase-L. Several host proteins have been shown to possess 2–5A-PDE activity, including AKAP7, ENPP1, and PDE12. AKAP7 was identified as a potential host 2′5′-PDE due to sequence homology with coronavirus NS2 and rotavirus VP3 proteins that belong to the HH family of nucleotide phosphodiesterases (15). Indeed, mouse AKAP7 was able to complement a defective mouse hepatitis virus NS2 gene to restore infectivity (16). This required deletion of the natural AKAP7 nuclear localization sequence to promote accumulation in the cytoplasm. PDE12 was the first 2′5′-PDE to be purified and sequenced (17). Small interfering RNA (siRNA) knockdown of PDE12 and an inhibitor of PDE12 were found to inhibit the replication of vaccinia virus in tissue culture (17). PDE12 belongs to the exonuclease-endonuclease-phosphatase (EEP) family of nucleases with members that are involved in regulating mRNA through cleavage of mRNA polyadenylate tails (18). ENPP1 was found to be a potential 2′5′-PDE through evaluation of EEP nuclease genes that increased 2–5A degrading activity in cell lysates following transient transfection (19). The role of these host 2′5′-PDEs in regulating viral infectivity is poorly understood. Analysis of mRNA expression databases indicates that PDE12 is likely to be abundantly expressed in tissues and cell lines. Moreover, PDE12 was purified from bovine spleen as the only major 2′5′-PDE detected in the tissue homogenate (17). Therefore, we chose to evaluate the role of PDE12 in the regulation of cellular 2–5A and viral infectivity.
We report that HeLa cells containing TALEN-mediated PDE12 gene inactivation have increased IFN induced 2–5A and resistance to viral infection. We identified a potent, selective compound series that inhibits PDE12 in a 2–5A competitive fashion. Treatment of host cells with these inhibitors mimics gene inactivation. The first crystal structure of apo-PDE12 and a structure of PDE12 bound to an inhibitor are presented. Further, refinement of this series may lead to promising broadly acting antiviral agents that augment host innate immune defense.
Experimental Procedures
Inactivation of the PDE12 Gene
A PDE12-deficient cell line was generated in a HeLa cell background using TALEN nuclease-directed genomic breaks (Transposagen Biopharmaceuticals, Inc., Lexington, KY). Nonhomologous end-joining of these breaks generates small deletions leading to frameshift mutations (20). A cell clone with frameshift mutations in all alleles of PDE12 was desired, although the exact number of alleles was unknown. Two TALEN-encoding cDNA constructs were designed to cleave the first exon of the human PDE12 gene downstream of the last alternate ATG start codon at base pair 646 (Homo sapiens chromosome 3, GRCh38 Primary Assembly, NCBI Reference Sequence: NC_000003.12). The target sites were as follows with the TALEN-binding sites underlined: (a) TCTTCTTCTTGGACTGAGACTGATGTGGAGGAGCGTGTCTACACCCCGTCCA; (b) TAGAGGCTGGGCCTGGCACCTGCACTTTTGACCACCGGCATCTCTACACGA. The TALEN constructs were co-transfected into HeLa cells (American Type Culture Collection, CCL-2). After 1 week of culture, the transfection was repeated, and the population was sorted to single cells and expanded for screening. Genomic DNA from candidate clones was isolated and subjected to quantitative PCR using a probe containing the targeted region of the TALENs. Clones with no quantitative PCR signal were further characterized. For these clones, a standard PCR using primers that span the region targeted by the TALENs was conducted to amplify the genomic DNA. Several bacterial clones from each cell clone were obtained and sequenced to determine the nature of the genomic alteration at the cut site. One cell clone was identified that showed four possible frameshift mutations in the PDE12 gene. This clone was selected for further genomic sequencing and Western blot analysis. PDE12 protein levels were determined using anti-PDE12 (Abcam, catalog no. ab87738, Cambridge, UK) at a 1:1000 dilution loading 40 μg of total cell protein per lane. This antibody was generated using a synthetic peptide representing PDE12 residues 560–609.
Determination of Cellular 2–5A and RNase-L Activation
Functional 2–5A was measured in cell lysates using purified RNase-L as a biosensor as described (21). To induce 2–5A production, cells were treated with IFNα and the double-stranded RNA mimetic polyinosinic-polycytidylic acid (poly(I-C)). IFNα (Sigma) at 15 units/ml was added to HeLa cells in Minimum Essential Media, 10% fetal bovine serum, nonessential amino acids, glutamine, sodium pyruvate, penicillin, and streptomycin. The following day, the cells were detached from the flask with Cell Dissociation Buffer (Gibco, Life Technologies, Inc.), counted, and diluted into antibiotic-free media to 500,000 cells/ml. The transfection mixture containing 0.8 μg/ml poly(I-C) (Sigma) and 5 μl/ml Lipofectamine 2000 in Opti-MEM (Gibco, Life Technologies, Inc.) media was mixed one to one with the cells, and 20 μl of this mixture was distributed to each well of a 384-well cell culture plate (Greiner Bio-One, Monroe, NC). The plate was placed in a 37 °C incubator for 3 h. The transfection mixture was aspirated, and 25 μl of ice-cold cell lysis buffer, 20 mm Tris (pH 7.5), 1% Triton X-100, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 2.5 mm sodium pyrophosphate, 1 mm sodium vanadate, 8 mm sodium fluoride was added to each well. 2–5A in the lysate was detected based upon the ability to activate purified RNase-L. The lysate was diluted 1 to 120 into RNase-L assay buffer, 25 mm HEPES (pH 7.5), 10 mm MgCl2, 100 mm NaCl, 0.5 mm CHAPS, 1 mm TCEP, 1 μm ATP, 50 μg/ml BSA and 5 μl was transferred to a 384-well assay plate (Greiner Bio-one). Purified RNase-L was diluted in assay buffer to 1.5 nm, and 5 μl was added to the diluted lysate and incubated at room temperature for 10 min. The reaction was started by adding 5 μl of RNase-L substrate, 5′-fluorescein phosphoramidite-UUAUCAAAUUCUAUUUGCCCCAUUUUUUUGGUUUA-black hole quencher 1®-3′ (Integrated DNA Technologies, Coralville, IA), diluted in assay buffer to 300 nm. Cleavage of the substrate was detected as a fluorescence increase at 530 nm (485 excitation) in an Analyst GT plate reader (Molecular Devices, Sunnyvale CA). The concentration of 2–5A in the lysate was estimated by comparing the rate of substrate cleavage to that produced by a standard curve of purified 2–5A.
RNase-L activation in whole cells was estimated by analyzing rRNA using an Agilent 2100 Bioanalyzer and RNA 6000 Nano LabChip kit (Agilent Technologies, Santa Clara CA). HeLa cells were plated at a density of 400,000 cells per well in a 12-well plate. The cells were treated with IFN and poly(I-C) for 2 h as described above in the presence or absence of PDE12 inhibitors. Total cellular RNA was extracted and analyzed following the manufacturer's recommended procedure.
Virus Infection Imaging Assays
Infection of small-airway epithelial cells (SAEC, Lonza, Basel, Switzerland, catalog no. SUI CC-2547) by human rhinovirus type A strain 16 (ATCC catalog no. VR-283HRV16) was measured by quantitative immunofluorescence microscopy using an antibody against HRV capsid protein (QED Bioscience, San Diego, catalog no.18758). SAEC were diluted into small airway cell growth media (SUI, Lonza, Basel, Switzerland) to a concentration of 100,000 cells per ml. HRV16 was added to the diluted cells at the indicated multiplicity of infection (m.o.i.). For compound testing the m.o.i. was typically 100. 50 μl of the infected cells were added to each well of a 384-well cell culture plate (Greiner Bio-one) and incubated at 33 °C, 5% CO2 for 5 days. The cells were fixed by adding 50 μl per well of Histochoice (Sigma) followed by sequential 50-μl treatments for 1 min each with 30, 60, and then 95% ethanol (Sigma). Plates were blocked for 1 h with Dulbecco's phosphate-buffered saline (DPBS) plus 5% goat serum (Gibco, Life Technologies, Inc.) and then incubated for 1 h with anti-HRV antibody diluted 1:1000 in DPBS plus 5% goat serum. All subsequent steps were conducted in 50 μl per well of DPBS plus 0.3% Triton X-100. The plates were washed three times (Sigma) and then incubated for 1 h with goat anti-mouse DyLight 594 labeled secondary antibody diluted 1:500 (Abcam, Cambridge MA, catalog no. AB96881). The plates were washed three times and then incubated for 10 min with HCS cell mask green diluted 1:15,000 (Invitrogen, Life Technologies, Inc.) and Hoechst 33342 diluted 1:10,000 (Invitrogen, Life Technologies, Inc.). Three final washes were conducted, and then the plates were imaged using an IN Cell Analyzer 2000 System (GE Healthcare). Images were collected for three IN Cell 2000 channel settings, nuclei (Hoechst), cell boundary (HCS cell mask green), and HRV capsid protein (DyLight 594) using a ×10 objective. Images were segmented using IN Cell Analyzer software. Cell boundaries were determined from the HCS cell mask image. The fraction of cells infected by HRV was determined as follows: median per cell of Texas Red signal in no HRV control wells was used to calculate a background cutoff value. All cells above the median background plus three median absolute deviations were counted as infected, and all cells below the background plus three median absolute deviations were counted as uninfected. Total cell number was determined by counting the number of nuclei visible in the DAPI channel.
RSV infection of SAEC was determined in a similar fashion with the following exceptions: RSV strain long (ATCC catalog no. VR-26) was used for infection at an m.o.i. of 0.01. Incubation following infection was 4 days. Infected cells were detected using an antibody to the RSV G glycoprotein at 1:1000 (Millipore, Darmstadt, Germany, catalog no. MAB858-2). HRV and RSV infection of HeLa and HeLaΔPDE12 cells were conducted and analyzed as above only using the indicated cell line, m.o.i., and time of incubation described in the figure legends.
EMCV Cytopathic Effect Assays (CPE)
For compound studies, HeLa Ohio cells (European Collection of Cell Cultures, catalog no. 84121901) were cultured in DMEM (Invitrogen, Life Technologies, Inc., catalog no. 12430), 10% HI FBS 1× Pen-Strep, 2 mm GlutaMAX (Gibco, Life Technologies, Inc.). The cells were suspended at a density of 20,000 cells per ml and mixed with EMCV (Advanced Bioscience Laboratories, Rockville, MD, catalog no. 7022) at m.o.i. of 0.01. 50 μl of this mixture was added to each well of a 384-well tissue culture-treated plate (Costar, Washington, D. C.). Cells without virus were used as an uninfected control. The plates were placed in a humidified incubator at 37 °C, 5% CO2 for 3 days, and the number of cells was determined using CellTiter Glo (Promega, Madison, WI). Evaluation of the effect of PDE12 gene inactivation was conducted in similar fashion only using HeLa and HeLaΔPDE12 at the indicated amount of virus.
Enzyme Expression and Purification
The following constructs were cloned in pET-24 vectors (Addgene, Cambridge, MA) by PCR-amplified oligonucleotide assembly (22). All of the sequences were human as follows: (a) PDE12(17–609), PDE12 amino acids 17–609 (NP_808881.3) fused to a FLAG tag (23), TEV protease cleavage site (24), and a C-terminal His6 tag; (b) PDE12(155–609), N-terminal His6 tag fused to TEV protease cleavage site and PDE12 amino acids 155–609; (c) PDE12(155–609Δ206–233), identical to PDE12 (155–609) with residues 206–233 deleted and Ala, Gly, Ser, inserted as a spacer at residue 206; (d) CNOT6(153–557), N-terminal His6 tag fused to TEV protease cleavage site and CNOT6 amino acids 153–557 (NP_056270.2); and (e) RNase-L(21–723), RNase-L amino acids 21–723 (NP_066956) followed by an AviTag (25), TEV protease cleavage site, FLAG tag, and a C-terminal His6 tag. All plasmid sequences were verified by DNA sequence analysis. Protein purification described below was conducted using the AKTA express system (GE Healthcare). Each protein was purified to greater than 90% purity as determined by stained SDS-PAGE analysis. Protein mass was determined by intact protein mass spectrometry.
The plasmid encoding PDE12(17–609) was transformed into Escherichia coli BL21(DE3) cells. Transformed cells were grown in 2-liter shake flasks in Luria-Bertani broth (Life Technologies, Inc.) at 37 °C to A600 = 0.8 and induced with 0.5 mm isopropyl β-d-1-thiogalactopyranoside. Incubation was continued overnight at 20 °C. Cells were pelleted and resuspended in 30 mm imidazole (pH 8.0), 150 mm NaCl, 10% glycerol, and lysed by sonication. The lysate was clarified by centrifuging at 24,000 × g, for 30 min at 4 °C, and the supernatant was loaded onto ProBond nickel affinity resin (Invitrogen, Life Technologies, Inc.). The resin was washed with 55 mm imidazole (pH 8.0), 150 mm NaCl, 10% glycerol, and the protein was eluted with a gradient of 55–500 mm imidazole. Fractions containing the expressed protein were determined by SDS-polyacrylamide gel staining with Gel-code Blue (Pierce, Life Technologies, Inc.). Fractions were pooled, and the NaCl concentration was reduced to 50 mm by dilution using 20 mm Tris-HCl (pH 8.0). The pooled fractions were loaded onto SP-Sepharose (GE Healthcare). The resin was washed with 20 mm Tris-HCl (pH 8.0), 30 mm NaCl, and the protein was eluted from the resin using a 30 mm to 1 m NaCl gradient. The fractions containing the PDE12 protein were pooled, and DTT and glycerol were added to final concentrations of 5 mm and 5%, respectively.
For crystallization, PDE12(155–609) protein was expressed in BL21(DE3) cells, lysed, and purified on ProBond nickel resin as described above. The fractions containing PDE12 protein were pooled, and the His6 tag was removed by TEV protease digestion overnight at 4 °C. The pooled fractions were dialyzed against 20 mm Tris-HCl (pH 8.0), 150 mm NaCl and passed across a second nickel affinity column to remove the cleaved tag. Unbound protein was pooled, concentrated, and applied to a HiLoad 26/60 Superdex 200 gel filtration column (GE Healthcare) equilibrated with 50 mm Tris-HCl (pH 8.0), 100 mm NaCl. Fractions containing the expressed protein were pooled, and DTT was added to a final concentration of 5 mm before concentrating the protein to 40 mg/ml. CNOT6(153–507) was expressed and purified as described above.
The plasmid encoding RNase-L was transformed into BL21(DE3) cells, and a 1-liter inoculum was added to a 20-liter fermenter (Applikon, Delft, The Netherlands) in 2× YT media, 0.5% glycerol to an initial A600 = 0.1. Growth was conducted at 37 °C, 20% dissolved oxygen, with stirring at 300–900 rpm. RNase-L expression was induced with 0.4 mm isopropyl β-d-1-thiogalactopyranoside at A600 = 3. Glycerol (5 g/liter) was added, and the temperature was reduced to 25 °C. Cells were harvested by centrifugation at A600 = 10. Purification was conducted with 50 g of cell paste resuspended in 500 ml of 25 mm HEPES (pH 7.4), 300 mm NaCl and lysed by two passages at 12,000 pounds per square inch through a Rannie pressure homogenizer (APV). The lysate was clarified by centrifugation at 20,000 × g for 45 min. The supernatant was loaded onto a 5-ml HisTrap column (GE Healthcare) equilibrated with base buffer, 25 mm HEPES (pH 7.4), 300 mm NaCl, 10% glycerol, 1 mm CHAPS, 50 mm imidazole. The bound protein was eluted with a 50–500 mm imidazole gradient. Fractions containing RNase-L were pooled and TCEP was added to 1 mm. The pooled sample was loaded onto a 5-ml HiTrap heparin column (GE Healthcare) equilibrated with base buffer and eluted with a gradient from 300 mm to 2 m NaCl. The pooled fractions were applied to a HiLoad 26/60 Superdex 200 gel filtration column (GE Healthcare) equilibrated with base buffer. RNase-L was eluted in two peaks, a higher molecular weight aggregate and a monomeric species. Monomeric fractions were pooled and stored at −80 °C.
Crystallization and Structure Solution of PDE12
PDE12 proteins were concentrated to 40 mg/ml in 20 mm Tris (pH 8.0), 500 mm NaCl, and 5 mm DTT. The protein or protein-inhibitor complex (1:5) was incubated on ice for 2 h. Crystals were grown by sitting-drop vapor diffusion. 600-nl drops were set with a Mosquito instrument (TTP LabTech; Melbourne, UK) in MRC 2-well Crystallization plates (Swissci; Zug, SUI) by combining 300 nl of PDE12 protein or protein-inhibitor complex with 300 nl of crystallization buffer. Initial apo-crystals were grown with the PDE12(155–609) in 0.2 m lithium acetate and 20% PEG3350. Apo-crystals of PDE12(155–609Δ206–233) were grown at 4 °C in 0.2 m MgCl2, 0.1 m Tris-HCl (pH 8.5), and 24% PEG3350. Inhibitor-bound crystals of PDE12(155–609Δ206–233) were grown at 22 °C in 0.1 m Tris (pH 8.5), 15% PEG20000, and 25% ethylene glycol. Crystals were harvested and cryo-protected with 25% glycerol, paraton, or 25% ethylene glycol followed by submersion in liquid nitrogen. Data were collected at beamline CLS-08-ID-1 on a Rayonics MX-300CCD detector for the initial apo-structure and 21-ID-F on a Rayonics MX225CCD detector for the loop-deleted structure. The inhibitor-bound structure was collected in-house on a FRE+ generator using a Saturn 944+ detector. All data were processed using HKL2000 (26).
The initial apo-PDE12 structure was solved in space group P212121 by molecular replacement using the program Phaser (27) as implemented in the Phenix software suite (28). A 279-residue portion of CNOT6 (Protein Data Bank code 3NGO) was used as the MR model that spanned residues 29–383 but included several major deletions, including all of residues 45–52, 111–122, 179–193, 233–246, and 278–301. The missing regions were built using the program Coot (29) by successive rounds of refinement in Autobuster (30) interleaved with DM averaging in the CCP4 suite (31) and manual building. Phenix autosolve was also used to assist with the building of the additional domain and to confirm manual building. A total of 441 residues were built for each of two copies of the protein in the asymmetric unit. Final refinements were completed using the Refmac program (32). Subsequent higher resolution structures were solved from this initial model similarly using Phaser for molecular replacement and iterative cycles of Buster and Coot.
Encoded Library Technology (ELT) Selections
DNA-tagged compounds present in our ELT library were selected for binding to purified PDE12 proteins essentially as described (33, 34). The indicated proteins were immobilized through the His6 tag to IMAC tips (PhyNexus, San Jose, CA). Before use, tips were prewashed in selection buffer, 5 mm MgCl2, 10 mm imidazole, 25 mm HEPES (pH 7.5), 100 mm NaCl, 1 mm TCEP with 1 mg/ml BSA (Ambion, Life Technologies, Inc.), and 0.1 mg/ml sheared salmon sperm DNA (Ambion, Life Technologies, Inc.). 20 μg of PDE12(272–609) and 10 μg of PDE12(17–609) were immobilized for 20 min at room temperature on the prewashed IMAC tips, and the tips were washed five times with selection buffer to remove excess protein. The ELT library (5 nmol) was diluted in 100 μl of selection buffer and passed over the IMAC tip for 1 h at room temperature. The tip was washed three times with selection buffer and two times with DNA and BSA-free selection buffer. Bound compounds were eluted by passing DNA- and BSA-free selection buffer heated to 72 °C over the tip for 10 min. The eluted material was passed over a fresh IMAC tip for 15 min at room temperature to remove any denatured protein. This procedure was repeated in the second round and analyzed in the third round. The DNA tags on the bound compounds were defined by sequencing using a Genome Analyzer II system (Illumina, San Diego). The sequence data describing the bound compounds were decoded to determine the chemical features of compounds that have affinity to PDE12.
PDE12 and CNOT6 Enzyme Assays
2–5A cleavage assays were conducted using substrate produced with purified porcine OAS1 and ATP as described (35). The enzymatic synthesis generates 5′-triphosphate-2′,5′-linked adenylate oligomers from 2 to 5 nucleotides in length. The different oligomers were separated by HPLC and identified by mass spectroscopy. The peak corresponding to the trimer pppA2′p5′A2′p5′A was pooled and is henceforth referred to as 2–5A substrate. PDE12(17–609) and CNOT6(153–507) cleave 2–5A into two AMP monomers and one ATP. The AMP product was detected using AMP-Glo (Promega). Assays were performed in 384-well low volume black plates (Greiner Bio-One) with compound present prior to reagent addition. The assay buffer was 25 mm HEPES (pH 7.5), 5 mm MgCl2, 100 mm NaCl, 0.5 mm CHAPS, 1 mm TCEP, 130 μg/ml nuclease-free BSA. Two μl of substrate mixture containing 10 μm 2–5A in assay buffer mixed 1 to 1 with AMP-Glo reagent was added to each well. Two μl of enzyme mixture containing 0.5 nm PDE12 or 2 nm CNOT6 in assay buffer was dispensed into each well, and the plate was incubated for 30 min at room temperature. The reaction was terminated with 2 μl of AMP detection reagent, and the plate was incubated at room temperature for 45–60 min. Luminescence was measured using a Viewlux (PerkinElmer Life Sciences).
Compound Handling, Kinetic Studies, and Data Analysis
All compounds were dissolved from solid stocks in DMSO. For IC50 determination, primary data were normalized and expressed as % inhibition as shown in Equation 1,
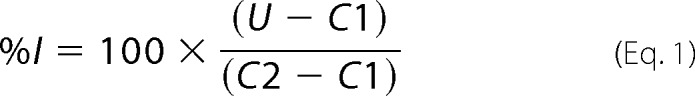 |
where U is the assay result in the presence of compound; C1 is the average of the high signal in the absence of compound, and C2 is the average of the low signal obtained with an appropriate negative control (no virus for viral assays, or no enzyme for enzyme assays). For IFN-poly(I-C)-induced 2–5A increase assays, the data were normalized and expressed % activation (%A) as shown in Equation 2,
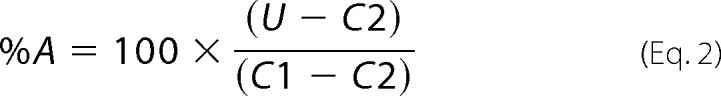 |
where U is the assay result in the presence of compound; C1 is the assay signal from IFN-poly(I-C)-treated cell lysates without compound; and C2 is the assay background (no added lysate). IC50 or EC50 values were determined by plotting the compound concentration (log M) versus %I or %A and fitting the four-parameter logistic equation to the normalized data as shown in Equation 3,
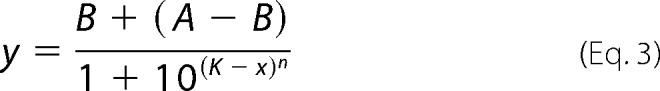 |
where y = %I or %A; B = minimum asymptote; A = maximum asymptote; x = compound concentration; K = log IC50 or log EC50; and n represents the slope factor of the inhibition curve (typically close to 1). The results for each compound are recorded as pIC50 or pEC50 values (−K from the above equation). All compounds were tested in each assay at least three times, and the reported results represent the mean ± S.D.
Steady state substrate and inhibitor kinetic experiments were conducted in triplicate. The initial rate of AMP production catalyzed by PDE12 or CNOT6 was determined from a linear fit of product formed as a function of time from the 20-min time course experiments read every 2 min. The initial rate was replotted versus the substrate concentration (Fig. 6). Vmax and Km values were determined by fitting Equation 4 to the initial rates,
 |
where v = initial rate; [S] = concentration of 2–5A or oligoA; Vmax = maximum velocity; and Km = apparent Michaelis constant. kcat value was calculated from Vmax based on the following relationship: Vmax = kcat × [E], where [E] is the concentration of enzyme used in the experiment based upon the protein concentration and purity estimate.
FIGURE 6.
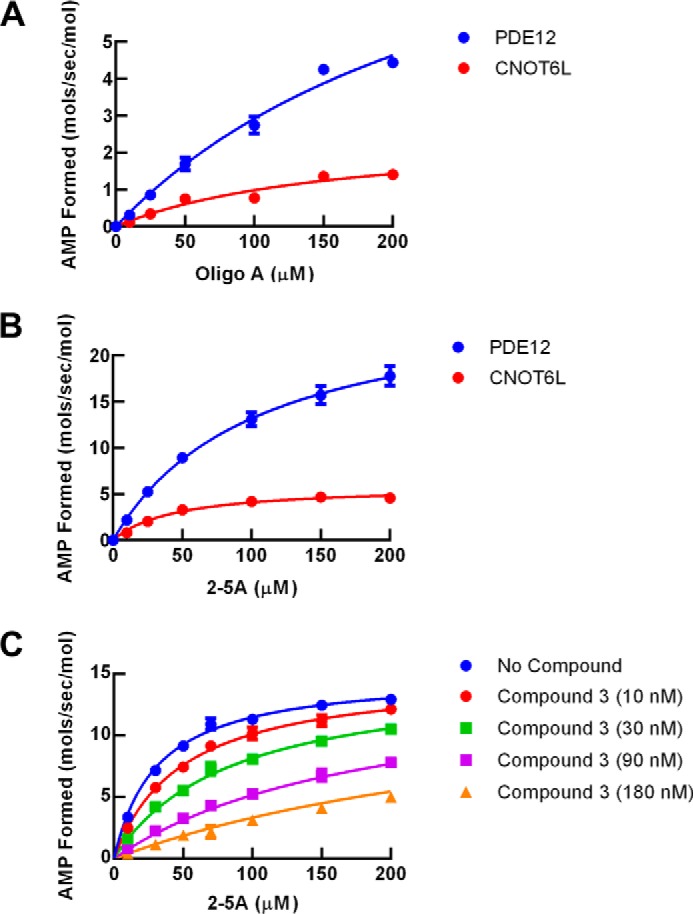
Kinetic characterization of PDE12 and CNOT6. Substrate and inhibitor steady state kinetic studies were determined using the indicated enzyme and nucleic acid substrate. AMP formed was determined as a function of time from 20-min progress curves, and the initial rates were replotted as shown. Kinetic parameters determined from these experiments are summarized in Table 1. A, reaction rate as a function of 2–5A concentration. Blue, PDE12; red, CNOT6. Lines represent fit of the data points to Equation 4. B, reaction rate as a function of oligoA; blue, PDE12; red, CNOT6. Line represents fit of the data points to Equation 3. C, inhibition of PDE12 by compound 3 as a function of 2–5A concentration. Blue, no compound; red, 10 nm compound 3; green, 30 nm compound 3; purple, 90 nm compound 3; orange, 180 nm compound 3. Lines represent a global fit of the data points to Equation 4 for 2–5A competitive inhibition.
The mode of inhibition analysis for compound 3 was conducted as described above at four fixed concentrations of inhibitor. The mode of inhibition with respect to 2–5A substrate was determined by fitting equations describing competitive (Equation 5), noncompetitive (Equation 6), and uncompetitive (Equation 7) modes of inhibition to the initial rates (36).
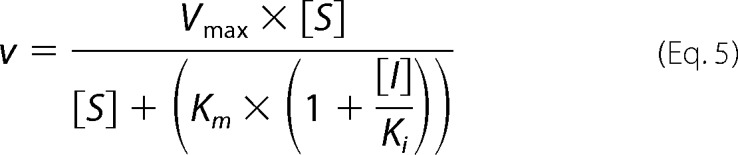 |
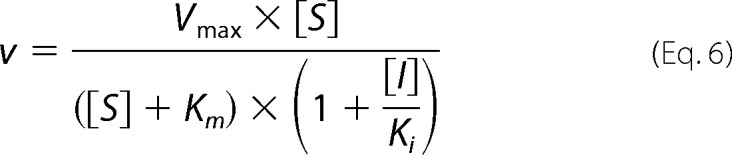 |
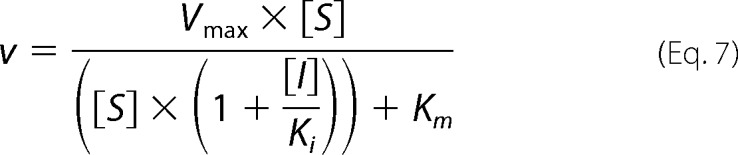 |
Nonlinear regressions were conducted using the program GraphPad Prism® (GraphPad Software, San Diego) for enzyme kinetic studies (Equations 4–7). Normalization and pEC50 determinations were conducted using ActivityBase XE® (IDBS, London, UK).
Results
Inactivation of PDE12 Gene Alleles in HeLa Cells
HeLa cells were chosen for PDE12 gene inactivation because they serve as a good host for a variety of viruses. Targeted short genomic deletions were created by multiple rounds of transient transfection of TALEN constructs. Clonal cells were obtained, and one cell line was selected that appeared to have four separate PDE12 alleles each containing a distinct out-of-frame deletion in the targeted region of exon 1. To confirm this change, the complete PDE12 gene was subjected to PCR amplification followed by Ion Torrent AmpliseqTM (Life Technologies, Inc.) sequencing. Approximately 2000 complete individually amplified PDE12 sequences were obtained from the line. Four different out-of-frame gene deletions were found with no wild-type sequence detected (Fig. 1A). These results confirm that the parent HeLa cells are tetraploid for the PDE12 locus and that the clone contained a frameshift mutation in the targeted coding region for each allele. Western blot analysis was conducted to confirm lack of PDE12 expression. Full-length PDE12 protein was readily detected in the parent HeLa line but not in the deletion clone (Fig. 1B). This line is subsequently referred to as HeLaΔPDE12.
FIGURE 1.

Characterization of frameshift mutations in HeLaΔPDE12 cell line. A, WT, structure of wild-type PDE12 mRNA. Exon 1, light blue; Exon 2, dark blue; Exon 3, purple; coding sequence of EEP nuclease domain, green bar; TALEN nuclease-targeted sequence, red. nt = nucleotide number; M1 = N-terminal methionine; T276 = threonine at the beginning of the catalytic domain. Alleles 1–4, length and positions of the first deleted nucleotide are shown in red; length of open reading frame is shown by a black arrow. Last encoded amino acid residue listed above. B, Western blot detection of PDE12. Lane 1, 40 μg of total cellular protein from HeLa cells. Lane 2, 40 μg of total cellular protein from HeLaΔPDE12 cells.
Effects of PDE12 Gene Inactivation on Cellular 2–5A Levels
Purified PDE12 has been shown to cleave 2–5A in vitro (17, 19), but it is unknown whether the enzyme controls cellular 2–5A. OAS1 activation was induced in two steps in HeLa cells and in HeLaΔPDE12. The cells were treated with IFNα overnight to induce OAS expression. Following IFNα treatment, the cells were transfected with the double-stranded RNA mimetic, poly(I-C), to activate OAS. The cells were lysed, and 2–5A in the lysate was quantified using purified RNase-L as a sensor. The HeLaΔPDE12 cells contained ∼5 times more 2–5A than the parental HeLa cells (Fig. 2A).
FIGURE 2.

Effect of PDE12 gene inactivation on cellular 2–5A levels and viral infection. HeLa (blue) or HeLaΔPDE12 (red) cells were evaluated as follows. A, 2–5A levels in lysates of cells treated with interferon-α and poly(I-C) are shown. B, cytopathic effect after 3 days of EMCV infection as a function of virus concentration was determined. C, fraction of cells infected by HRV16 was determined by evaluating cells for the presence of HRV capsid protein by immunofluorescence and imaging at the indicated m.o.i. and time of incubation for infection. D, fraction of cells infected by RSV-L was determined by evaluating cells for the presence of RSV G protein by immunofluorescence and imaging at the indicated m.o.i. and time of incubation for infection.
Effects of PDE12 Gene Inactivation on Viral Infection
The OAS/RNase-L pathway has been shown to impact viruses with RNA positive-strand genomes such as EMCV, hepatitis C, and West Nile (2, 4). Thus, we evaluated the effect of PDE12 gene inactivation on EMCV and HRV, which have RNA positive-strand genomes and replicate in HeLa cells. EMCV infection was determined indirectly by measuring virus-induced CPE. HeLa CPE after 3 days of infection was dependent on the amount of virus (Fig. 2B). The fraction of cells remaining as a function of m.o.i. for each cell line was fit to a four-parameter logistic curve (see Equation 3). The inflection point (m.o.i. EC50) for HeLa cell killing was (2.92 ± 0.02) × 10−4 compared with (1.91 ± 0.07) × 10−2 for HeLaΔPDE12. Thus, HeLaPΔE12 cells required 65 times more virus to induce the same degree of CPE after 3 days of infection. We evaluated HRV infection using a cellular imaging assay that measures the expression of HRV capsid protein VP2. Interestingly, the fraction of cells infected by HRV was low on the 1st day of infection but similar for HeLa and HeLaΔPDE12 at every level of virus. By day 5, the fraction of cells infected had progressed significantly in HeLa, but it had declined slightly in HeLaΔPDE12 (Fig. 2C). Thus, PDE12 gene inactivation resulted in significant resistance to infection by HRV.
The effect of the OAS/RNase-L pathway on infection by viruses outside the RNA positive-strand group is not as well studied. RSV has a negative-strand RNA genome and efficiently infects HeLa cells. We evaluated RSV infection using an imaging assay that measures the accumulation of protein G of the viral envelope. HeLa cells were significantly more sensitive to infection by RSV than HRV as indicated by the lower m.o.i. required to establish the infection. By day 2, both HeLa and HeLaΔPDE12 were similarly infected in an m.o.i.-dependent manner (Fig. 2D). However, with longer incubation, the fraction of cells infected by RSV progressed significantly at a low m.o.i. in HeLa cells. In contrast, the fraction of cells infected at a low m.o.i. in HeLaΔPDE12 did not progress. Thus, PDE12 inactivation did not affect the initial infection but appeared to block the spread of RSV.
Crystal Structure of PDE12
The effects of PDE12 gene inactivation suggest that inhibiting PDE12 cleavage of 2–5A may enhance the innate immune response to a variety of viruses such as the important respiratory pathogens HRV and RSV. PDE12 is a member of the EEP nuclease family of enzymes, which have not been pursued as drug targets. To provide a greater understanding of the enzyme structure and to help inform a drug discovery strategy, we pursued the crystal structure of human PDE12 because no structures of any PDE12 isoforms were available. An initial crystal structure was determined for PDE12 residues 155–609 at 2.7 Å, which included two molecules/asymmetric unit in space group P21 (Protein Data Bank code 4Z0V). The structure revealed the majority of the enzyme is well ordered with electron density observed for nearly all residues with the exception of residues 207–222, which appear to be on a disordered loop connecting two adjacent anti-parallel β-strands. A construct was subsequently created that removed these disordered residues that allowed us to obtain crystals in space group P212121, which has a smaller unit cell that contains only one molecule per asymmetric unit and diffracts to beyond 1.8 Å (Table 1). It is unlikely that this deletion influences the binding of substrate or inhibitors due to the location of these residues on the face opposite the catalytic domain. Moreover, the activity of this construct was similar to that of our other PDE12 constructs (data not shown).
TABLE 1.
X-ray structure data collection and refinement statistics
Statistics for the highest resolution shell are shown in parentheses r.m.s. means root mean square deviation.
| PDE12 with compound 3 | PDE12 apo | |
|---|---|---|
| Wavelength | 1.54178 | 0.97872 |
| Resolution range | 26.03–1.8 (1.864–1.8) | 25.73–1.78 (1.844–1.78) |
| Space group | P 21 21 21 | P 1 21 1 |
| Unit cell | 56.309 66.532 124.88 90 90 90 | 56.688 62.554 65.393 90 110.398 90 |
| Total reflections | 191,057 | 155,459 |
| Unique reflections | 39,837 (2513) | 41,197 (4117) |
| Multiplicity | 4.8 (2.1) | 3.8 (3.8) |
| Completeness (%) | 0.92 (66.1) | 1.00 (1.00) |
| Mean I/σ(I) | 24.45 (2.84) | 16.69 (3.61) |
| Wilson B-factor | 18.04 | 16.21 |
| R-merge | 0.054 (0.266) | 0.073 (0.402) |
| R-meas | 0.060 (0.332) | 0.085 (0.242) |
| CC1/2 | (0.904) | (0.863) |
| Reflections used in refinement | 39,836 (2513) | 41,193 (4117) |
| Reflections used for R-free | 1254 (67) | 1291 (131) |
| R-work | 0.1743 (0.2218) | 0.1624 (0.2113) |
| R-free | 0.2114 (0.2410) | 0.1975 (0.2428) |
| No. of non-hydrogen atoms | 4113 | 3851 |
| Macromolecules | 3580 | 3454 |
| Ligands | 69 | 37 |
| Protein residues | 449 | 434 |
| r.m.s. (bonds) | 0.014 | 0.014 |
| r.m.s. (angles) | 1.63 | 1.62 |
| Ramachandran favored (%) | 97 | 98 |
| Ramachandran allowed (%) | 2.6 | 1.8 |
| Ramachandran outliers (%) | 0.22 | 0.23 |
| Rotamer outliers (%) | 0.75 | 1.1 |
| Clash score | 1.82 | 0.87 |
| Average B-factor | 21.83 | 21.15 |
| Macromolecules | 20.78 | 19.93 |
| Ligands | 17.57 | 41.59 |
| Solvent | 30.55 | 30.76 |
The structure of PDE12 shows the protein forms two clear domains, an N-terminal domain composed of residues 161–274 and a C-terminal domain composed of residues 275–609 (Fig. 3A). The N-terminal domain of the enzyme is primarily composed of extended loop regions around a core composed of a six β-strand meander that forms a two-layer β-sandwich. This domain most closely matches the immunoglobulin portion of the plant actin-binding protein SCAB1 (37). The C-terminal domain contains the potential catalytic domain and forms a mixed multilayer α-helix–β-strand structure similar to the other EEP family members. This domain most closely resembles that seen in the CNOT6 enzyme structure (38).
FIGURE 3.

X-ray crystal structure of PDE12. A, ribbon diagram of the overall structure of PDE12 residues 161–609 colored with a gradient from blue to red beginning at the N terminus. B, surface representation of PDE12 colored by charge density. Blue, positive charge; red, negative charge; white, neutral. C, comparison between the PDE12 (yellow) and CNOT6 (cyan) overall structure. The Mg2+ ion common between the two structures is shown in gray. The Mg2+ ion unique to CNOT6 is shown in green. D, electron density around the Mg2+ ion in the active site.
The active site cleft seen in CNOT6 is apparent in the structure of the PDE12 catalytic domain and contains the positively charged region expected for enzymes that interact with RNAs (Fig. 3B). An overlay of the CNOT6 catalytic domain with the C-terminal domain of PDE12 (Fig. 3C) illustrates the overall structural similarity between the two enzymes particularly in the core and active site. The metal-chelating residues in the active site are conserved between PDE12 and CNOT6 and overlay well within the structure. CNOT6-Glu-240 and PDE12-Glu-351 form similar direct coordinating interactions to a Mg2+ ion, and CNOT6-Asp-528 and PDE12-Asp-598 are both hydrogen bonding to water ions that in turn directly coordinate with a Mg2+ ion at the same position in both enzymes. Although the primary Mg2+ ion seen in the active site of CNOT6 has a corresponding ion readily seen in the electron density of the active site cleft of PDE12 (Fig. 3D), the second more loosely bound ion seen in the CNOT6 structure does not appear to be present in PDE12. PDE12 appears to have a water molecule near the corresponding location of the second Mg2+ ion seen in CNOT6 because there is no clear six-way coordination expected for a Mg2+ ion in PDE12.
There are several other differences between the PDE12 and CNOT6 structures. In CNOT6, a loop encompassing residues 267–280 extends up and away from the active site forming a well defined end to the active site cleft. In PDE12, a single tight β-turn at residues 378–379 bridges the β-strands forming a much less defined end to the active site cleft, which may allow for processing of different or longer substrates. Additionally, residues 202–204, which help to form one side of the active site cleft in CNOT6, are replaced by a longer region, residues 308–315, in PDE12, which form two additional helical turns resulting in significant differences for that side of the pocket. These changes along with several individual amino acid substitutions (CNOT6-Leu-414:PDE12-Thr-500; CNOT6-Ile-488:PDE12-Leu-560; CNOT6-Gln-205:PDE12-Val-316; and CNOT6-Asp-483:PDE12-Gly-555) likely form the basis for differences in substrate selection and inhibitor binding (Tables 2 and 3).
TABLE 2.
Substrate kinetics and compound 3 inhibition
Substrate and inhibitor steady state kinetic studies were performed using the indicated enzyme and nucleic acid substrate. AMP formed was determined as a function of time from 20-min progress curves conducted in triplicate, and Equation 4 was fit to the initial rates to determine kcat and Km values, or Equation 5 was fit to determine Ki values for compound 3. The best fit parameter estimates are shown ± the standard error of the fit.
| 2–5A |
OligoA-12 |
Compound 3, Ki | |||||
|---|---|---|---|---|---|---|---|
| Km | kcat | kcat/Km | Km | kcat | kcat/Km | ||
| μm | s−1 | m−1 s−1 | μm | s−1 | m−1 s−1 | nm | |
| PDE12 | 103 ± 4 | 27 ± 0.5 | 2.6 × 105 | 280 ± 100 | 11 ± 3 | 0.39 × 105 | 17.5 ± 0.8 |
| CNOT6 | 45 ± 7 | 6 ± 0.3 | 1.3 × 105 | 164 ± 86 | 2.6 ± 0.7 | 0.16 × 105 | >10,000 |
TABLE 3.
Structure-activity relationships for PDE12 inhibitors
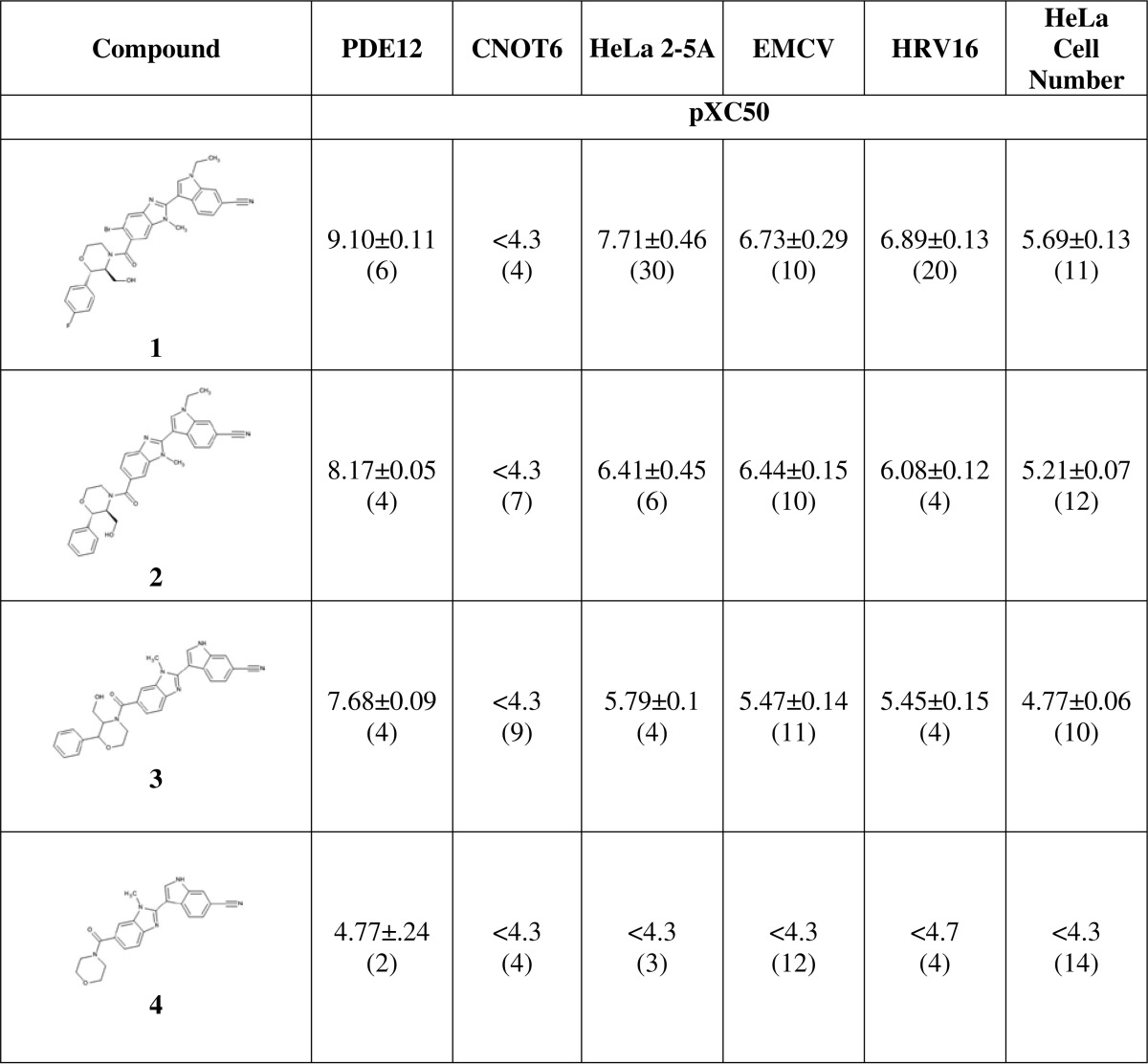
Compound dose-response experiments were conducted and analyzed as described under “Experimental Procedures.”PDE12, PDE12 enzyme inhibition; CNOT6, CNOT6 enzyme inhibition; HeLa 2–5A, IFNα and poly(IC)-stimulated 2–5A levels in cell lysate; EMCV, EMCV CPE in HeLa Ohio cells; HRV16, inhibition of HRV16 infection of SAEC measured by imaging capsid protein. For inhibition assays, pXC50 represents the negative log of the IC50 in molar (pIC50). For HeLa 2–5A, pXC50 represents negative log of the inflection point of the increase (pEC50). The values presented are the mean ± S.D. from the number of experiments shown in parentheses. For some of the replicate experimental determinations, a parameter fit was not obtained because the inflection point was outside the compound concentration range. These values had to be excluded from the reported mean: compound 1, PDE12 pIC50 >9.1 n = 1; HeLa 2–5A pEC50 >8.1 n = 2. Compound 2, HeLa 2–5A pEC50 <4.8 n = 2; compound 4, PDE12 pIC50 <4.4 n = 1.
The two domains within our PDE12 construct have a contact area involving the loop between residues 168 and 182 on the N-terminal domain, residues on the interconnecting loop, and the helical face of the C-terminal domain. The contact has a surface area of ∼400 Å2 and is primarily hydrophilic between the two domains; however, a small hydrophobic core is formed by residues Phe-172, Met-174, Phe-177, and Pro-274 of the N-terminal domain with residues Cys-277, Phe-279, Tyr-324, Tyr-330, and Leu-334 of the C-terminal domain.
Discovery of PDE12 Inhibitors Using ELT
The structure of PDE12 identified a unique cleft in a catalytic domain shared by the EEP nuclease family that would provide a potential binding site for inhibitors. We searched for such inhibitors using ELT, a novel hit identification platform (39). The technology uses ultra-large collections of chemotypically diverse DNA-encoded small molecule libraries. The protein target is immobilized on beads and incubated with a compound library in solution. Protein-bound compounds are separated from nonbound compounds and identified by sequencing the DNA tags. The platform features two major benefits, access to a broad set of chemotypes with vast structural diversity and use of negligible amounts of target protein. Since its inception, ELT has successfully identified tractable hit series for diverse targets (33, 34, 40). Purified PDE12 was immobilized, and selections were performed to screen a three-cycle library. The DNA-encoded library (Fig. 4A) utilized two fluorinated aromatic acids in cycle 1, followed by benzimidazole formation with 1456 aldehydes (cycle 2), and then the acid functionality was capped with 3157 amines (cycle 3) to afford an ELT library with 9.19 million enumerated compounds. After selections, DNA sequencing was conducted, and the sequences were translated to identify the structures of the putative PDE12 binders. Several potential active compounds were designed and synthesized. Compound 3 represents one of the first active molecules designed and was chosen as the starting point for our hit optimization effort (Fig. 4B).
FIGURE 4.

Encoded library structure and initial PDE12 inhibitor. A, schematic of the ELT library used for PDE12 inhibitor structure. The helix represents the position of DNA tags used to identify protein-bound compounds following DNA sequencing. C1 represents the variants in the core structure. C2 represents the variants found at position R (blue). C3 represents the variants found at position R (green). B, chemical structure of key active compound discovered by ELT screening, compound design, and synthesis (compound 3). The biological activity for compound 3 is described in Table 3.
Structure-Activity Relationships for PDE12 Inhibitors
An x-ray crystal structure of a complex of PDE12 with compound 3 was determined using the 155–609 construct (Fig. 5A, Protein Data Bank code 4Z2B). Strong electron density for the compound could be seen in the active site, and we were able to fit the compound unambiguously by confirming the chirality of the bound species directly (Fig. 5B). Compound 3 shows a very optimized fit and displays several important ligand-protein interactions (Fig. 5C). The 6-CN moiety on the indole forms a favorable hydrogen bonding interaction (2.2 Å) with the backbone NH of Thr-500. Any attempts to replace this nitrile with bioisosteres proved deleterious to potency (data not shown). The carbonyl at 4-C of the benzimidazole has proximity to the bottom of the protein pocket making a strong interaction with the nearby Mg2+ ion, which is stabilized by interactions with several water molecules in the region. Similar to the apo-structure, we observed only one Mg2+ ion in the active site. The primary alcohol at the 2-position on morpholine forms a crucial H-bonding interaction (1.4 Å) with Glu-351. Any capping of this moiety results in a dramatic drop in potency (data not shown). The structure also reveals a π-stacking interaction between the neighboring phenyl group and Tyr-307. Removal of any or both of the phenyl and hydroxyl groups results in a drastic drop in potency (compound 4). Alkylation of the indole with ethyl (compound 2) improves the potency due to a favorable hydrophobic interaction of the ethyl at the edge of the receptor. Preparation of the 5-bromobenzimidazole analog (compound 1) boosted potency compared with compound 2. We hypothesized that substitutions at the 5-benzimidazole position impart a steric effect causing the 4-CO to adopt a perpendicular orientation with respect to the benzimidazole ring maximizing the interaction between the CO and the magnesium.
FIGURE 5.

Co-crystal structure of compound 3 bound to PDE12. A, structure of compound 3 bound to the PDE12 catalytic domain construct, PDE12 (155–609Δ206–233). B, electron density of the inhibitor in the active site. C, schematic representation of the contacts between the inhibitor and PDE12. D, overlay of inhibited PDE12 (shown in yellow with a green inhibitor) with CNOT6 enzyme (shown in cyan with its poly(A) substrate).
Mode of Action of PDE12 Inhibitors
The PDE12-compound 3 co-crystal structure indicates that the compound binds in the active site cleft that was identified by molecular modeling (41). A comparison between the crystal structure of CNOT6 with a polyadenylate RNA substrate and our PDE12 compound 3 complex shows that our inhibitor is occupying nearly the equivalent volume and position in PDE12 as the substrate occupies in CNOT6 (Fig. 3D). This implies that the compound may inhibit by competing with 2–5A. To evaluate this hypothesis further, we conducted steady state substrate kinetic experiments to determine the mode of inhibition of compound 3. The initial rate of AMP produced was analyzed at five fixed concentrations of compound 3 using a range of 2–5A as substrate from 1 to 250 μm (Fig. 6C). The initial rates were analyzed by global curve fitting to equations representing competitive (Equation 5), noncompetitive (Equation 6), or uncompetitive (Equation 7) modes of inhibition. The competitive model provided the best fit (R2 = 0.997) compared with the noncompetitive model (R2 = 0.976) and the uncompetitive model (R2 = 0.940). The competitive Ki value for compound 1 inhibition was 17.5 (± 8) nm. Taken together with the crystal structure and modeling results, we conclude that compound 3 inhibits PDE12 by competing with 2–5A substrate.
PDE12 is a member of the EEP nuclease family. CNOT6 is the most similar enzyme to PDE12 with 31% amino acid sequence identity in the EEP nuclease domain (41). PDE12 has been shown to be able to cleave typical 3′,5′-linked oligoA substrates (19, 41, 42), but CNOT6 has not been shown to cleave 2–5A substrates. We compared the steady state substrate kinetics using 2–5A (Fig. 6A) and oligoA (Fig. 6B) for PDE12 and CNOT6. kcat and Km values were determined by fitting the initial rate of AMP formation as a function of substrate concentration to Equation 4. kcat/Km value is generally regarded as the key measure of substrate efficiency. PDE12 and CNOT6 cleave both substrates efficiently (kcat/Km near 105 m−1 s−1). PDE12 prefers 2–5A with a 2-fold higher kcat/Km compared with oligoA. CNOT6 also prefers 2–5A. These results suggest that both enzymes have a slight preference for cleaving 2′,5′-oligoadenylate. However, chain length differences between 2–5A (3 nucleotides) and oligoA (12 nucleotides) may also contribute to the observed kinetic parameters. The IC50 values for compounds 1–4 were determined for CNOT6 (Table 2). Importantly, the compounds in this series did not inhibit CNOT6 even at the highest concentration tested, 50 μm (Table 3).
Effect of PDE12 Inhibitors on Cellular 2–5A, RNase-L Activation, and Virus Infection
PDE12 gene inactivation resulted in increased 2–5A induced by IFN and poly(I-C) (Fig. 2A). We evaluated the effect of PDE12 inhibitors in HeLa cells to determine whether inhibition of enzyme activity produced the same effect as gene inactivation. Compound 1 is one of our most potent PDE12 inhibitors with a pIC50 value for enzyme inhibition of 9.1. Compound 1 produced a maximum 4-fold increase in 2–5A levels, which is similar to what was seen with gene inactivation. The pEC50 value for this effect was 7.7 (Fig. 7A). This 1.4 log decrease in potency from the pIC50 value for PDE12 enzyme inhibition is consistent for all compounds tested in this series (Table 3). Compounds in the PDE12 inhibitor series did not affect CNOT6, the most closely related enzyme to PDE12. Nevertheless, it remains possible that they could produce biological effects through unknown proteins. We evaluated the effect of compound 1 in the HeLaΔPDE12 cells. The IFN-poly(I-C)-induced level of 2–5A was increased compared with the parent HeLa line as expected. Compound 1 did not increase the 2–5A level further at any concentration (Fig. 7A). Thus, the ability of compounds in this series to increase 2–5A is dependent upon the presence of a functional PDE12 enzyme.
FIGURE 7.
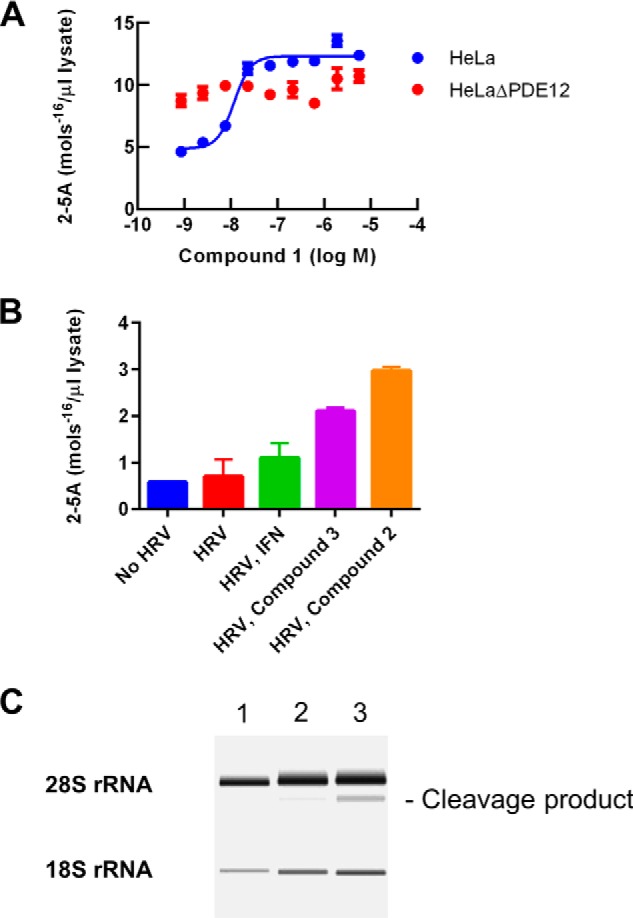
Cellular mechanism of PDE12 inhibitors. A, 2–5A concentrations were determined following IFN and poly(I-C) treatment of the indicated cell line as a function of compound concentration. Blue, HeLa cells; red, HeLaΔPDE12 cells. B, 2–5A concentrations were determined in HeLa cells infected with HRV (m.o.i. = 30) after 2 days of infection in the presence or absence of IFN (100 units/ml) or PDE12 inhibitors (5 μm). C, rRNA from HeLa cells was extracted and analyzed using an Agilent Bioanalyzer. Lane 1, untreated HeLa cells. Lane 2, HeLa cells treated for 2 h with IFN and poly(I-C). Lane 3, HeLa cells treated for 2 h with IFN and poly(I-C) in the presence of 1 μm compound 1. A major cleavage product used to estimate the increase in RNase-L activation is indicated.
IFN-poly(I-C) induces relatively high levels of cellular 2–5A. This allows for accurate determination of 2–5A but may differ from a viral infection. We evaluated the ability of PDE12 inhibitors to affect 2–5A in HRV-infected HeLa cells (Fig. 7B). HRV alone resulted in a 15% increase in 2–5A compared with background levels observed in uninfected cells. Prior treatment with IFN resulted in a slightly greater increase. Addition of PDE12 inhibitors to HRV-infected cells resulted in a 4–6-fold increase above the background level. By comparison, 2–5A levels in the compound-treated, IFN-poly(I-C)-induced cells were typically 20-fold higher than background levels (Fig. 7A).
Activation of RNase-L is the primary mechanism for the antiviral effect of 2–5A. High level activation of RNase-L results in cleavage of rRNA and reduced protein synthesis (2–4). Cellular rRNA was analyzed using an Agilent Bioanalyzer (Fig. 7C). IFN-poly(I-C) treatment of HeLa cells for 2 h resulted in the appearance of a major cleavage product representing 10% of the detectable 28S rRNA. Addition of compound 1 resulted in a further increase in the proportion of this cleavage product to 23% indicative of increased RNase-L activation in response to PDE12 inhibition.
PDE12 inhibitors were evaluated for their antiviral activity (Table 3). Compound 1 inhibited the EMCV-induced cytopathic effect in HeLa Ohio cells with a pIC50 of 6.7. The compound inhibited HeLa Ohio cell proliferation in the absence of EMCV infection with a pIC50 of 5.7. Because PDE12 gene inactivation is tolerated in the HeLa cell background, it is possible the effect on cell proliferation is due to a PDE12-independent effect. We evaluated the effect of compound 1 on the proliferation of HeLaΔPDE12 cells. Compound 1 inhibited the proliferation of both HeLa and HeLaΔPDE12 cells with a pIC50 of 5.7. Thus, the compounds in this series appear to have some anti-proliferative PDE12-independent activities at higher concentrations. Typically, the antiviral activity of these compounds is 10–30-fold more potent than the anti-proliferative activity (Table 3). The mechanism of the PDE12-independent anti-proliferative activity is unknown.
Our results suggest that PDE12 inhibitors exert an antiviral effect through modulation of the IFN-induced OAS/RNase-L effector pathway. This pathway may be altered in the HeLa cell background due to changes associated with oncogenesis. HRV replicates well in human small-airway epithelial cells so we used the HRV imaging assay to measure the effect of PDE12 inhibitors in a primary cell host. PDE12 inhibitors blocked the replication of HRV in these cells (Table 3). For example, compound 1 inhibited HRV infection with a pIC50 of 6.9. In general, the potency of compounds in this series for 2–5A modulation, EMCV inhibition, and HRV inhibition correlated with the potency of PDE12 enzyme inhibition.
Discussion
PDE12 is a widely expressed, abundant 2–5A-degrading enzyme that may have potential as an antiviral target. Two previous lines of evidence support the antiviral role for PDE12, siRNA-mediated knockdown of protein expression and the effects of an inhibitor of PDE12 enzyme activity (17). The proposed mechanism for this activity is modulation of the IFN-induced OAS/2–5A/RNase-L pathway, although direct evidence supporting this has not been presented. In addition to PDE12, virus-encoded 2–5A nucleases and other host enzymes have been described that possess 2–5A nuclease activity, and modulation of these genes has been found to influence viral infectivity (12). Therefore, the role of PDE12 in regulation of host antiviral defense remains unclear.
We employed TALEN nuclease-mediated gene inactivation to create a HeLa-derived cell line, HeLaΔPDE12, with no functional PDE12 expression. HeLaΔPDE12 cells demonstrated increased 2–5A compared with the parent HeLa cell line when treated with IFN and the double-stranded RNA mimic poly(I-C). This confirms that PDE12 controls cellular 2–5A levels in response to stimuli known to activate OAS. PDE12 gene inactivation also resulted in increased resistance to infection by EMCV, HRV, and RSV. The lack of PDE12 activity did not prevent the expression of HRV or RSV structural proteins in the early stages of HeLaΔPDE12 infection, but the spread of infection observed at lower levels of virus was significantly reduced compared with infection of the parental HeLa cells (Fig. 2, C and D). The delayed antiviral response may be attributed to the time required for the host cells to respond to the initial infection, induce IFN, induce OAS, and then accumulate 2–5A to activate the antiviral protein RNase-L.
Host targets like PDE12 may provide a means for breakthrough discoveries of therapeutic agents with broadly acting antiviral potential. Phosphodiesterases like PDE4 and PDE5 have proven to be tractable drug targets for a variety of indications, including inflammation and erectile dysfunction, respectively (43). Despite its name based upon an ability to break the phosphodiester bonds linking the adenylate monomers of 2–5A, PDE12 is not a member of the cyclic nucleotide phosphodiesterase superfamily that contains the enzymes PDE1 through PDE11. PDE12 is a member of the EEP superfamily of deadenylases based upon amino acid sequence homology of active sites. Other EEP enzymes that include CNOT6, Nocturnin, and ANGEL control a range of biological activities by regulating typical 3′,5′-poly(A) tail length and subsequent RNA stability (18). Small molecule inhibitors of enzymes in the EEP nuclease family have not been reported. Therefore, we elected to initiate our drug discovery effort using ELT, an approach that selects enzyme-binding small molecules from extremely diverse chemical libraries (39). Based upon an analysis of ELT compounds that bound PDE12, we generated a series of small molecule inhibitors with representatives described in Table 3. Treatment of host cells with these compounds modulates cellular 2–5A, activates RNase-L, and imparts resistance to viral infection in a fashion that mimics PDE12 gene inactivation. These molecules may represent attractive starting points for host-directed antiviral drug discovery.
To further our understanding of the enzyme and aid compound design, we determined the x-ray crystal structures of PDE12 in the apo-state and with the enzyme bound to compound 3. The structures of PDE12 reveal similarities with other members of the nuclease family. Nearly all of the residues determined to play a role in the catalytic mechanism for APE1 (44) are conserved in similar hydrogen bond networks and structural alignment in PDE12. These residues include Asp-561, His-599, a water molecule, the Mg2+ ion, Glu-351, Asn-301, Asn-498, and Asp-496 leading us to believe that the catalytic mechanism is the same as what was elucidated for other members of this family. Comparison of PDE12 and CNOT6 reveals important differences that may account for substrate preference and inhibitor binding specificity within the EEP subfamily of nucleases. PDE12 has an insertion of four residues to extend two short helices on one side of the substrate pocket and a number of single residue differences at positions 316, 500, 555, and 560. These changes result in overall differences in the shapes of the PDE12 and CNOT6 substrate pockets presenting unique surfaces involved in substrate and inhibitor binding. However, the pockets of both enzymes are large and open relative to the conserved geometry of the catalytic residues that may explain the observed flexibility in substrate selection (Table 2).
Both ends of the active site appear to be more open in PDE12 relative to CNOT6. PDE12 contains a double glycine at residues 554–555 compared with Phe-482–Asp-483 in cNOT6 at one end of the pocket. On the opposite end, CNOT6 contains an 11-residue insertion that provides a well defined end to the active site cleft. The absence of these residues in PDE12 opens up the cleft. The significance of these differences is unknown but may affect cleavage of longer substrates.
At the N terminus, PDE12 contains an immunoglobulin (Ig)-like domain that interacts with the surface of the catalytic domain. It is interesting to note that other EEP family enzymes do not appear to contain this domain. Although the function of the N-terminal Ig domain remains unknown, we speculate that it may function like other Ig domains as an interaction region to anchor the enzyme to another protein or lipid membrane. Alternatively, the basic patch on the surface of the Ig domain (Fig. 3C) could interact with the phosphate backbone of longer substrates.
PDE12 has been shown to be located in the mitochondrial matrix and found to have the ability to cleave 3′,5′-oligoadenylate in addition to 2–5A (19, 41, 42). Moreover, overexpression of PDE12 reduced the levels of key mitochondrial proteins and rates of oxidative phosphorylation; thus, it appears that PDE12 functions as a mitochondrion-specific deadenylase controlling gene expression in a fashion similar to other EEP enzymes (41). In contrast, we have found that PDE12 prefers 2–5A to 3′,5′-oligoA as a substrate with a 6.5-fold higher kcat/Km value. In addition, PDE12 gene inactivation or inhibition with small molecules results in higher cellular 2–5A in response to OAS activation. Thus, PDE12 appears to be a biologically relevant regulator of the 2–5A-mediated IFN response as well as a regulator of mitochondrial gene expression. The localization of PDE12 to the mitochondrial matrix confounds our understanding of the subcellular compartmentalization of the OAS/2–5A/RNase-L pathway, which is typically thought of as being a cytoplasmic process. Perhaps a subpopulation of PDE12 is present in the cytoplasm. In contrast, OAS and RNase-L have been shown to be associated with mitochondria, so PDE12 may be part of a larger mitochondrion-associated innate defense response (45–47). We have found that PDE12 gene inactivation and inhibitors are generally well tolerated by HeLa cells. The effect of gene inactivation and inhibitors on mitochondrial biology has not yet been studied, but these effects will clearly be important in evaluating the potential of PDE12 inhibition for antiviral therapy.
As discussed in the Introduction, other host enzymes in the LigT-like group of 2H phosphodiesterases, including AKAP7 have been shown to degrade 2–5A (14). Moreover, EEP nucleases, including EPP1 (19) and CNOT6, have been shown to possess this activity. Thus, it seems that mammalian cells have evolved multiple mechanisms for down-regulating the OAS/2–5A/RNase-L pathway. The alternative subcellular localization of these enzymes or other levels of regulation may be important for preventing excessive RNA degradation and subsequent cellular damage. From a practical perspective, the redundancy within the host cell as well as virus-encoded 2–5A-cleaving enzymes may complicate the utility of future agents targeting these enzymes.
Despite the uncertainty around the role of PDE12 in mitochondrial biology or the role of other 2–5A-cleaving enzymes in the regulation of the IFN-mediated antiviral response, it is clear that blocking PDE12 activity through gene inactivation or with small molecules enhances cellular resistance to viral pathogens, including EMCV, HRV, and RSV. Whether or not this effect will extend to other viruses is not known, but a variety of important pathogens are affected by modulation of OAS and RNase-L, including West Nile virus, Chikungunya virus, Dengue virus, and hepatitis C virus (5, 48–52). To test whether the PDE12 inhibitor-induced cellular resistance will translate to a beneficial effect in vivo will require the production of PDE12 knock-out animals or the development of compounds with sufficient potency and exposure for in vivo antiviral studies.
Author Contributions
E. R. W. was responsible for study design and is the primary author. R. B., G. T., B. W., and J. D. T. designed and conducted expression and purification of essential reagents. J. N., S. H. G., T. S., S. Y., D. S., and L. H. K. conducted cellular 2–5A and antiviral studies. C. R. conducted deep sequencing of the HeLaΔPDE12 line. L. W. and R. T. N. conducted crystallization and solved the x-ray structures. E. N. conducted enzyme assays for compound SAR. J. N. conducted enzyme kinetic studies. J. C., P. D., H. D., Y. D., J. B. S., V. T., B. X., and N. B. designed and synthesized the compounds described. H. D. led the design of compounds. All authors analyzed the data, contributed to the writing by describing their contributions, and approved the final version of the manuscript.
Acknowledgments
The HeLaΔPDE12 cell line was genetically engineered by Transposagen Biopharmaceuticals, Inc. We thank Warren Rocque and Kendra Hightower for critically reading the manuscript.
All authors were employees of GlaxoSmithKline at the time the work was performed.
The atomic coordinates and structure factors (code 4Z0V) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- OAS
- 2′,5′-oligoadenylate synthetase
- 2–5A
- 2′,5′-oligoadenylate
- TALEN
- transcription activator-like effector nuclease
- EMCV
- encephalomyocarditis virus
- HRV
- human rhinovirus
- RSV
- respiratory syncytial virus
- m.o.i.
- multiplicity of infection
- EEP
- exonuclease-endonuclease-phosphatase
- TCEP
- tris(2-carboxyethyl phosphine)
- ELT
- encoded library technology
- CPE
- cytopathic effect
- oligoA
- 3′,5′-linked oligoadenylate
- 2′5′-PDE
- 2′,5′-specific phosphodiesterase
- SSAEC
- small-airway epithelial cell
- TEV
- tobacco etch virus.
REFERENCES
- 1. Lou Z., Sun Y., Rao Z. (2014) Current progress in antiviral strategies. Trends Pharmacol. Sci. 35, 86–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li X.-L., Ezelle H. J., Hsi T. Y., Hassel B. A. (2011) A central role for RNA in the induction and biological activities of type 1 interferons. Wiley Interdiscip. Rev. RNA 2, 58–78 [DOI] [PubMed] [Google Scholar]
- 3. Sadler A. J., Williams B. R. (2008) Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 8, 559–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Silverman R. H. (2007) Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 81, 12720–12729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ezelle H. J., Hassel B. A. (2012) Pathologic effects of RNase-L dysregulation in immunity and proliferative control. Front. Biosci. 4, 767–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Williams B. R., Kerr I. M., Gilbert C. S., White C. N., Ball L. A. (1978) Synthesis and breakdown of pppA2′p5′A2′p5′A and transient inhibition of protein synthesis in extracts from interferon-treated and control cells. Eur. J. Biochem. 92, 455–462 [DOI] [PubMed] [Google Scholar]
- 7. Torrence P. F., Imai J., Johnston M. I. (1983) Assay of 2′,5′-oligoadenylate phosphodiesterase activity in mouse L-cell extracts. Anal. Biochem. 129, 103–110 [DOI] [PubMed] [Google Scholar]
- 8. Drappier M., Sorgeloos F., Michiels T. (2014) The OAS/RNase L pathway and its inhibition by viruses. Virologie 18, 1026–1036 [DOI] [PubMed] [Google Scholar]
- 9. Han J.-Q., Townsend H. L., Jha B. K., Paranjape J. M., Silverman R. H., Barton D. J. (2007) A phylogenetically conserved RNA structure in the poliovirus open reading frame inhibits the antiviral endoribonuclease RNase L. J. Virol. 81, 5561–5572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sorgeloos F., Jha B. K., Silverman R. H., Michiels T. (2013) Evasion of antiviral Innate immunity by Theiler's virus L* protein through direct inhibition of RNase L. PLoS Pathog. 9, e1003474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Min J.-Y., Krug R. M. (2006) The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 2′-5′ oligoA synthetase/RNase L pathway. Proc. Natl. Acad. Sci. U.S.A. 103, 7100–7105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang R., Jha B. K., Ogden K. M., Dong B., Zhao L., Elliott R., Patton J. T., Silverman R. H., Weiss S. R. (2013) Homologous 2′,5′-phosphodiesterases from disparate RNA viruses antagonize antiviral innate immunity. Proc. Natl. Acad. Sci. U.S.A. 110, 13114–13119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhao L., Jha B. K., Wu A., Elliott R., Ziebuhr J., Gorbalenya A. E., Silverman R. H., Weiss S. R. (2012) Antagonism of the interferon-induced OAS-RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Microbe 11, 607–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Silverman R. H., Weiss S. R. (2014) Viral phosphodiesterases that antagonize double-stranded RNA signaling to RNase L by degrading 2–5A. J. Interferon Cytokine Res. 34, 455–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mazumder R., Iyer L. M., Vasudevan S., Aravind L. (2002) Detection of novel members, structure-function analysis and evolutionary classification of the 2H phosphoesterase superfamily. Nucleic Acids Res. 30, 5229–5243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gusho E., Zhang R., Jha B. K., Thornbrough J. M., Dong B., Gaughan C., Elliott R., Weiss S. R., Silverman R. H. (2014) Murine AKAP7 has a 2,5-phosphodiesterase domain that can complement an inactive murine coronavirus ns2 gene. mBio 5, e01312–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kubota K., Nakahara K., Ohtsuka T., Yoshida S., Kawaguchi J., Fujita Y., Ozeki Y., Hara A., Yoshimura C., Furukawa H., Haruyama H., Ichikawa K., Yamashita M., Matsuoka T., Iijima Y. (2004) Identification of 2′-phosphodiesterase, which plays a role in the 2–5A system regulated by interferon. J. Biol. Chem. 279, 37832–37841 [DOI] [PubMed] [Google Scholar]
- 18. Goldstrohm A. C., Wickens M. (2008) Multifunctional deadenylase complexes diversify mRNA control. Nat. Rev. Mol. Cell Biol. 9, 337–344 [DOI] [PubMed] [Google Scholar]
- 19. Poulsen J. B., Andersen K. R., Kjær K. H., Vestergaard A. L., Justesen J., Martensen P. M. (2012) Characterization of human phosphodiesterase 12 and identification of a novel 2′-5′ oligoadenylate nuclease-The ectonucleotide pyrophosphatase/phosphodiesterase 1. Biochimie 94, 1098–1107 [DOI] [PubMed] [Google Scholar]
- 20. Gaj T., Gersbach C. A., Barbas C. F., 3rd (2013) ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 31, 397–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Elbahesh H., Jha B. K., Silverman R. H., Scherbik S. V., Brinton M. A. (2011) The Flvr-encoded murine oligoadenylate synthetase 1b (Oas1b) suppresses 2–5A synthesis in intact cells. Virology 409, 262–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miklos A. E., Hughes R. A., Ellington A. D. (2012) Design and assembly of large synthetic DNA constructs. Curr. Protoc. Mol. Biol. Chapter 3, Unit 3.23 [DOI] [PubMed] [Google Scholar]
- 23. Einhauer A., Jungbauer A. (2001) The FLAG(trademark) peptide, a versatile fusion tag for the purification of recombinant proteins. J. Biochem. Biophys. Methods 49, 455–465 [DOI] [PubMed] [Google Scholar]
- 24. Parks T. D., Leuther K. K., Howard E. D., Johnston S. A., Dougherty W. G. (1994) Release of proteins and peptides from fusion proteins using a recombinant plant virus proteinase. Anal. Biochem. 216, 413–417 [DOI] [PubMed] [Google Scholar]
- 25. Kay B. K., Thai S., Volgina V. V. (2009) High-throughput biotinylation of proteins. Methods Mol. Biol. 498, 185–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Otwinowski Z., Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 27. McCoy A. J. (2007) Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63, 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smart O. S., Womack T. O., Flensburg C., Keller P., Paciorek W., Sharff A., Vonrhein C., Bricogne G. (2012) Exploiting structure similarity in refinement: Automated NCS and target-structure restraints in BUSTER. Acta Crystallogr. D Biol. Crystallogr. 68, 368–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 33. Deng H., O'Keefe H., Davie C. P., Lind K. E., Acharya R. A., Franklin G. J., Larkin J., Matico R., Neeb M., Thompson M. M., Lohr T., Gross J. W., Centrella P. A., O'Donovan G. K., Bedard K. L., et al. (2012) Discovery of highly potent and selective small molecule ADAMTS-5 inhibitors that inhibit human cartilage degradation via encoded library technology (ELT). J. Med. Chem. 55, 7061–7079 [DOI] [PubMed] [Google Scholar]
- 34. Disch J. S., Evindar G., Chiu C. H., Blum C. A., Dai H., Jin L., Schuman E., Lind K. E., Belyanskaya S. L., Deng J., Coppo F., Aquilani L., Graybill T. L., Cuozzo J. W., Lavu S., et al. (2013) Discovery of thieno[3,2-d]pyrimidine-6-carboxamides as potent inhibitors of SIRT1, SIRT2, and SIRT3. J. Med. Chem. 56, 3666–3679 [DOI] [PubMed] [Google Scholar]
- 35. Morin B., Rabah N., Boretto-Soler J., Tolou H., Alvarez K., Canard B. (2010) High yield synthesis, purification and characterisation of the RNase L activators 5′-triphosphate 2′-5′-oligoadenylates. Antiviral Res. 87, 345–352 [DOI] [PubMed] [Google Scholar]
- 36. Copeland R. A. (2000) Enzymes: A Practical Introduction to Structure, Mechanism and Data Analysis, 2nd Ed., John Wiley and Sons, New York [Google Scholar]
- 37. Zhang W., Zhao Y., Guo Y., Ye K. (2012) Plant actin-binding protein SCAB1 is dimeric actin cross-linker with atypical pleckstrin homology domain. J. Biol. Chem. 287, 11981–11990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang H., Morita M., Yang X., Suzuki T., Yang W., Wang J., Ito K., Wang Q., Zhao C., Bartlam M., Yamamoto T., Rao Z. (2010) Crystal structure of the human CNOT6L nuclease domain reveals strict poly(A) substrate specificity. EMBO J. 29, 2566–2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Clark M. A., Acharya R. A., Arico-Muendel C. C., Belyanskaya S. L., Benjamin D. R., Carlson N. R., Centrella P. A., Chiu C. H., Creaser S. P., Cuozzo J. W., Davie C. P., Ding Y., Franklin G. J., Franzen K. D., Gefter M. L., et al. (2009) Design, synthesis and selection of DNA-encoded small-molecule libraries. Nat. Chem. Biol. 5, 647–654 [DOI] [PubMed] [Google Scholar]
- 40. Thalji R. K., McAtee J. J., Belyanskaya S., Brandt M., Brown G. D., Costell M. H., Ding Y., Dodson J. W., Eisennagel S. H., Fries R. E., Gross J. W., Harpel M. R., Holt D. A., Israel D. I., Jolivette L. J., et al. (2013) Discovery of 1-(1,3,5-triazin-2-yl)piperidine-4-carboxamides as inhibitors of soluble epoxide hydrolase. Bioorg. Med. Chem. Lett. 23, 3584–3588 [DOI] [PubMed] [Google Scholar]
- 41. Rorbach J., Nicholls T. J., Minczuk M. (2011) PDE12 removes mitochondrial RNA poly(A) tails and controls translation in human mitochondria. Nucleic Acids Res. 39, 7750–7763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Poulsen J. B., Andersen K. R., Kjær K. H., Durand F., Faou P., Vestergaard A. L., Talbo G. H., Hoogenraad N., Brodersen D. E., Justesen J., Martensen P. M. (2011) Human 2′-phosphodiesterase localizes to the mitochondrial matrix with a putative function in mitochondrial RNA turnover. Nucleic Acids Res. 39, 3754–3770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lugnier C. (2006) Cyclic nucleotide phosphodiesterase (PDE) superfamily: a new target for the development of specific therapeutic agents. Pharmacol. Ther. 109, 366–398 [DOI] [PubMed] [Google Scholar]
- 44. Mol C. D., Izumi T., Mitra S., Tainer J. A. (2000) DNA-bound structures and mutants reveal abasic DNA binding by APE1 DNA repair and coordination. Nature 403, 451–456 [DOI] [PubMed] [Google Scholar]
- 45. Kjær K. H., Pahus J., Hansen M. F., Poulsen J. B., Christensen E. I., Justesen J., Martensen P. M. (2014) Mitochondrial localization of the OAS1 p46 isoform associated with a common single nucleotide polymorphism. BMC Cell Biol. 15, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Le Roy F., Silhol M., Salehzada T., Bisbal C. (2007) Regulation of mitochondrial mRNA stability by RNase L is translation-dependent and controls IFNα-induced apoptosis. Cell Death Differ. 14, 1406–1413 [DOI] [PubMed] [Google Scholar]
- 47. Domingo-Gil E., Esteban M. (2006) Role of mitochondria in apoptosis induced by the 2–5A system and mechanisms involved. Apoptosis 11, 725–738 [DOI] [PubMed] [Google Scholar]
- 48. Lin R.-J., Yu H.-P., Chang B.-L., Tang W.-C., Liao C.-L., Lin Y.-G. (2009) Distinct antiviral roles for human 2′,5′-oligoadenylate synthetase family members against dengue virus infection. J. Immunol. 183, 8035–8043 [DOI] [PubMed] [Google Scholar]
- 49. Flodström-Tullberg M., Hultcrantz M., Stotland A., Maday A., Tsai D., Fine C., Williams B., Silverman R., Sarvetnick N. (2005) RNase L and double-stranded RNA-dependent protein kinase exert complementary roles in islet cell defense during coxsackievirus infection. J. Immunol. 174, 1171–1177 [DOI] [PubMed] [Google Scholar]
- 50. Zhou A., Paranjape J., Brown T. L., Nie H., Naik S., Dong B., Chang A., Trapp B., Fairchild R., Colmenares C., Silverman R. H. (1997) Interferon action and apoptosis are defective in mice devoid of 2′,5′-oligoadenylate-dependent RNase L. EMBO J. 16, 6355–6363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Samuel M. A., Whitby K., Keller B. C., Marri A., Barchet W., Williams B. R., Silverman R. H., Gale M., Jr., Diamond M. S. (2006) PKR and RNase L contribute to protection against lethal West Nile virus infection by controlling early viral spread in the periphery and replication in neurons. J. Virol. 80, 7009–7019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Scherbik S. V., Paranjape J. M., Stockman B. M., Silverman R. H., Brinton M. A. (2006) RNase L plays a role in the antiviral response to West Nile virus. J. Virol. 80, 2987–2999 [DOI] [PMC free article] [PubMed] [Google Scholar]