

Background: Myelin-associated glycoprotein (MAG) mediates varicella-zoster virus (VZV) infection by associating with glycoprotein B (gB).
Results: Analyses of glycans on VZV gB revealed that sialic acids (SAs) on gB are required for membrane fusion and infection against MAG-expressing cells.
Conclusion: SA-containing glycans on gB are necessary for VZV membrane fusion.
Significance: The role of SAs during VZV infection is elucidated.
Keywords: herpesvirus, membrane fusion, N-linked glycosylation, sialic acid, virus entry, O-linked glycosylation, myelin-associated glycoprotein (MAG), sialic acid-binding immunoglobulin-type lectin 4 (Siglec-4), varicella-zoster virus (VZV)
Abstract
Varicella-zoster virus (VZV) is a member of the human Herpesvirus family that causes varicella (chicken pox) and zoster (shingles). VZV latently infects sensory ganglia and is also responsible for encephalomyelitis. Myelin-associated glycoprotein (MAG), a member of the sialic acid (SA)-binding immunoglobulin-like lectin family, is mainly expressed in neural tissues. VZV glycoprotein B (gB) associates with MAG and mediates membrane fusion during VZV entry into host cells. The SA requirements of MAG when associating with its ligands vary depending on the specific ligand, but it is unclear whether the SAs on gB are involved in the association with MAG. In this study, we found that SAs on gB are essential for the association with MAG as well as for membrane fusion during VZV infection. MAG with a point mutation in the SA-binding site did not bind to gB and did not mediate cell-cell fusion or VZV entry. Cell-cell fusion and VZV entry mediated by the gB-MAG interaction were blocked by sialidase treatment. N-glycosylation or O-glycosylation inhibitors also inhibited the fusion and entry mediated by gB-MAG interaction. Furthermore, gB with mutations in N-glycosylation sites, i.e. asparagine residues 557 and 686, did not associate with MAG, and the cell-cell fusion efficiency was low. Fusion between the viral envelope and cellular membrane is essential for host cell entry by herpesviruses. Therefore, these results suggest that SAs on gB play important roles in MAG-mediated VZV infection.
Introduction
The human herpesvirus family comprises eight viruses, including herpes simplex virus (HSV)3 and varicella-zoster virus (VZV). VZV causes varicella (chicken pox) in most children, zoster (shingles) in adults or immunocompromised hosts, as well as encephalomyelitis and cranial neuritis (1–3). Latently infected VZV may be observed in the sensory ganglia, and it is reactivated in an immunocompromised state (4). Therefore, it is important to elucidate the mechanism employed by VZV to infect nerve tissues. Membrane fusion between the viral envelope and cellular membrane is an essential process for enveloped viruses, such as herpesviruses (5, 6), when entering host cells (7–9). Therefore, elucidating the mechanism of membrane fusion is important for understanding the entry mechanism of enveloped viruses. Viral fusion proteins associate with membrane proteins on their host cells to induce membrane fusion in enveloped viruses. Viruses that belong to Herpesviridae typically require glycoprotein B (gB) and the gH-gL complex for membrane fusion. In the case of HSV, in addition to gB and the gH-gL complex, the association of HSV gD with nectin-1,2 and the herpesvirus entry mediator on host cells is essential for membrane fusion. On the other hand, VZV does not have gD, unlike HSV, and gB and the gH-gL complex are minimum components for VZV membrane fusion, as we have reported previously (10).
In VZV, the cell surface receptors that mediate membrane fusion by associating with envelope proteins have been unclear for a long time. However, we demonstrated that myelin-associated glycoprotein (MAG) binds to VZV gB, where this interaction mediates membrane fusion during VZV infection (10). MAG belongs to the sialic acid (SA)-binding Ig-like lectin (Siglec) family, and it is also known as Siglec-4. Most Siglecs are expressed on hematopoietic cells, but the expression of MAG is restricted to neural tissues. It has been suggested that Siglec family molecules recognize their ligands in an SA-dependent manner (11, 12). However, it has been reported that MAG can associate with the Nogo-66 receptor (NgR) in an SA-independent manner (13–15) or an SA-dependent manner (16). Although MAG also binds to the mouse paired immunoglobulin receptor B, human leukocyte Ig-like receptor B2 (LILRB2), and β1-integrin, it has remained unclear whether SAs are required for MAG binding to these ligands (17, 18). In addition, MAG associates with fibronectin and some gangliosides, such as GD1a and GT1b, in an SA-dependent manner (19–21). These observations suggest that the requirement for SAs to allow MAG to associate with its ligands appears to differ depending on the nature of the ligands (22, 23). Therefore, it is important to clarify whether SA is required for VZV infection mediated by the MAG-gB interaction.
It has been known that SAs on host cells are targets for various viruses to attach to the host cells during infection (24). Particularly, some viruses, such as the influenza virus, use SAs as their entry receptors. On the other hand, most of the glycoproteins produced in mammalian cells are modified with SAs. Viruses utilize the host cell systems to synthesize their components, so viral envelope glycoproteins are also modified with SAs (24). For example, the SAs on HSV envelope glycoproteins are involved in viral infection. Sialidase treatment of HSV decreases its infectivity (25). Furthermore, mutations of the O-glycosylation sites in HSV gB abrogate the interaction with paired immunoglobulin-like type 2 receptor α, one of the entry receptors for HSV (26). VZV gB, the gE-gI complex, and the gH-gL complex are also sialylated (27–32). However, the specific SAs on VZV envelope proteins that are involved in membrane fusion and infection remain unclear. Therefore, it is important to clarify whether SAs are required for the MAG-gB interaction during membrane fusion and infection of VZV.
Cell-cell fusion assays are powerful virus-free models for examining the machinery that is essential for membrane fusion during viral entry because it is not necessary to consider the effects of other viral components (33–37). Cell-cell fusion assays using gB, gH, gL, and MAG are the only systems available for assaying the function of VZV membrane fusion. In this study, we used these assays to investigate the roles of SAs on VZV gB during infection and performed viral infection experiments.
Experimental Procedures
Antibodies and Reagents
Mouse anti-VZV gB (clone SG2) and anti-MAG (clone 513) mAbs were purchased from Santa Cruz Biotechnology and Millipore, respectively. Tunicamycin (Wako, 0.10 μg/ml), deoxynojirimycin (DNJ) (Wako, 2.0 mm), benzyl-α-GalNac (Sigma, 5.0 mm), and neuraminidase (sialidase, Clostridium perfringens, Roche, 0.05 units/ml) were used in assay medium in this study.
Sialidase Treatment
The cell lysates used for Western blotting analysis and the cells utilized for analyzing the association with WT-MAG-Ig were treated with sialidase at 37 °C for 10 and 30 min. VZV was treated for 10 min in the infection analysis and for 30 min in the VZV binding assay.
Treatment with Glycosylation Inhibitors
293T cells were cultured in medium containing tunicamycin, DNJ, or benzyl-α-GalNac and then transfected with WT-gB or mock-transfected. 24 h after transfection, the cells were stained with WT-MAG-Ig or anti-gB mAb (SG2), followed by flow cytometry analysis.
Cell Lines and Viruses
Human melanoma MeWo cells (provided by Dr. A. M. Arvin, Stanford University), 293T cells (purchased from Riken), Plat-E cells (provided by Dr. T. Kitamura, The University of Tokyo), and human oligodendroglial cell line (OL cells) (provided by Dr. K. Ikuta, Biken) were cultured in DMEM (Nacalai Tesque). All cells were cultured at 37 °C in 5% CO2 in medium supplemented with 10% FCS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 50 μm 2-mercaptoethanol. The VZV Oka strain and a recombinant Oka strain carrying the GFP reporter gene (GFP-VZV, provided by Dr. A. M. Arvin, Stanford University) were used in the VZV infection analyses (38). Because GFP is expressed via the CMV promoter in the recombinant VZV, the virus particle itself does not contain GFP. The cell-free virus was prepared from VZV-infected MeWo cells by harvesting cells with 0.25% EDTA (2.5 ml/100-mm dish of infected cells) and resuspending the harvested cells in 0.4 ml of SPGA buffer (pH 8.0) (218 mm sucrose, 3.8 mm KH2PO4, 4.9 mm sodium glutamate, and 1% bovine serum albumin). The suspended cells were sonicated twice on ice for 15 s with a 1-min interval, followed by centrifugation at 12,000 × g for 5 min at 4 °C. The resulting supernatant was passed through a 0.45-μm filter and stored at −80 °C. The frozen supernatant was thawed immediately before use as the cell-free virus. The viral titers were determined using MAG-transfected OL cells. MeWo cells, cultured at a density of 2 × 105 cells/well in 24-well tissue culture plates, were infected with GFP-VZV in a cell-associated manner and cultured with N- and O-glycosylation inhibitors simultaneously. Culture supernatants were collected after more than 80% of cells showed cytopathic effect. Each supernatant, after the removal of cells and debris, was used as a cell-free virus.
Plasmids
The plasmids encoding human wild-type MAG (WT-MAG), WT-MAG-Ig, VZV gB, gH, gL, and gB-Ig have been described previously (10). Plasmids for the mutated MAG and MAG-Ig fusion protein, i.e. the mutation of arginine at position 118 to alanine (R118A-MAG and R118A-MAG-Ig, respectively), were engineered using a QuikChange site-directed mutagenesis kit (Agilent Technologies) and a primer pair (sense, 5′-GGGAAGTACTACTTCGCTGGGGACCTGGGCGGC-3′; antisense, 5′-GCCGCCCAGGTCCCCAGCGAAGTAGTACTTCCC-3′). The gB mutants were cloned by recombinant PCR using the WT-gB plasmid as a template as follows: cloning the upper portion using a primer pair (sense, IO2045 5′-aataatGAATTCCACCatgtccccttgtggct-3′; antisense, each antisense primer substituting Ser/Thr or Asn with Ala (Figs. 4 and 6); cloning the lower portions using a primer pair (sense, each sense primer substituting Ser/Thr or Asn with Ala (Figs. 4 and 6); antisense IO3230, 5′-aataatctcgagttacacccccgttacat-3′); and cloning the full-length gB with a mutation using the upper and lower portions as templates with the primer pair IO2045 and IO3230. The mutated gB was inserted into the pCAGGS-MCS vector at the EcoRI and XhoI sites. A plasmid expressing the extracellular domain of gB fused with the glycosylphosphatidylinositol (GPI) anchor of decay-accelerating factor (CD55) was cloned by recombinant PCR as follows: cloning the upper portion using a primer pair (sense, IO2045; antisense, 5′-tttggggttgtttcatgaaaCTCGAGcccaaatgggttagataaaa-3′) with the WT-gB plasmid as a template; cloning the lower portion using a primer pair (sense, 5′-ttttatctaacccatttgggCTCGAGtttcatgaaacaaccccaaa-3′; antisense, IO3025 5′-aataatGTCGACctaagtcagcaagcccatgg-3′) with human peripheral blood mononuclear cell cDNA as a template; the upper and lower portions were connected with IO2045 and IO3025. WT-gB-GPI was digested with the restriction enzymes EcoRI and SalI and inserted into the pCAGGS-MCS vector at the EcoRI and XhoI sites. The extracellular domain of gB (N147A, T129A, and S559A) was cloned from the full-length gB (N147A, T129A, and S559A) as described above using a primer pair (sense, IO2045; antisense, aataatCTCGAGaaatgggttagataaaaa). The extracellular domain of WT-gB in WT-gB-GPI inserted into pCAGGS-MCS was replaced by the extracellular domain of gB (N147A, T129A, and S559A) using the restriction enzymes EcoRI and XhoI.
FIGURE 4.
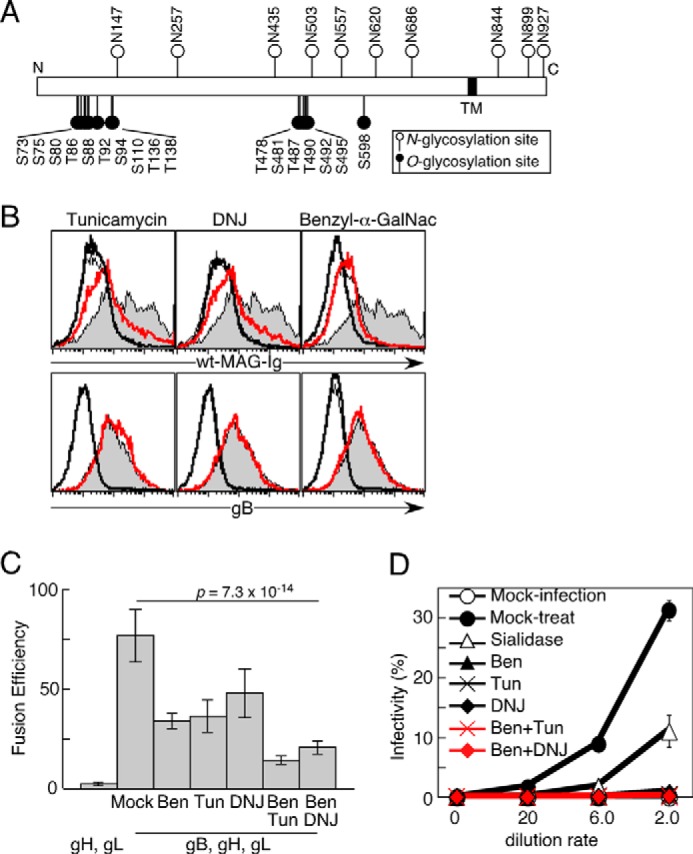
The requirement of N- and O-glycosylations of gB for the association with MAG and fusion with MAG-expressing cells. A, schematic of gB showing the predicted N- and O-glycosylation sites. TM, transmembrane lesion of gB; N, N terminus of gB; C, C terminus of gB. B, mock- or gB-transfected 293T cells were cultured with medium containing vehicle or a glycosylation inhibitor: tunicamycin, DNJ, or benzyl-α-GalNac. The cells were stained with WT-MAG-Ig (top panels) and anti-gB mAb (bottom panels), followed by flow cytometry analysis. Relative cell numbers are shown for mock-transfected cells treated with vehicle (thin line), gB-transfected cells treated with vehicle (gray area), mock-transfected cells treated with each inhibitor (bold line), and gB-transfected cells treated with each inhibitor (red line). C, 293T effector cells cultured with medium containing tunicamycin (Tun), DNJ, benzyl-α-GalNac (Ben), both tunicamycin and benzyl-α-GalNac (Ben/Tun), both DNJ and benzyl-α-GalNac (Ben/DNJ) or mock and transfected with gH and gL, or gB, gH, and gL were cocultured with other target 293T cells transfected with MAG. The relative fusion efficiencies are shown on the basis of representative data from three independent experiments. The error bars represent the mean ± S.D. on the basis of six replicates. Statistical differences were determined using one-way analysis of variance. D, OL cells stably transfected with WT-MAG were infected for 24 h with GFP-VZV produced by cells that were cultured with medium containing the indicated combination of glycosylation inhibitor(s). The proportions of VZV-infected cells (GFP+) are shown. Representative data from three independent experiments are shown. The error bars represent the mean ± S.D. on the basis of duplicate samples.
FIGURE 6.
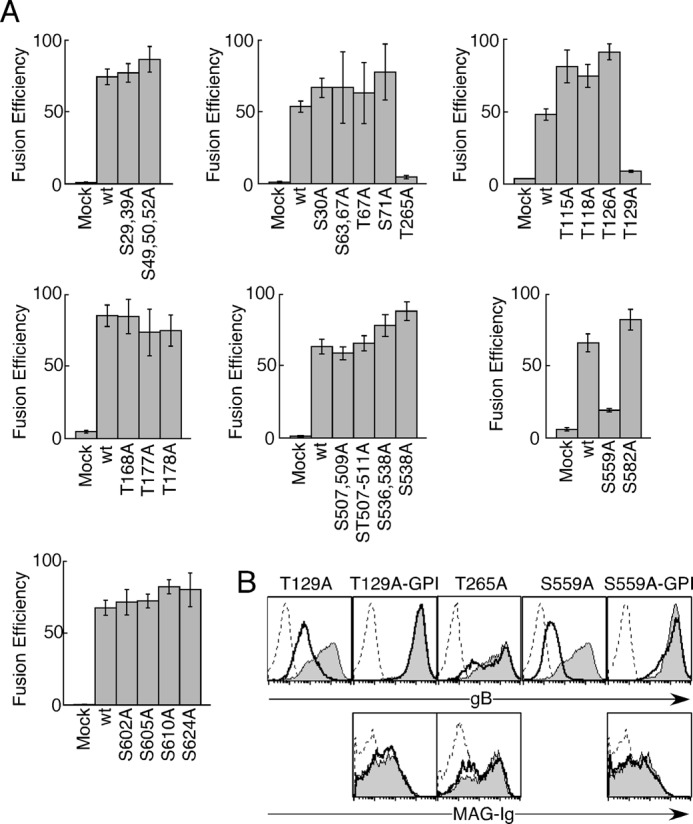
Little contribution of several Ser/Thr residues on gB to cell-cell fusion. A, WT-gB or each mutated gB was cotransfected into 293T effector cells with gH and gL. The effector cells were cocultured with other 293T target cells transfected with MAG. The relative fusion efficiencies are shown on the basis of representative data from three independent experiments followed by luminescence measurements. The error bars represent the mean ± S.D. on the basis of six replicate samples. Statistical differences were determined using the Student's t test. Each mutated gB except for T129A, T265A, and S559A did not differ significantly compared with wild-type gB. p < 0.05 was considered significant. B, mock-transfected (dotted line), WT-gB-(GPI)-transfected (gray area), or mutated gB-(GPI)-transfected (bold line) 293T cells were stained with anti-gB mAb (top panels) and MAG-Ig (bottom panels).
Ig Fusion Protein
The plasmids for Ig fusion proteins were constructed as described above. 293T cells were transfected transiently with expression vectors for Ig fusion proteins, and the culture supernatants were collected. The empty Ig fusion protein containing the signal peptide of mouse signaling lymphocyte activation molecule (SLAM, CD150) and the human IgG1 Fc portion was used as a control (39).
Transfection
Plasmid DNA (0.8 μg in 50 μl of Opti-MEM (Life Technologies) mixed with polyethylenimine “Max” (molecular mass, 25,000) (Polysciences) (4 μg in 50 μl of Opti-MEM) was incubated at room temperature for 20 min. This diluent was transfected into 293T cells at a density of 2 × 105 cells/well in 24-well tissue culture plates. Stable transfectants that expressed human MAG or R118A-MAG were generated with a retroviral transfection system using pMxs-puro retroviral vector Plat-E packaging cells and puromycin (1 μg/ml, Nacalai Tesque), as described previously (40, 41).
Immunoprecipitation and Immunoblotting
Infected melanoma cells were disrupted in lysis buffer (20 mm Tris, 150 mm NaCl (pH 7.5)) containing 1% Brij 98 (Sigma), and the lysates were immunoprecipitated with human WT-MAG-Ig, R118A-MAG-Ig, or control Ig and protein A-Sepharose beads (GE Healthcare). The 293T transfectants were lysed in buffer (1% Nonidet P-40, 1 mm phenylmethylsulfonyl fluoride, protease inhibitor mixture (Sigma), 2 mm EDTA, 10 mm Tris, and 150 mm NaCl (pH 7.5)). After treatment with or without sialidase, the immunoprecipitates of melanoma cells, total lysates of melanoma cells, and lysates of 293T transfectants were eluted by boiling in non-reducing SDS-PAGE sample buffer and separated on 5–20% polyacrylamide gels (ATTO). The proteins were transferred onto PVDF membranes (Millipore) and blotted with anti-gB mAb (SG2) and HRP-conjugated anti-mouse antibody (Thermo). The blotted membrane was incubated with a stripping buffer (101 mm 2-mercaptoethanol, 2% SDS, and 62.5 mm Tris-HCl (pH 6.7)) at 50 °C for 30 min and then reblotted with anti-human-Fc antibody (GE Healthcare). Each membrane was analyzed with LAS1000 and Imagegauge software (Millipore) or ChemiDoc Touch and Image Lab software (Bio-Rad).
Flow Cytometry Analysis
Cells were incubated with Ig fusion proteins or primary mAbs, followed by staining with allophycocyanin-conjugated anti-human IgG or anti-mouse IgG Ab (Jackson ImmunoResearch Laboratories) before analysis with a flow cytometer (FACSCalibur, BD Biosciences). MAG, R118A-MAG, gB, gB mutants, or mock were transfected with GFP into 293T cells. GFP+ cells were analyzed as transfected cells using CellQuest Pro (BD Biosciences). In the analysis of VZV binding, cells were incubated with cell-free VZV (Oka strain) suspension for 30 min on ice, followed by washing and staining with anti-gB mAb and allophycocyanin-conjugated anti-mouse IgG antibody.
Viral Infection
Cell-free VZV infection was analyzed by mixing 2 × 104 cells with various amounts of cell-free VZV in 96-well tissue culture plates. The plates were centrifuged at 2500 rpm for 2 h at 32 °C, followed by 24-h culture. The cells were analyzed by flow cytometry or by fluorescence microscopy (Carl Zeiss). Photographs were obtained using a D3 digital camera (Nikon), and the images were then processed using Canvas software (ACD Systems). GFP+ cells were detected as infected cells.
Cell-Cell Fusion Assay
Plasmids coding VZV-gB, VZV-gH, and VZV-gL as well as a plasmid encoding T7 RNA polymerase (pCAGT7) were cotransfected into 293T cells, and the transfectants were used as effector cells. A plasmid coding WT-MAG, R118A-MAG, or mock as well as a plasmid carrying the firefly luciferase gene under the control of the T7 promoter (pT7EMCLuc) was cotransfected into 293T cells, and the transfectants were used as target cells (42). As an internal control, the Renilla luciferase gene driven by the SV40 promoter (pRL-SV40, Promega) was also cotransfected into the effector cells or target cells. 24 h after transfection, the effector cells (4 × 104 cells) were cocultured with target cells (4 × 104 cells) in 96-well tissue culture plates for 18 h, and the efficiency of cell-cell fusion was quantified using a Dual-Luciferase reporter assay system (Promega) and luminometer (TriStar LB941, Berthold), as reported previously (10, 42). Relative firefly luciferase activity was calculated as follows: (firefly luciferase activity / Renilla luciferase activity) × 100) / maximum (firefly luciferase activity / Renilla luciferase activity). The cells were transfected with VZV glycoproteins and cultured with medium containing a combination of tunicamycin, DNJ, or benzyl-α-GalNac. Thereafter, VZV glycoproteins-transfected effector cells were cocultured with 293T target cells transfected with MAG in the presence of respective inhibitors. In the other assay, effector cells transfected with VZV glycoproteins were treated with sialidase for 30 min before coculture with target cells. Thereafter, effector cells were cocultured with target cells in the presence of sialidase. Significant differences between the results were determined using Student's t test or one-way analysis of variance (each significant p value is shown in the figures), where p < 0.05 was considered significant.
Metabolic Labeling
293T cells transfected with WT-gB, mutant gBs, or mock were cultured in DMEM containing 50 μm N-azidoacetylmannosamine (ManNAz) (Life Technologies) for 48 h. The cells were collected and incubated with PBS containing 1% FCS and phosphine conjugated with biotin (250 μm) (Cayman) at room temperature for 1 h. The cells were stained with streptavidin conjugated with allophycocyanin and subjected to flow cytometry analysis.
Results
SAs on gB Are Required to Interact with MAG
To analyze the role of SAs in the interaction between VZV gB and MAG, we first determined whether gB contains SAs in VZV-infected and gB-transfected cells. The cell lysates of VZV-infected and gB-transfected cells were treated with sialidase or mock, and the molecular weight of gB was determined by SDS-PAGE. We found two forms of gB with different apparent molecular weights in VZV-infected cells using anti-gB mAb (Fig. 1A). This result was consistent with a previous study where the immunoprecipitant obtained from the lysate of VZV-infected cells with anti-gB mAb contained 140- and 124-kDa gB molecules (29). The molecular weight of the 140-kDa gB treated with sialidase was significantly lower than that of the mock-treated gB (Fig. 1A). The 124-kDa band also shifted to a molecular mass lower than 100 kDa. These data suggest that both forms of gB are sialylated. At least, these results also suggest that the gB in VZV-infected and gB-transfected cells was sialylated. Next we used flow cytometry to examine whether the SAs on gB are required for the association with MAG. The MAG-Ig fusion protein that comprised the MAG extracellular domain and the Fc fragment of human immunoglobulin bound to the gB-transfectants, as reported previously (Fig. 1B, top panel) (10). MAG-Ig binding to the gB transfectants was decreased by sialidase treatment. On the other hand, MAG-Ig bound to the mock-transfected cells weakly. The binding was also decreased by sialidase treatment. The analysis using anti-gB mAb demonstrated that the cell surface expression level of gB was not changed by sialidase treatment (Fig. 1B, bottom panel). These results suggest that SAs are required for gB to associate with MAG and that gB possesses certain sialylated glycan structures that are preferentially recognized by MAG compared with other molecules on 293T cells.
FIGURE 1.
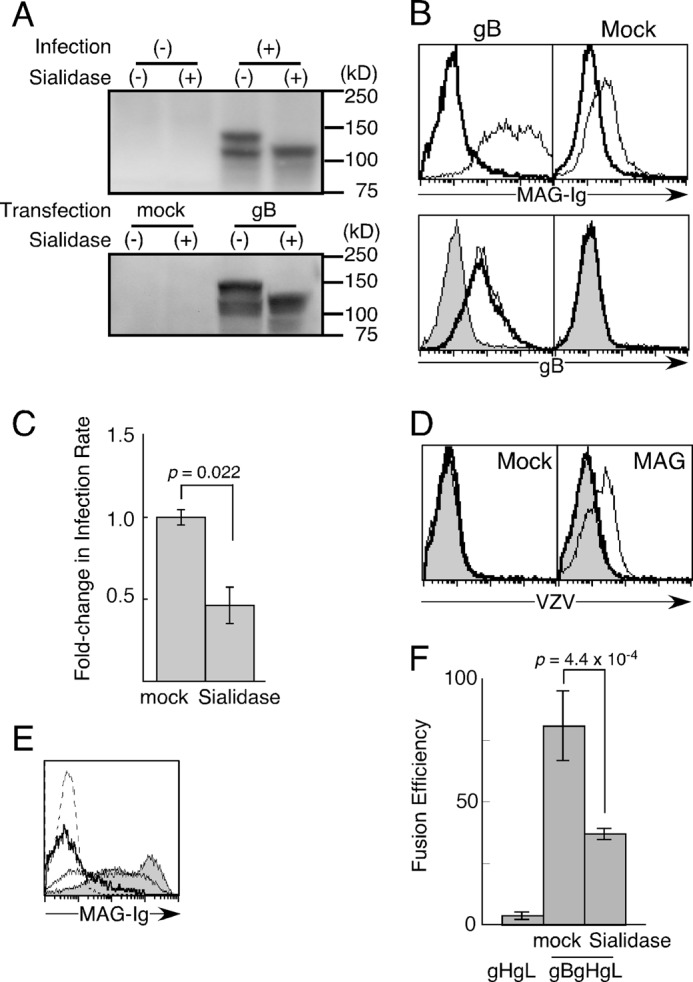
There is a requirement for sialic acids to interact with MAG. A, MeWo cells were infected with (+) or without VZV (−) (top panel). 293T cells were transfected with gB or mock-transfected (bottom panel). Cell lysates were analyzed by non-reducing SDS-PAGE after treatment with (+) or without sialidase (−). Proteins were blotted with anti-gB mAb. B, mock- or gB-transfected 293T cells were treated with sialidase (bold line) or vehicle (thin line) and stained with MAG-Ig or anti-gB mAb, followed by flow cytometry analysis. Cells were stained with secondary antibody only (gray area). C, OL cells stably transfected with WT-MAG were exposed to GFP-VZV at a multiplicity of infection of 0.2 for 24 h. Fold-changes in the infection rate are shown. Fold-changes in the infection rate were calculated by dividing the percentage of GFP+ cells in each indicated treatment by the percentage of GFP+ cells infected with mock-treated VZV (%GFP+ cells among the total cells exposed to mock- or sialidase-treated VZV/%GFP+ cells among the total cells exposed to mock-treated VZV). Representative data from three independent experiments are shown. The error bars represent the mean ± S.D. on the basis of triplicate samples, and the p values were calculated using Student's t test. D, 293T cells transfected with MAG and mock-transfected were incubated with VZV virions after mock treatment (thin lines) or treatment with sialidase for 30 min (bold lines), respectively, followed by staining with anti-gB mAb and secondary antibody. Cells were also stained with antibodies only (gray area). E, 293T cells transfected with gB were stained with MAG-Ig after treatment with sialidase for 0 (gray area), 10 (thin line), and 30 min (bold line). Cells were also stained with secondary antibodies only (dotted line). F, 293T effector cells transfected with gH, gL, T7 polymerase, and Renilla luciferase as an internal control as well as gB (gBgHgL) or mock-transfected (gHgL) were cocultured with other 293T target cells transfected with WT-MAG and firefly luciferase driven by the T7 polymerase promoter. gB-transfected cells were treated with sialidase or mock-treated. The relative fusion efficiencies are shown on the basis of representative data from three independent experiments. The error bars represent the mean ± S.D. on the basis of triplicate samples, and p values were calculated using Student's t test.
We then examined whether sialylated gB is involved in VZV entry into MAG-expressing cells. MAG-transfected oligodendrocytes (OL cells) were exposed to cell-free recombinant VZV carrying the GFP reporter gene (GFP-VZV). GFP-VZV virions were treated with sialidase or mock-treated before infection. The proportion of VZV-infected cells among the MAG-transfectants decreased by 50% after sialidase treatment compared with mock treatment (Fig. 1C). Indeed, virion binding to MAG-expressing cells was also decreased by treatment with sialidase (Fig. 1D). Therefore, the SAs in VZV appear to be involved in viral binding and entry. However, the treatment of virions, even mock treatment, for more than 15 min resulted in no infectivity (data not shown). Therefore, we treated VZV virions just with sialidase for 10 min. Sialidase treatment of VZV virions significantly inhibited VZV infection compared with mock treatment, although the inhibition was incomplete. MAG-Ig binding to gB-transfected cells was not decreased completely with sialidase treatment for 10 min, although MAG-Ig binding to gB-expressing cells was almost completely decreased after sialidase treatment for 30 min (Fig. 1E). Therefore, the incomplete inhibition of VZV infection by sidalidase treatment for 10 min seems to be due to the insufficient removal of SAs from VZV.
Therefore, we also examined the involvement of SAs in membrane fusion using a cell-cell fusion assay. 293T cells were transfected with gB, gH, and gL and then treated with sialidase for 30 min before coculture. Thereafter, the sialidase-treated effector cells were cocultured with 293T target cells transfected with MAG in the presence of sialidase. Cell-cell fusion was significantly but incompletely inhibited by sialidase (Fig. 1F). These observations suggested that the SAs appeared to be involved in cell-cell fusion. In addition, newly synthesized and sialylated gB on effector cells might have mediated weak cell-cell fusion, even in the presence of sialidase, although there is a possibility that sialidase activity was not enough to remove SAs completely.
Arg-118 in MAG Plays an Important Role in the Association with gB
Furthermore, we employed an alternative approach to determine whether the SAs on VZV are involved in the infection process. Siglecs possess a conserved Arg residue that is essential for the recognition of SAs. Indeed, Arg-118 is known to be essential for MAG to associate with SA-containing molecules (11). To investigate whether Arg-118 is involved in the interaction between MAG and gB, we generated a mutated MAG-Ig fusion protein by mutating Arg-118 to Ala (R118A-MAG-Ig). As shown in Fig. 2A, gB-transfected 293T cells were stained strongly with wild-type MAG-Ig (WT-MAG-Ig) but not with R118A-MAG-Ig or control Ig. Furthermore, when the cell lysates of melanoma cells infected with VZV were immunoprecipitated with WT- and R118A-MAG-Ig fusion proteins, gB was precipitated with WT-MAG-Ig but not with R118A-MAG-Ig (Fig. 2B). By contrast, 293T cells transfected with WT-MAG were stained with gB-Ig, but 293T cells transfected with R118A-MAG were not stained with gB-Ig (Fig. 2C). R118A-MAG transfectants also showed less binding to VZV virions than WT-MAG transfectants (Fig. 2D). These results suggest that Arg-118 in MAG is essential for the association between MAG and VZV.
FIGURE 2.
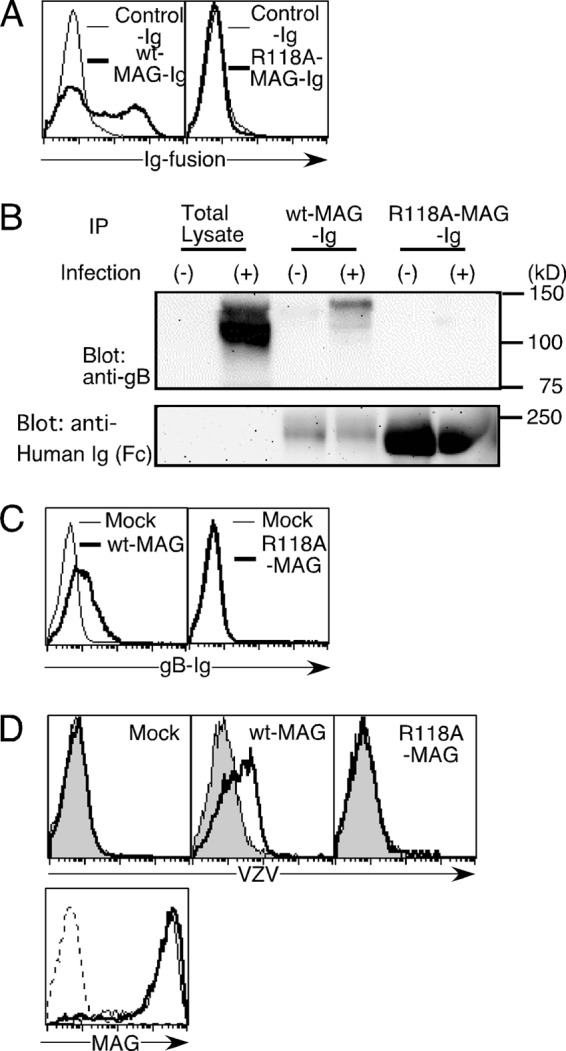
The role of the arginine residue at MAG position 118 in MAG association with gB. A, 293T cells transfected with gB were stained with WT-MAG-Ig (bold line, left panel), R118A-MAG-Ig (bold line, right panel), or control Ig (thin lines, both panels). B, VZV-infected MeWo cells (+) or non-infected MeWo cells (−) were lysed, and the cell lysates were immunoprecipitated (IP) with the indicated Ig fusion proteins. The precipitants and total lysates were analyzed by non-reducing SDS-PAGE, followed by blotting with anti-gB mAb (top panel) or anti-human-Fc Ab (bottom panel). C, 293T cells transfected with WT-MAG (bold line) or mock-transfected (thin line) were stained with gB-Ig (left panel). 293T cells transfected with R118A-MAG (bold line) or mock-transfected (thin line) were stained with gB-Ig (right panel). D, 293T cells transfected with WT-MAG and R118A-MAG and mock-transfected were incubated with VZV virions, followed by staining with anti-gB mAb and secondary antibody. (bold lines). Cells were also stained with antibodies only (gray area, top panels). The relative cell numbers of MAG expression of 293T cells transfected with WT-MAG (thin line) and R118A-MAG (bold line) and mock-transfected (dotted line) are shown (bottom panel).
Arg-118 in MAG Is Required for MAG-mediated Cell-Cell Fusion and VZV Entry
Membrane fusion is necessary for VZV entry into host cells, and the interaction between MAG and gB mediates membrane fusion during VZV infection (10). Using a cell-cell fusion assay, we analyzed whether the SA-dependent recognition of gB by MAG is involved in membrane fusion. The WT-MAG-transfected cells exhibited efficient cell-cell fusion with cells transfected with VZV envelope glycoproteins (gB, gH, and gL), whereas R118A-MAG-transfected and mock-transfected cells exhibited little fusion (Fig. 3A).
FIGURE 3.
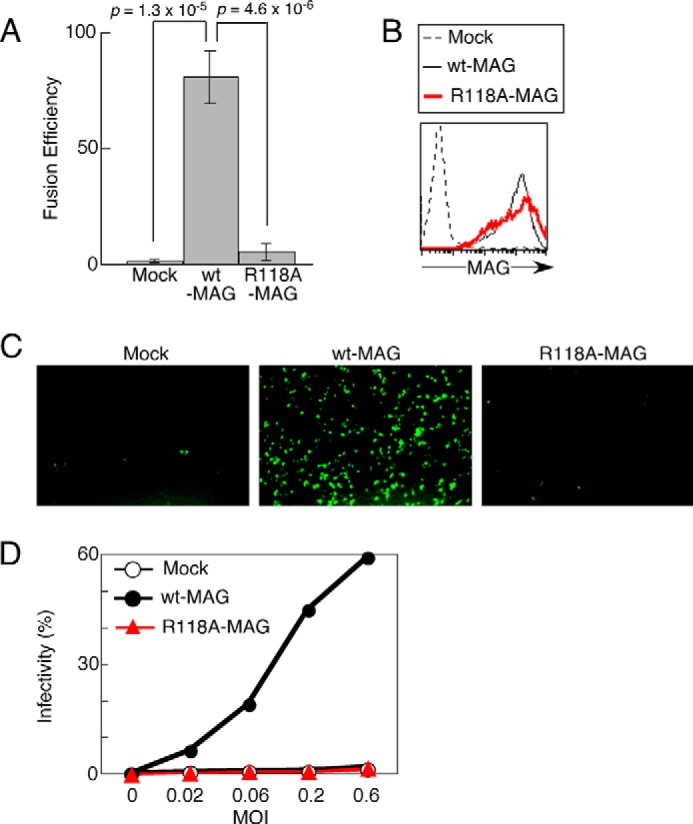
The requirement of sialic acids for MAG-mediated cell-cell fusion and VZV entry. A, effector 293T cells transfected with gB, gH, gL, and T7 polymerase were cocultured with target 293T cells transfected with WT-MAG or R118A-MAG or mock-transfected as well as transfected with firefly luciferase driven by the T7 polymerase promoter and Renilla luciferase as an internal control. The relative fusion efficiencies are shown on the basis of representative data from three independent experiments. The error bars represent the mean ± S.D. on the basis of six replicates. B, OL cells stably transfected with WT-MAG and R118A-MAG or mock-transfected were stained with anti-MAG mAb. C, OL cells transfected with WT-MAG, R118A-MAG, or mock-transfected were infected with GFP-VZV at a multiplicity of infection of 0.2 for 24 h, followed by fluorescence microscopy analysis at ×100 magnification. D, OL cells transfected with WT-MAG and R118A-MAG or mock-transfected were infected with GFP-VZV at the indicated multiplicity of infection for 24 h. The proportions of VZV-infected cells (GFP+) are shown. Representative data from three independent experiments are shown. The error bars represent the mean ± S.D. on the basis of duplicate samples.
Next, we examined whether MAG recognition of sialylated gB is involved in VZV infection of MAG-expressing cells. WT-MAG-, R118A-MAG-, or mock-transfected oligodendrocytes were exposed to GFP-VZV. As shown in Fig. 3B, the expression levels of R118A-MAG on cell surfaces were comparable with that of WT-MAG when they were analyzed using an anti-MAG monoclonal antibody. As expected, both the R118A-MAG transfectants and mock transfectants were resistant to GFP-VZV, whereas the WT-MAG transfectants were infected efficiently with GFP-VZV when the GFP expression levels in the infected cells were analyzed by fluorescence microscopy (Fig. 3C). Similar results were obtained by flow cytometry. The proportions of infected cells among WT-MAG-transfected cells increased in a virus dose-dependent manner but not those among R118A-MAG-transfected cells or mock-transfected cells (Fig. 3D). These results suggest that MAG recognition of SA on gB is required for membrane fusion during VZV infection of MAG-expressing cells.
Both N- and O-glycosylation of gB Are Required for the Association of gB with MAG and Cell-Cell Fusion
It is known that SAs typically exist at the termini of N- or O-linked glycans or glycosphingolipids in mammalian cells (43). We predicted the N- or O-glycosylation sites on VZV gB using the NetNGlyc 1.0 server and NetOGlyc 4.0 server. The extracellular domain of gB was predicted to have seven N-glycosylation sites and 17 O-glycosylation sites (Fig. 4A). gB-transfected 293T cells were treated with inhibitors of N-glycan synthesis, tunicamycin and DNJ, or an inhibitor of O-glycan synthesis, benzyl-α-GalNac. The gB-transfectants treated with tunicamycin and DNJ associated less efficiently with WT-MAG-Ig compared with mock-treated cells (Fig. 4B). Treatment with benzyl-α-GalNac also decreased the binding of WT-MAG-Ig to gB-transfected cells (Fig. 4B). WT-MAG-Ig also exhibited a lower affinity for mock transfectants after treatment with benzyl-α-GalNac than after mock treatment. These results suggest that WT-MAG-Ig recognizes not only sialylated N-glycans on gB but also sialylated O-glycans on certain molecules, including gB. In addition, benzyl-α-GalNac also seems to affect glycan structures on certain MAG-binding molecules of 293T cells. Using a cell-cell fusion assay, we then analyzed whether SAs on the N- and O-glycans of gB-transfected cells were involved in membrane fusion with MAG-expressing cells. 293T effector Cells transfected with VZV glycoproteins or mock-transfected were incubated with a medium that contained combination of tunicamycin, DNJ, benzyl-α-GalNac, or mock before subsequent coculture with MAG-expressing cells. Thereafter, effector cells were cocultured with target cells in the presence of inhibitors. The treatment with tunicamycin, DNJ, or benzyl-α-GalNac decreased fusion efficiency compared with mock treatment (Fig. 4C). Furthermore, culture with medium containing N-glycosylation inhibitors along with benzyl-α-GalNac showed lower fusion efficiencies than culture with medium containing N-glycosylation inhibitors or O-glycosylation inhibitor (Fig. 4C). VZV produced by cells cultured in the presence of N-glycosylation and/or O-glycosylation inhibitors showed a lower infectivity to MAG-expressing cells than VZV produced by cells cultured in the absence of these inhibitors (Fig. 4D). The infectivity of VZV derived from cells incubated with either of the two N-glycosylation inhibitors along with benzyl-α-GalNac was not significantly different from the infectivity of VZV derived from cells treated with either of these inhibitors (Fig. 4D). These results suggest that SAs capping N- and O-glycans of gB are involved in cell-cell fusion and VZV infection mediated by MAG.
Mutation of Putative O-glycosylation Sites on gB Does Not Significantly Affects Cell-Cell Fusion
It is important to identify specific amino acid residues or domains of envelope glycoproteins that contribute to membrane fusion to elucidate the mechanism of viral entry. We investigated which of the seven N-glycosylation sites or 17 O-glycosylation sites are required for cell-cell fusion with cells that express MAG. We generated gBs where Asn residue or Ser/Thr residues were substituted with Ala at each putative N-glycosylated or O-glycosylated site.
As shown in Fig. 5, cells that expressed gBs with mutations of putative O-glycosylation sites exhibited comparable fusion efficiency as WT-gB-expressing cells. We performed cell-cell fusion assays using gBs where other Ser/Thr residues were substituted with Ala. We thought that the SAs on O-glycans might be involved in the recognition of gB by MAG in the same way that SAs on the O-glycans of HSV gB are required for PILRα to bind to HSV gB (26, 44). We selected Ser/Thr residues in gB that were predicted to have enhancement value product values of >2.00 using the ISOGlyP server. However, the fusion efficiency of each mutated gB-transfected cell type was not decreased significantly compared with that of the WT-gB-transfected cells, except for cell types transfected with mutants of Thr-129, Thr-265, and Ser-559 (Fig. 6A). The cell surface expression of gBs where Thr-129 or Ser-559 was substituted with Ala (T129A-gB or S559A-gB) was impaired severely (Fig. 6B). We expressed T129A-gB, S559A-gB, and WT-gB as GPI-anchored forms (T129A-gB-GPI, S559A-gB-GPI, and WT-gB-GPI) to express them equally on the cell surface. The expression levels of T129A-gB-GPI and S559A-gB-GPI were comparable with the expression level of WT-gB-GPI, and the interaction between MAG-Ig and S559A-gB-GPI was similar to the interaction with WT-gB-GPI (Fig. 6B). T265A-gB was expressed on the transfected cell surface as well as WT-gB. MAG-Ig binding to T265A-gB-transfectants was also as efficient as MAG-Ig binding to WT-gB-transfectants (Fig. 6B). Therefore, Thr-129, Thr-265, and Ser-559 did not appear to be involved in cell-cell fusion mediated by binding to MAG. Overall, none of the O-glycosylation site-mutated gBs exhibited significantly decreased fusion efficiencies compared with WT-gB.
FIGURE 5.
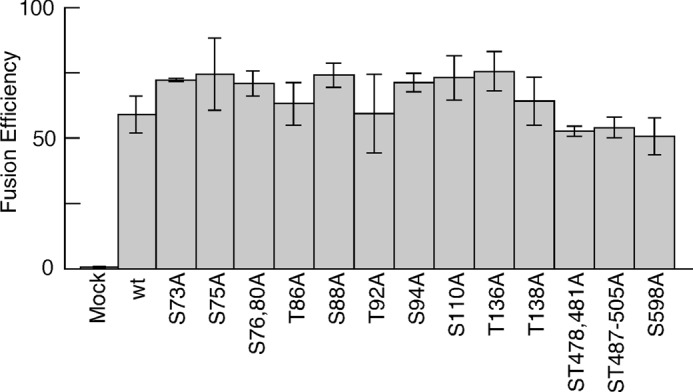
Comparable cell-cell fusion efficiency of WT-gB-transfected effector cells with that of effector cells transfected with gB carrying mutation(s) of putative O-glycosylation sites. WT-gB or each mutated gB in which putative O-glycosylation sites were mutated was cotransfected into 293T effector cells with gH and gL. The effector cells were cocultured with other 293T target cells transfected with MAG, followed by luminescence measurements. The relative fusion efficiencies are shown on the basis of representative data from three independent experiments. The error bars represent the mean ± S.D. on the basis of six replicate samples. Statistical differences were determined using Student's t test. Each mutated gB without a p value did not differ significantly compared with wild-type gB (WT). p < 0.05 was considered significant.
Asn-557 and Asn-686 in gB Are Involved in Cell-Cell Fusion Mediated by the Association with MAG
Among Cells that expressed gB with mutations in putative N-glycosylation sites, the binding efficiencies to MAG-Ig of cells that expressed gB with a mutation in Asn-147, Asn-557, or Asn-686 (N147A-gB, N557A-gB, or N686A-gB) were lower than that of WT-gB (Fig. 7A, top panel). However, the binding of anti-gB mAb to N147A-gB also decreased (Fig. 7A, bottom panel). When we generated the GPI-anchored form of N147A-gB, the expression level of N147A-gB-GPI was comparable with that of WT-gB-GPI and the binding of MAG-Ig to N147A-gB-GPI was similar to the binding to WT-gB-GPI (Fig. 7B). Cells that expressed gB with mutations in putative N-glycosylation sites, Asn-147, Asn-557, or Asn-686 (N147A-gB, N557A-gB, or N686A-gB), exhibited reduced cell-cell fusion with MAG-expressing cells compared with cells that expressed WT-gB (Fig. 7C). Therefore, the decreased fusion efficiency caused by the mutation of Asn-147 appeared to be attributable to the decreased surface expression of mutant gB (Fig. 7A). On the other hand, Asn-686 might additionally contribute to cell-cell fusion because N686A-gB significantly but slightly decreased fusion efficiency. Therefore, Asn-557 in gB mainly appears to be the key N-glycosylation site involved in membrane fusion with MAG-expressing cells.
FIGURE 7.
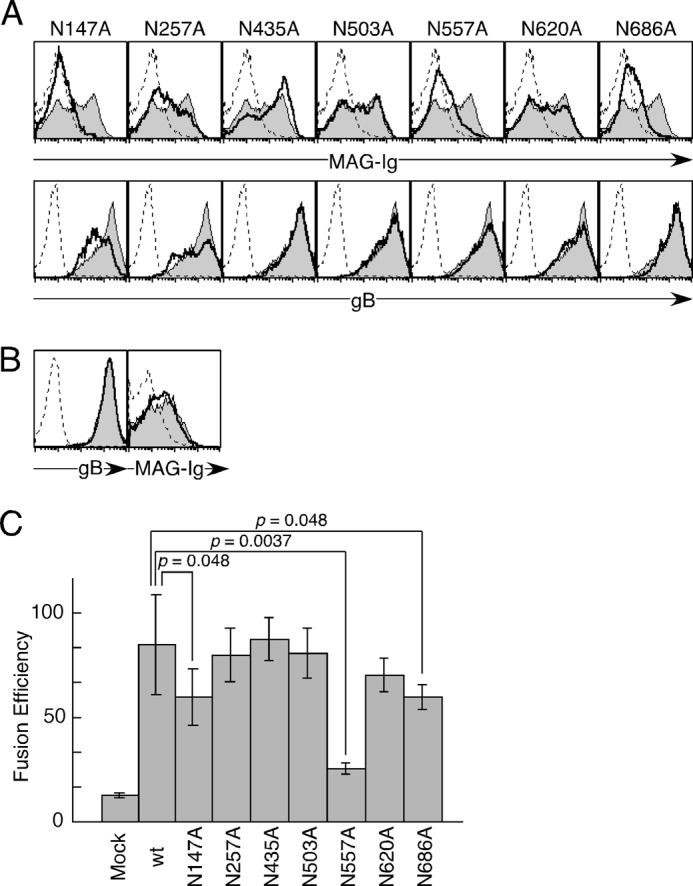
The involvement of several Asn residues of gB in cell-cell fusion. A, mock-transfected (dotted line), WT-gB-transfected (gray area), or mutated gB-transfected (bold line) 293T cells were stained with MAG-Ig (top panels) and anti-gB mAb (bottom panels), followed by flow cytometry analysis. B, mock-transfected (dotted line), WT-gB-GPI-transfected (gray area), or gB (N147A)-GPI-transfected (bold line) 293T cells were stained with anti-gB mAb (left panel) or MAG-Ig (right panel), followed by flow cytometry analysis. C, WT-gB or mutated gB in which putative N-glycosylation sites were mutated was cotransfected into 293T effector cells with gH and gL. The effector cells were cocultured with other 293T target cells transfected with MAG, followed by luminescence measurements. The relative fusion efficiencies are shown on the basis of representative data from three independent experiments. The error bars represent the mean ± S.D. on the basis of six replicate samples. Statistical differences were determined using Student's t test. Each mutated gB without a p value did not differ significantly compared with wild-type gB (WT). p < 0.05 was considered significant.
Asn-557 and Asn-686 Are Involved in gB Sialylation
To confirm whether Asn-557 and Asn-686 are sialylated, 293T cells transfected with WT-gB, N557A-gB, N686A-gB, and mock transfectants were labeled with ManNAz (Fig. 8, A and B). Using this labeling method, 5–40% of the total SAs in glycoproteins and glycolipids were labeled with ManNAz, which was detected by biotin-labeled phosphine (45, 46). In this analysis, the overexpression of gB in 293T cells significantly increased SAs on the cell surface compared with mock transfectants. Expression levels of N557A-gB and N686A-gB on the cell surface were almost the same as that of WT-gB (data not shown). The cell surfaces of the N557A-gB- and N686A-gB-transfected cells were weakly labeled with ManNAz compared with WT-gB, although it was labeled more than the tunicamycin-treated WT-gB transfectants. The cell surfaces of N686A-gB-transfected cells were significantly but weakly labeled with ManNAz compared with WT-gB-transfected cells. These results suggest that both N557 and N686 are involved in sialylation of gB.
FIGURE 8.
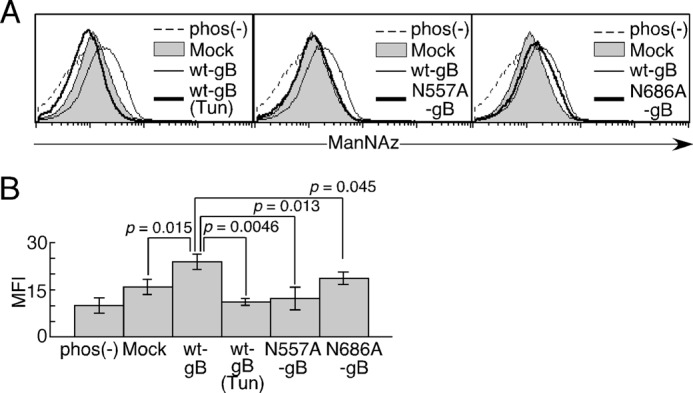
The sialylation of Asn-557 and Asn-686 in gB. A, 293T cell transfectants were labeled with ManNAz. The relative cell numbers are shown for mock transfectants (gray area), WT gB transfectants (thin line), mutated gB transfectants, and WT gB transfectants treated with tunicamycin (Tun) (bold line). gB transfectants labeled with ManNAz but not incubated with phosphine-biotin were stained with SA-allophycocyanin (dotted line). B, the mean fluorescence intensity (MFI) of each cell is shown. The error bars represent the mean ± S.D. on the basis of triplicate samples. Statistical differences were determined using Student's t test. p < 0.05 was considered significant. Representative data from three independent experiments are shown.
Discussion
It has been suggested that the SAs of envelope proteins are involved in VZV infection, although the mechanism has remained unclear (31, 32). Sialylation of gB, the gE-gI complex, and the gH-gL complex have been reported previously (27–32). In a previous study, we also demonstrated that MAG associates with gB and gE but not with the gH-gL complex (10). It has been suggested that gE is essential for VZV survival (27) but that gE and gI are not involved in MAG-mediated cell-cell fusion (10). Therefore, we investigated whether the SAs on gB are involved in membrane fusion during VZV infection of MAG-expressing cells. In addition, MAG has been reported to bind ligands in an SA-dependent or SA-independent manner (22, 23). In this study, we demonstrated that the SAs on VZV gB are required for the interaction between gB and MAG. In addition, we observed that WT-MAG-Ig immunoprecipitated 140-kDa gB more efficiently than 124-kDa gB from VZV-infected cells. It has been reported that 140-kDa gB is the sialylated form of 124-kDa sulfated gB (29). These suggest that WT-MAG tends to recognize sialylated gB more strongly than unsialylated gB. Without sialidase treatment, VZV also lost its infectivity as time passed, but the infectivity of VZV treated with sialidase was lower than that of untreated VZV. In addition, sialidase treatment abrogated cell-cell fusion incompletely probably because newly synthesized and sialylated gB on effector cells might mediate weak fusion.
To determine whether SAs are involved in VZV infection, we employed a MAG mutant where Arg-118 was substituted with Ala. R118A-MAG did not mediate membrane fusion with VZV envelope glycoproteins. Although R118A-MAG was expressed on the cell surface as well as WT-MAG, there is a possibility that substitution of Arg-118 with Ala caused a conformational change in MAG and affected cell-cell fusion. Because the structure of MAG has not been reported, we do not know whether Arg-118 affects the conformation of MAG. However, Arg-118, which forms an important salt bridge with the negatively charged carboxyl group of SA, is conserved among other Siglec family molecules. The structures of other Siglec family molecules suggest that Arg-118 in MAG is directly involved in binding to SAs (11, 12, 47–54). Therefore, mutation in Arg-118 in MAG seems to have affected the association with SAs rather than the conformation of MAG. PILRα, which has a structure similar to the structures of Siglec family molecules, recognizes both the sialylated sugar chain and polypeptides (44). Therefore, there is a possibility that MAG also recognizes both sialylated sugar chains and polypeptides, although the binding of MAG to fibronectin and gangliosides such as GD1a and GT1b was blocked by SA-containing glycans (19–21).
Previously, it has been reported that MAG binds to both N-glycans and O-glycans in an SA-dependent manner. Some studies have shown that MAG binds to O-glycans more strongly than N-glycans (55), whereas other studies have demonstrated that MAG has a higher affinity for N-glycans than O-glycans (56, 57). In addition, fibronectin, which possesses more N-glycans than O-glycans, is suggested to be one of the MAG ligands that binds in an SA-dependent manner (19). Similarly, MAG binds to gB with seven putative N-glycosylation sites and 17 putative O-glycosylation sites in the extracellular domain. The treatment of gB-transfected cells with tunicamycin, DNJ, or benzyl-α-GalNAc decreased the binding of MAG-Ig to the gB-transfected cells. The fusion efficiency of cells treated with tunicamycin, DNJ, or benzyl-α-GalNac was higher than that of cells treated with sialidase. Furthermore, N- and O-glycosylation inhibitors additively inhibited cell-cell fusion. These results suggest that both the N- and O-glycans on gB are involved in cell-cell fusion. Anti-gB mAb (151) inhibits VZV infection, but this mAb failed to precipitate gB from tunicamycin-treated VZV-infected cells (29), thereby supporting the hypothesis that the N-glycans on gB are required for VZV infection. In this study, VZV infection was blocked by N- or O-glycosylation inhibitors, although there is a possibility that not only gB but also other essential molecules of VZV might be modified by these inhibitors, resulting in the impairment of VZV replication or survival.
In fact, we demonstrated that two of seven putative N-glycosylation sites (Asn-557 and Asn-686) are sialylated and that they are involved in binding to gB, thereby mediating significant cell-cell fusion. Weak labeling of the cell surface of N686A-gB-transfected cells by ManNAz might result in a trivial decrease of cell-cell fusion by N686A-gB-transfected cells. The mutation of a single glycosylation site, Asn-557, on gB decreased the amount of SAs compared with WT gB on the cell surface. This suggests that more sialylated N-glycans are attached on Asn-557 than other glycosylation sites, including Asn-686. N-glycans differ in branch number, composition, length, capping arrangements, and core modifications, resulting in the generation of N-glycans possessing a various number of SAs (58). Indeed, glycoproteins often have a range of different N-glycans on a particular N-glycosylation site (58, 59). Different sites in a molecule have different subsets of N-glycans with different numbers of SAs (58, 60, 61). Therefore, the amount of SAs on each gB glycosylation site might be different, and Asn-557 of gB seems to be the major glycosylation site for sialylated N-glycans.
On the other hand, although expression levels of N557A-gB and N686A-gB on the cell surface were almost the same as that of WT-gB, there is a possibility that the substitution of Asn-557 and Asn-686 with Ala might have caused a conformational change in gB, resulting in loss of cell-cell fusion. Indeed, T265A-gB lost fusion function, although the expression on the cell surface and binding to MAG-Ig were not impaired. Thr-265 is localized in a gB fusion loop that is essential for membrane fusion (62). In addition, Asn-557 and Asn-686 are localized in domain III and domain V of gB, respectively, although their functions are unclear (62). Therefore, there is a possibility that the conformational changes of gB might be caused by point mutations, N557A and N686A as well as T265A, and influence fusion function.
On the other hand, the predicted O-glycosylation sites were not accurate compared with the predicted N-glycosylation sites because many enzymes are involved in O-glycan synthesis, whereas a single oligosaccharyltransferase enzyme transfers an oligosaccharide to the target protein during N-glycan generation (63, 64). The fusion efficiencies of cells transfected with gB that possessed mutations in each putative O-glycosylation site were not specifically decreased compared with the fusion efficiency of cells transfected with WT-gB. However, O-glycosylation of gB appears to be necessary for cell-cell fusion because the treatment with benzyl-α-GalNAc significantly impaired cell-cell fusion. It is possible that there are other O-glycosylated Ser/Thr residues or that O-glycans generated on multiple O-glycosylation sites might be synchronously involved in cell-cell fusion.
Cell-cell fusion assays are powerful tools for the direct examination of the molecules essential for membrane fusion during viral entry because it is not necessary to consider the effects of other viral components. The cell-cell fusion assay using VZV gB, gH, gL, and MAG is the only system available for analyzing the mechanism of VZV membrane fusion. In this study, we investigated the involvement of SAs in membrane fusion during VZV entry using cell-cell fusion assays. This system is useful for investigating the roles of SAs but also other molecular modifications of the virus and host during VZV infection. Our study provides novel insights into the molecular mechanisms involved in membrane fusion during VZV infection.
Author Contributions
T. S. designed the study, performed experiments, analyzed and discussed the data, and wrote the manuscript. M. M. and F. A. assisted with experiments and discussed the data. M. K. and K. H assisted with experimental design and discussed the data. Y. M. discussed the data. H. A. designed the study, analyzed the data, and wrote the manuscript.
Acknowledgments
We thank K. Furuno, K. Shida, S. Matsuoka, and R. Hirohata for technical assistance and C. Kita for secretarial assistance.
This research was supported by JSPS KAKENHI grants 15H02545, 24115005, and 25460565 Japan (to T. S. and H. A.) and partly by grants from the Takeda Science Foundation (to T. S.) and from the Terumo Life Science Foundation, the Uehara Memorial Foundation, the Tokyo Biochemical Research Foundation, and the Naito Foundation (to H. A.). The authors declare that they have no conflicts of interest with the contents of this article.
- HSV
- herpes simplex virus
- VZV
- varicella-zoster virus
- gB
- glycoprotein B
- MAG
- myelin-associated glycoprotein
- SA
- sialic acid
- Siglec
- sialic acid-binding immunoglobulin-type lectin
- DNJ
- deoxynojirimycin
- GPI
- glycosylphosphatidylinositol
- ManNAz
- N-azidoacetylmannosamine.
REFERENCES
- 1. Arvin A. M. (2001) Varicella-zoster virus: molecular virology and virus-host interactions. Curr. Opin. Microbiol. 4, 442–449 [DOI] [PubMed] [Google Scholar]
- 2. Cohen J. I., Straus S. E., Arvin A. M. (2006) Varicella-zoster virus: replication, pathogenesis, and management. in Fields VIROLOGY, 5th Ed., Vol. 2, pp. 2773–2818, Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 3. Zerboni L., Sen N., Oliver S. L., Arvin A. M. (2014) Molecular mechanisms of varicella zoster virus pathogenesis. Nat. Rev. Microbiol. 12, 197–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Steiner I., Kennedy P. G., Pachner A. R. (2007) The neurotropic herpes viruses: herpes simplex and varicella-zoster. Lancet Neurol. 6, 1015–1028 [DOI] [PubMed] [Google Scholar]
- 5. Connolly S. A., Jackson J. O., Jardetzky T. S., Longnecker R. (2011) Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 9, 369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sedý J. R., Spear P. G., Ware C. F. (2008) Cross-regulation between herpesviruses and the TNF superfamily members. Nat. Rev. Immunol. 8, 861–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Smith A. E., Helenius A. (2004) How viruses enter animal cells. Science 304, 237–242 [DOI] [PubMed] [Google Scholar]
- 8. Marsh M., Helenius A. (2006) Virus entry: open sesame. Cell 124, 729–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Steven A. C., Spear P. G. (2006) Biochemistry: viral glycoproteins and an evolutionary conundrum. Science 313, 177–178 [DOI] [PubMed] [Google Scholar]
- 10. Suenaga T., Satoh T., Somboonthum P., Kawaguchi Y., Mori Y., Arase H. (2010) Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc. Natl. Acad. Sci. U.S.A. 107, 866–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Varki A., Angata T. (2006) Siglecs: the major subfamily of I-type lectins. Glycobiology 16, 1R–27R [DOI] [PubMed] [Google Scholar]
- 12. Crocker P. R., Paulson J. C., Varki A. (2007) Siglecs and their roles in the immune system. Nat. Rev. Immunol. 7, 255–266 [DOI] [PubMed] [Google Scholar]
- 13. Domeniconi M., Cao Z., Spencer T., Sivasankaran R., Wang K., Nikulina E., Kimura N., Cai H., Deng K., Gao Y., He Z., Filbin M. (2002) Myelin-associated glycoprotein interacts with the Nogo66 receptor to inhibit neurite outgrowth. Neuron 35, 283–290 [DOI] [PubMed] [Google Scholar]
- 14. Liu B. P., Fournier A., GrandPré T., Strittmatter S. M. (2002) Myelin-associated glycoprotein as a functional ligand for the Nogo-66 receptor. Science 297, 1190–1193 [DOI] [PubMed] [Google Scholar]
- 15. Wörter V., Schweigreiter R., Kinzel B., Mueller M., Barske C., Böck G., Frentzel S., Bandtlow C. E. (2009) Inhibitory activity of myelin-associated glycoprotein on sensory neurons is largely independent of NgR1 and NgR2 and resides within Ig-like domains 4 and 5. PLoS ONE 4, e5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Venkatesh K., Chivatakarn O., Lee H., Joshi P. S., Kantor D. B., Newman B. A., Mage R., Rader C., Giger R. J. (2005) The Nogo-66 receptor homolog NgR2 is a sialic acid-dependent receptor selective for myelin-associated glycoprotein. J. Neurosci. 25, 808–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Atwal J. K., Pinkston-Gosse J., Syken J., Stawicki S., Wu Y., Shatz C., Tessier-Lavigne M. (2008) PirB is a functional receptor for myelin inhibitors of axonal regeneration. Science 322, 967–970 [DOI] [PubMed] [Google Scholar]
- 18. Goh E. L., Young J. K., Kuwako K., Tessier-Lavigne M., He Z., Griffin J. W., Ming G. L. (2008) β1-integrin mediates myelin-associated glycoprotein signaling in neuronal growth cones. Mol. Brain 1, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Strenge K., Brossmer R., Ihrig P., Schauer R., Kelm S. (2001) Fibronectin is a binding partner for the myelin-associated glycoprotein (siglec-4a). FEBS Lett. 499, 262–267 [DOI] [PubMed] [Google Scholar]
- 20. Vinson M., Strijbos P. J., Rowles A., Facci L., Moore S. E., Simmons D. L., Walsh F. S. (2001) Myelin-associated glycoprotein interacts with ganglioside GT1b. A mechanism for neurite outgrowth inhibition. J. Biol. Chem. 276, 20280–20285 [DOI] [PubMed] [Google Scholar]
- 21. Vyas A. A., Patel H. V., Fromholt S. E., Heffer-Lauc M., Vyas K. A., Dang J., Schachner M., Schnaar R. L. (2002) Gangliosides are functional nerve cell ligands for myelin-associated glycoprotein (MAG), an inhibitor of nerve regeneration. Proc. Natl. Acad. Sci. U.S.A. 99, 8412–8417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Quarles R. H. (2009) A hypothesis about the relationship of myelin-associated glycoprotein's function in myelinated axons to its capacity to inhibit neurite outgrowth. Neurochem. Res. 34, 79–86 [DOI] [PubMed] [Google Scholar]
- 23. Schnaar R. L. (2010) Brain gangliosides in axon-myelin stability and axon regeneration. FEBS Lett. 584, 1741–1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vigerust D. J., Shepherd V. L. (2007) Virus glycosylation: role in virulence and immune interactions. Trends Microbiol. 15, 211–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Teuton J. R., Brandt C. R. (2007) Sialic acid on herpes simplex virus type 1 envelope glycoproteins is required for efficient infection of cells. J. Virol. 81, 3731–3739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang J., Fan Q., Satoh T., Arii J., Lanier L. L., Spear P. G., Kawaguchi Y., Arase H. (2009) Binding of herpes simplex virus glycoprotein B (gB) to paired immunoglobulin-like type 2 receptor α depends on specific sialylated O-linked glycans on gB. J. Virol. 83, 13042–13045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Berarducci B., Rajamani J., Reichelt M., Sommer M., Zerboni L., Arvin A. M. (2009) Deletion of the first cysteine-rich region of the varicella-zoster virus glycoprotein E ectodomain abolishes the gE and gI interaction and differentially affects cell-cell spread and viral entry. J. Virol. 83, 228–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Litwin V., Jackson W., Grose C. (1992) Receptor properties of two varicella-zoster virus glycoproteins, gpI and gpIV, homologous to herpes simplex virus gE and gI. J. Virol. 66, 3643–3651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Montalvo E. A., Grose C. (1987) Assembly and processing of the disulfide-linked varicella-zoster virus glycoprotein gpII(140). J. Virol. 61, 2877–2884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Montalvo E. A., Parmley R. T., Grose C. (1985) Structural analysis of the varicella-zoster virus gp98-gp62 complex: posttranslational addition of N-linked and O-linked oligosaccharide moieties. J. Virol. 53, 761–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shiraki K., Sato H., Yamamura J., Li Z. H., Yokoyama T., Hasegawa T., Okuno T., Kurokawa M., Kageyama S. (1997) Functions of purified gB, gE:gI, and gH:gL, and their sialyl residues in varicella-zoster virus infection. Arch. Virol. 142, 2295–2301 [DOI] [PubMed] [Google Scholar]
- 32. Matsui S., Okuno T., Shiraki K. (1994) Functional roles of terminal glycomoieties in varicella-zoster virus infection. Virology 198, 50–58 [DOI] [PubMed] [Google Scholar]
- 33. Haan K. M., Lee S. K., Longnecker R. (2001) Different functional domains in the cytoplasmic tail of glycoprotein B are involved in Epstein-Barr virus-induced membrane fusion. Virology 290, 106–114 [DOI] [PubMed] [Google Scholar]
- 34. Pertel P. E. (2002) Human herpesvirus 8 glycoprotein B (gB), gH, and gL can mediate cell fusion. J. Virol. 76, 4390–4400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tanaka Y., Suenaga T., Matsumoto M., Seya T., Arase H. (2013) Herpesvirus 6 glycoproteins B (gB), gH, gL, and gQ are necessary and sufficient for cell-to-cell fusion. J. Virol. 87, 10900–10903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Turner A., Bruun B., Minson T., Browne H. (1998) Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J. Virol. 72, 873–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vanarsdall A. L., Ryckman B. J., Chase M. C., Johnson D. C. (2008) Human cytomegalovirus glycoproteins gB and gH/gL mediate epithelial cell-cell fusion when expressed either in cis or in trans. J. Virol. 82, 11837–11850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zerboni L., Sommer M., Ware C. F., Arvin A. M. (2000) Varicella-zoster virus infection of a human CD4-positive T-cell line. Virology 270, 278–285 [DOI] [PubMed] [Google Scholar]
- 39. Shiratori I., Yamaguchi M., Suzukawa M., Yamamoto K., Lanier L. L., Saito T., Arase H. (2005) Down-regulation of basophil function by human CD200 and human herpesvirus-8 CD200. J. Immunol. 175, 4441–4449 [DOI] [PubMed] [Google Scholar]
- 40. Nosaka T., Kawashima T., Misawa K., Ikuta K., Mui A. L., Kitamura T. (1999) Stat5 as a molecular regulator of proliferation, differentiation and apoptosis in hematopoietic cells. EMBO J. 18, 4754–4765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kitamura T., Koshino Y., Shibata F., Oki T., Nakajima H., Nosaka T., Kumagai H. (2003) Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Exp. Hematol. 31, 1007–1014 [PubMed] [Google Scholar]
- 42. Okuma K., Nakamura M., Nakano S., Niho Y., Matsuura Y. (1999) Host range of human T-cell leukemia virus type I analyzed by a cell fusion-dependent reporter gene activation assay. Virology 254, 235–244 [DOI] [PubMed] [Google Scholar]
- 43. Paulson J. C., Macauley M. S., Kawasaki N. (2012) Siglecs as sensors of self in innate and adaptive immune responses. Ann. N.Y. Acad. Sci. 1253, 37–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kuroki K., Wang J., Ose T., Yamaguchi M., Tabata S., Maita N., Nakamura S., Kajikawa M., Kogure A., Satoh T., Arase H., Maenaka K. (2014) Structural basis for simultaneous recognition of an O-glycan and its attached peptide of mucin family by immune receptor PILRα. Proc. Natl. Acad. Sci. U.S.A. 111, 8877–8882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Laughlin S. T., Bertozzi C. R. (2007) Metabolic labeling of glycans with azido sugars and subsequent glycan-profiling and visualization via Staudinger ligation. Nat. Protoc. 2, 2930–2944 [DOI] [PubMed] [Google Scholar]
- 46. Prescher J. A., Bertozzi C. R. (2006) Chemical technologies for probing glycans. Cell 126, 851–854 [DOI] [PubMed] [Google Scholar]
- 47. Alphey M. S., Attrill H., Crocker P. R., van Aalten D. M. (2003) High resolution crystal structures of Siglec-7: insights into ligand specificity in the Siglec family. J. Biol. Chem. 278, 3372–3377 [DOI] [PubMed] [Google Scholar]
- 48. Attrill H., Imamura A., Sharma R. S., Kiso M., Crocker P. R., van Aalten D. M. (2006) Siglec-7 undergoes a major conformational change when complexed with the α(2,8)-disialylganglioside GT1b. J. Biol. Chem. 281, 32774–32783 [DOI] [PubMed] [Google Scholar]
- 49. Attrill H., Takazawa H., Witt S., Kelm S., Isecke R., Brossmer R., Ando T., Ishida H., Kiso M., Crocker P. R., van Aalten D. M. (2006) The structure of siglec-7 in complex with sialosides: leads for rational structure-based inhibitor design. Biochem. J. 397, 271–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bukrinsky J. T., St Hilaire P. M., Meldal M., Crocker P. R., Henriksen A. (2004) Complex of sialoadhesin with a glycopeptide ligand. Biochim. Biophys. Acta 1702, 173–179 [DOI] [PubMed] [Google Scholar]
- 51. Dimasi N., Moretta L., Biassoni R., Mariuzza R. A. (2003) Expression, crystallization and preliminary crystallographic analysis of the extracellular IgV-like domain of the human natural killer cell inhibitory receptor p75/AIRM1. Acta Crystallogr. D Biol. Crystallogr. 59, 1856–1858 [DOI] [PubMed] [Google Scholar]
- 52. Zaccai N. R., Maenaka K., Maenaka T., Crocker P. R., Brossmer R., Kelm S., Jones E. Y. (2003) Structure-guided design of sialic acid-based Siglec inhibitors and crystallographic analysis in complex with sialoadhesin. Structure 11, 557–567 [DOI] [PubMed] [Google Scholar]
- 53. Zaccai N. R., May A. P., Robinson R. C., Burtnick L. D., Crocker P. R., Brossmer R., Kelm S., Jones E. Y. (2007) Crystallographic and in silico analysis of the sialoside-binding characteristics of the Siglec sialoadhesin. J. Mol. Biol. 365, 1469–1479 [DOI] [PubMed] [Google Scholar]
- 54. Zhuravleva M. A., Trandem K., Sun P. D. (2008) Structural implications of Siglec-5-mediated sialoglycan recognition. J. Mol. Biol. 375, 437–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kelm S., Pelz A., Schauer R., Filbin M. T., Tang S., de Bellard M. E., Schnaar R. L., Mahoney J. A., Hartnell A., Bradfield P. (1994) Sialoadhesin, myelin-associated glycoprotein and CD22 define a new family of sialic acid-dependent adhesion molecules of the immunoglobulin superfamily. Curr. Biol. 4, 965–972 [DOI] [PubMed] [Google Scholar]
- 56. Strenge K., Schauer R., Bovin N., Hasegawa A., Ishida H., Kiso M., Kelm S. (1998) Glycan specificity of myelin-associated glycoprotein and sialoadhesin deduced from interactions with synthetic oligosaccharides. Eur. J. Biochem. 258, 677–685 [DOI] [PubMed] [Google Scholar]
- 57. Strenge K., Schauer R., Kelm S. (1999) Binding partners for the myelin-associated glycoprotein of N2A neuroblastoma cells. FEBS Lett. 444, 59–64 [DOI] [PubMed] [Google Scholar]
- 58. Stanley P., Schachter H., Taniguchi N. (2009) Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., Etzler M. E., eds), 2nd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 59. An H. J., Froehlich J. W., Lebrilla C. B. (2009) Determination of glycosylation sites and site-specific heterogeneity in glycoproteins. Curr. Opin. Chem. Biol. 13, 421–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sareneva T., Mørtz E., Tölö H., Roepstorff P., Julkunen I. (1996) Biosynthesis and N-glycosylation of human interferon-γ: Asn25 and Asn97 differ markedly in how efficiently they are glycosylated and in their oligosaccharide composition. Eur. J. Biochem. 242, 191–200 [DOI] [PubMed] [Google Scholar]
- 61. Sutton C. W., O'Neill J. A., Cottrell J. S. (1994) Site-specific characterization of glycoprotein carbohydrates by exoglycosidase digestion and laser desorption mass spectrometry. Anal. Biochem. 218, 34–46 [DOI] [PubMed] [Google Scholar]
- 62. Oliver S. L., Sommer M., Zerboni L., Rajamani J., Grose C., Arvin A. M. (2009) Mutagenesis of varicella-zoster virus glycoprotein B: putative fusion loop residues are essential for viral replication, and the furin cleavage motif contributes to pathogenesis in skin tissue in vivo. J. Virol. 83, 7495–7506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gerken T. A. (2012) O-glycoprotein biosynthesis: site localization by Edman degradation and site prediction based on random peptide substrates. Methods Mol. Biol. 842, 81–108 [DOI] [PubMed] [Google Scholar]
- 64. Ten Hagen K. G., Fritz T. A., Tabak L. A. (2003) All in the family: the UDP-GalNac:polypeptide N-acetylgalactosaminyltransferases. Glycobiology 13, 1R–16R [DOI] [PubMed] [Google Scholar]