

Background: Rad9 C-tail interacts with various checkpoint factors.
Results: Intramolecular folding of C-tail in 9-1-1 interfered with its DNA binding. The sequence for the interaction coincided with that for TopBP1 binding.
Conclusion: The intramolecular folding regulates DNA binding of 9-1-1 with DNA and competes with TopBP1 for binding to the C-tail.
Significance: We identified a novel intramolecular folding in 9-1-1 for DNA-damage checkpoint activation.
Keywords: checkpoint control, DNA damage response, DNA-protein interaction, protein phosphorylation, protein/protein interaction, clamp protein
Abstract
The human checkpoint clamp Rad9-Hus1-Rad1 (9-1-1) is loaded onto chromatin by its loader complex, Rad17-RFC, following DNA damage. The 120-amino acid (aa) stretch of the Rad9 C terminus (C-tail) is unstructured and projects from the core ring structure (CRS). Recent studies showed that 9-1-1 and CRS bind DNA independently of Rad17-RFC. The DNA-binding affinity of mutant 9ΔC-1-1, which lacked the Rad9 C-tail, was much higher than that of wild-type 9-1-1, suggesting that 9-1-1 has intrinsic DNA binding activity that manifests in the absence of the C-tail. C-tail added in trans interacted with CRS and prevented it from binding to DNA. We narrowed down the amino acid sequence in the C-tail necessary for CRS binding to a 15-aa stretch harboring two conserved consecutive phenylalanine residues. We prepared 9-1-1 mutants containing the variant C-tail deficient for CRS binding, and we demonstrated that the mutant form restored DNA binding as efficiently as 9ΔC-1-1. Furthermore, we mapped the sequence necessary for TopBP1 binding within the same 15-aa stretch, demonstrating that TopBP1 and CRS share the same binding region in the C-tail. Indeed, we observed their competitive binding to the C-tail with purified proteins. The importance of interaction between 9-1-1 and TopBP1 for DNA damage signaling suggests that the competitive interactions of TopBP1 and CRS with the C-tail will be crucial for the activation mechanism.
Introduction
The checkpoint pathway is highly conserved among eukaryotes, and it is essential for the coordination of cell cycle progression with various cellular key events (1, 2). Phosphatidylinositol 3-kinase-related proteins, such as ataxia telangiectasia mutated (ATM)3 and ATM and Rad3-related (ATR), play key roles in the DNA-damage checkpoint (3–6). In human cells, ATM mainly reacts to double strand breaks (DSBs) (7, 8) and activates nonhomologous end-joining or homologous recombination (HR) (9, 10). ATR, in complex with the ATR-interacting protein (ATRIP) (11), reacts to single-stranded (ss) DNA exposed by a variety of DNA-damaging agents and replication stresses (12), and it activates the DNA repair pathway (13). The ssDNA binding replication protein A coats ssDNA (14) and recruits ATR-ATRIP (15) and Rad17-RFC (16), which consists of Rad17 and four replication factor C (RFC) small subunits (17). Three checkpoint proteins, Rad9, Hus1, and Rad1, form a heterotrimeric complex, Rad9-Hus1-Rad1 (9-1-1) (18), and Rad17-RFC loads it on DNA (19–21). Human 9-1-1 interacts with the topoisomerase IIβ-binding protein 1 (TopBP1) (22, 23) through Ser-341 and Ser-387 of Rad9, which are phosphorylated by casein kinase 2 (CK2) (24). These two Ser residues are crucial for the DNA-damage response in HeLa cells (25), as well as for the successful formation of the damage-sensing complex (including ATR) following exposure to DNA-damaging agents (26). Furthermore, TopBP1 interacts with ATR-ATRIP through its ATR activation domain, thereby activating ATR (27–29). However, the actual mechanism by which ATR is activated remains to be elucidated.
9-1-1 consists of a core ring structure (CRS) highly similar to proliferating cell nuclear antigen (PCNA) (30–32) and an intrinsically disordered region of the Rad9 C terminus (C-tail); the latter region contains a nuclear localization signal (NLS) (33) and multiple serine and threonine residues (34, 35), phosphorylation of which is involved in interaction of the C-tail with other factors, including replication protein A or TopBP1 (22–24, 36, 37).
Recent studies showed that 9-1-1 or mutant 9-1-1 lacking the C-tail can bind DNA in a Rad17-RFC-independent manner (31, 38). We found that the intramolecular binding of C-tail to CRS suppressed this intrinsic DNA binding. In addition, the region of the C-tail required for CRS binding was partly coincident with the region required for TopBP1 binding, and the interactions of TopBP1 and CRS with C-tail were competitive. Considering the importance of 9-1-1/TopBP1 interactions for DNA-damage signaling, we conclude that the intramolecular folding and unfolding of the CRS to the C-tail is a crucial event to regulate the signaling pathway.
Experimental Procedures
DNA Substrates
DNA substrates for EMSA were prepared using oligonucleotides shown in Table 1. Some oligonucleotides were obtained from Dr. Y. Ishino (Kyushu University, Japan) and were described previously (39). TEMP90, TEMP70, TEMP50, TEMP30, and 49N were labeled with [γ-32P]ATP (PerkinElmer Life Sciences) using T4 polynucleotide kinase (Takara), and 500 fmol of labeled DNA was annealed with equimolar amounts of complementary oligonucleotide (5′ end DNAs, 3′-TEMP/TEMP90 and d22/49N; 3′ end DNAs, 5′-TEMP/TEMP90 and d27/49N; dsDNAs, RTEMP90/TEMP90, RTEMP70/TEMP70, RTEMP50/TEMP50, RTEMP30/TEMP30, and 49R/49N; fork DNA, F5-d42/49N) in annealing buffer (500 mm NaCl, 10 mm Tris-HCl (pH 8.0), and 1 mm EDTA) by gradual cooling to room temperature for 5 h following heating at 95 °C for 5 min.
TABLE 1.
DNA substrates for EMSA
| Name | Sequence (5′-3′) |
|---|---|
| TEMP90 | TGAGGTTCAGCAAGGTGATGCTTTAGATTTTTCATTTGCTGCTGGCTCTCAG- |
| CGTGGCACTGTTGCAGGCGGTGTTAATACTGACCGCCT | |
| RTEMP90 | AGGCGGTCAGTATTAACACCGCCTGCAACAGTGCCACGCTGAGAGCCAGC- |
| AGCAAATGAAAAATCTAAAGCATCACCTTGCTGAACCTCA | |
| 3′-TEMP | ATCTAAAGCATCACCTTGCTGAACCTCA |
| 5′-TEMP | AGGCGGTCAGTATTAACACCGCCTGCAACAGT |
| TEMP70 | TGAGGTTCAGCAAGGTGATGCTTTAGATTCAGCGTGGCACTGTTGCAGGCG- |
| GTGTTAATACTGACCGCCT | |
| RTEMP70 | AGGCGGTCAGTATTAACACCGCCTGCAACAGTGCCACGCTGAATCTAAAGC- |
| ATCACCTTGCTGAACCTCA | |
| TEMP50 | TGAGGTTCAGCAAGGTGATGCTTTAGATGCGGTGTTAATACTGACCGCCT |
| RTEMP50 | AGGCGGTCAGTATTAACACCGCATCTAAAGCATCACCTTGCTGAACCTCA |
| TEMP30 | TGAGGTTCAGGGTGTTAATACTGACCGCCT |
| RTEMP30 | AGGCGGTCAGTATTAACACCCTGAACCTCA |
| 49N | AGCTACCATGCCTGCACGAATTAAGCAATTCGTAATCATGGTCATAGCT |
| 49R | AGCTATGACCATGATTACGAATTGCTTAATTCGTGCAGGCATGGTAGCT |
| d22 | AATTCGTGCAGGCATGGTAGCT |
| d27 | ATACGAATTCGTGCAGGCATGGTAGCT |
| F5-d42 | GATGTCAAGCAGTCCTAAGGAATTCGTGCAGGCATGGTAGCT |
Antibodies
Rabbit anti-Rad9 polyclonal antibody (Lifespan Biosciences), mouse anti-FLAG monoclonal antibody (Sigma), rabbit anti-TopBP1 polyclonal antibody (Bethyl Laboratories), and HRP-conjugated goat anti-rabbit and anti-mouse IgGs (Bio-Rad) antibodies were commercially purchased. Rabbit antibody specific for Rad9 phosphorylated at Ser-387 was prepared by Sigma Genosys Japan. Rabbit anti-Hus1 and anti-Rad1 polyclonal antibodies were obtained from Dr. K. Sugimoto (New Jersey Medical School, Newark).
Plasmid Construction and Baculovirus Preparation
Plasmids and baculoviruses for expression of FLAG-Rad9, Hus1, Rad1, and His-TopBP1 were described previously (24). Recombinant baculoviruses were produced using the Bac-to-Bac expression system (Invitrogen). Plasmids for expression of FLAG-Rad9ΔC were prepared by inserting PCR-amplified Rad9 sequence containing the corresponding truncations into pFastBac1 (Invitrogen). Plasmids for expression of FLAG-Rad9Δ351d5 and FLAG-Rad9Δ351 were prepared by site-directed mutagenesis using the QuikChange site-directed mutagenesis kit (Stratagene) and pFastBac1 plasmid harboring FLAG-tagged Rad9 cDNA and the corresponding primers (Table 2). Mutated Rad9 cDNA fragments were digested with BamHI and NotI and inserted into the corresponding sites of pFastBac1. Recombinant baculoviruses were prepared using bacmids derived from the plasmids described above.
TABLE 2.
Primers
| Name | Sequence (5′-3′) |
|---|---|
| FFAA_Fw | GAAGTTCCGCTCACTGGCCGCCGGCTCCATCCTGGCa |
| FFAA_Rv | GCCAGGATGGAGCCGGCGGCCAGTGAGCGGAACTTCa |
| S387A_Rv | AAGCTTATCAGCCTTCACCCTCAGCGa |
| R9-Cter_Fw | GGATCCACCATGTCCCAGGACCTGGGCTCC |
| R9–272_Fw | GGATCCCAGGACCTGGGCTCCCCAGA |
| R9–272_Rv | CGAAAGCGGCCGCTAGGAGTGCGAGTCGGTGTCTG |
| R9–391_Rv | AAGCTTATCAGCCTTCACCCTCACTG |
| R9–333_Fw | GGATCCATGGACTACAAGGATGACGATGACAAGCCCCCCAAGAGCCCCGGT |
| R9–332_Rv | AAAAGCGGCCGCTCACTGGGGGCCAGGTGAAAGG |
| Δ336–345_Fw | GATGACGATGACAAGCCCCCCAAGGAGGCTGAGCCCAGTACAGTG |
| Δ336–345_Rv | CACTGTACTGGGCTCAGCCTCCTTGGGGGGCTTGTCATCGTCATC |
| Δ341–350_Fw | CCCCAAGAGCCCCGGTCCCCACACAGTGCCTGGGACTCCCCCACC |
| Δ341–350_Rv | GGTGGGGGAGTCCCAGGCACTGTGTGGGGACCGGGGCTCTTGGGG |
| Δ351–360_Fw | GGAAGATGAGGCTGAGCCCAGTTTCCGCTCACTGTTCTTCGGCTC |
| Δ351–360_Rv | GAGCCGAAGAACAGTGAGCGGAAACTGGGCTCAGCCTCATCTTCC |
| Δ351–355_Fw | GAGGCTGAGCCCAGTCCCCCACCCAAGAAG |
| Δ351–355_Fw | CTTCTTGGGTGGGGGACTGGGCTCAGCCTC |
| Δ356–365_Fw | CCAGTACAGTGCCTGGGACTTTCGGCTCCATCCTGGCCCCTGTAC |
| Δ356–365_Rv | GTACAGGGGCCAGGATGGAGCCGAAAGTCCCAGGCACTGTACTGG |
| Δ361–370_Fw | CTGGGACTCCCCCACCCAAGAAGGCCCCTGTACGCTCCCCCCAGG |
| Δ361–370_Rv | CCTGGGGGGAGCGTACAGGGGCCTTCTTGGGTGGGGGAGTCCCAG |
| Δ371–380_Fw | CACTGTTCTTCGGCTCCATCCTGCCTGTGCTGGCGGAAGACAGTG |
| Δ371–380_Rv | CACTGTCTTCCGCCAGCACAGGCAGGATGGAGCCGAAGAACAGTG |
a The underlined nucleotides in FFAA_Fw, FFAA_Rv, and S387A_Rv created amino acid substitutions in site-directed mutagenesis.
To express GST- or GST/FLAG-tagged Rad9 fragments in Escherichia coli, we used pGEX-6P-3 (GE Healthcare). The Rad9 cDNA fragments harboring BamHI and HindIII ends were amplified by PCR with primers in Table 2 and inserted into the plasmid DNA digested with BamHI and SalI. The HindIII and SalI ends were ligated after blunting with T4 DNA polymerase (Takara). Non-FLAG-tagged Rad9 C-tail cDNA was amplified from the plasmid encoding Rad9 C-tail using R9-Cter_Fw and R9–391_Rv primers. FLAG-tagged C-N and C-C fragments of Rad9 were amplified from the plasmid encoding FLAG-tagged Rad9 C-tail using the corresponding primers (R9–272_Fw and R9–332_Rv for C-N and R9–333_Fw and R9-391_Rv for C-C). The cDNA fragments for Δ336, Δ341, Δ351, Δ351d5, Δ356, Δ361, Δ371, C-C2A, C-tailFFAA, C-tailΔ351d5, and C-tailΔ351 were prepared by site-directed mutagenesis as described above.
Preparation of Cell Lysate and Protein Purification
Purified FLAG-tagged 9-1-1, His-tagged TopBP1, and His-tagged CK2 were prepared as described previously (24), and FLAG-tagged 9ΔC-1-1 was prepared in the same manner as FLAG-tagged 9-1-1.
GST, GST/FLAG-tagged C-tail, and its derivatives were expressed in E. coli, Rosetta(DE3)pLysS (Novagen) grown in 200 ml of L broth containing 100 μg/ml ampicillin at 37 °C to A600 = 0.2, followed by addition of final 0.1 mm isopropyl β-d-thiogalactopyranoside and incubation at 32 °C for 4 h. The cells were collected, lysed by sonication in 2 ml of buffer H (25 mm HEPES-NaOH (pH 8.1), 1 mm EDTA, 10% glycerol, 0.01% Nonidet P-40, 100 μm PMSF, and 2 μg/ml leupeptin) containing 150 mm NaCl, and centrifuged at 7.2 × 104 g for 30 min at 4 °C. The supernatant was recovered as the cell lysate and used for GST pulldown assay.
For inhibition of DNA binding of 9ΔC-1-1 using the C-tail, we expressed the GST/FLAG-tagged C-tail in E. coli and purified it from lysates as described above. The lysates were loaded onto DEAE-Sepharose (10 ml; GE Healthcare) in buffer H containing 500 mm NaCl, and the unbound fractions were successively loaded onto glutathione-Sepharose (500 μl; GE Healthcare) in the same buffer, washed with buffer H containing 50 mm NaCl, and eluted with the same buffer containing 10 mm reduced glutathione. Fractions containing GST/FLAG-tagged C-tail were pooled and further purified with Mono Q (5/5; GE Healthcare) using a 16-ml linear gradient of NaCl (50–600 mm) in buffer H.
EMSA
Labeled DNA substrate (5 fmol) and various purified 9-1-1 complexes as indicated were incubated at 4 °C for 20 min in a 5-μl reaction mixture (10 mm HEPES-NaOH (pH 7.8), 10 mm MgCl2, 0.4 mm EDTA, 10% glycerol, 150 mm NaCl, 1 mm DTT, and 0.1 mg/ml BSA). The reaction products were electrophoresed in 7.5% polyacrylamide gel in TAEG buffer (40 mm Tris acetate (pH 7.8), 2.5 mm EDTA, and 5% glycerol) at 240 V at 4 °C for 23 min. The gel was dried, and the labeled DNA was visualized on a FLA-5000 phosphorimager (GE Healthcare).
GST Pulldown Assay
A cell lysate expressing GST, GST/FLAG-tagged C-tail, or its derivatives was incubated with glutathione-Sepharose beads (2.5 μl; GE Healthcare) for 1 h at 4 °C. The beads were washed three times with buffer H containing 0.15 m NaCl. Then, the various purified FLAG-tagged 9-1-1 complexes were incubated with the beads at 4 °C for 1 h in the same buffer. The bound proteins were eluted with SDS sample buffer (50 μm Tris-HCl (pH 6.8), 0.1 m DTT, 2% SDS, 0.05% bromphenol blue, and 10% glycerol) after three or five washes with 100 μl of the same buffer, and then analyzed by SDS-PAGE followed by staining with Coomassie Brilliant Blue or Ponceau S and immunoblotting with the indicated antibodies.
Competitive Binding Assay
Anti-FLAG beads (2 μl; Sigma) were incubated with 10 pmol of purified FLAG-tagged 9ΔC-1-1 in buffer H containing 150 mm NaCl at 4 °C for 1 h. The beads were washed three times with the same buffer and incubated with 60 pmol of GST-tagged C-tail with or without CK2 phosphorylation at 4 °C for 1 h. After washing the beads three times, 0, 3, or 5 pmol of purified TopBP1 were added and further incubated at 4 °C for 1 h. After washing three times, the bound proteins were eluted with SDS sample buffer and analyzed by immunoblotting with the indicated antibodies.
Results
9-1-1 Exhibited DNA Binding, Which Was Enhanced by the Absence of the C-tail
To study the DNA binding of 9-1-1 in detail, we prepared FLAG-tagged human 9-1-1 and 9ΔC-1-1; in the latter complex, Rad9 was replaced with the C-tail deletion mutant of Rad9 (amino acids (aa) 1–272). These complexes were expressed with baculoviruses, highly purified, and subjected to EMSA using a 90-mer primer-template DNA with 5′ recessed ends (5′ end). Upon incubation of the DNA with increasing amounts of both complexes, the intensity of the mobility-shifted band was more intense in the presence of 9ΔC-1-1 than 9-1-1 (Fig. 1A). The dissociation constant (Kd) of 9ΔC-1-1 to the DNA was calculated as 2.2 × 10−7 from the quantified graph, whereas that of 9-1-1 was much higher and unable to estimate the value precisely from this quantified range. This result indicated that 9-1-1 can bind to DNA intrinsically, but the C-tail suppresses this binding. Thus, 9ΔC-1-1 exhibits the primary DNA-binding property of 9-1-1. We confirmed that the band shift was indeed caused by direct binding of 9ΔC-1-1 to DNA by the addition of anti-FLAG antibody to the reaction, which resulted in a supershift (Fig. 1B).
FIGURE 1.

DNA binding of 9ΔC-1-1 analyzed by EMSA. Autoradiographs of EMSA with 9-1-1 or 9ΔC-1-1 under various conditions are shown. A, increasing amounts of 9-1-1 or 9ΔC-1-1 (0, 0.06, 0.17, 0.5, 1.5, 3, and 6 pmol each) were incubated with 5′ end DNA. B, 9ΔC-1-1 (2 pmol) was incubated with 5′ end DNA, with or without 250 ng of anti-FLAG monoclonal antibody. A super-shifted band appeared in the presence of the antibody. C, 9ΔC-1-1 (0, 0.75, or 1.5 pmol) was incubated with the 5′ end, 3′ end, ssDNA, and dsDNA. D, 9ΔC-1-1 (0, 1.5, or 3 pmol) was incubated with ssDNA or dsDNA with lengths ranging from 30- to 90-mer. E, 9ΔC-1-1 (0, 2.5, and 5 pmol) was incubated with various 49-mer DNAs (ss, ds, 5′ end, 3′ end, and fork). It should be noted that differences among experiments in DNA-binding efficiencies were the result of different specific activities of 9ΔC-1-1 preparations. Intensities of the shifted bands in A, C, D, and E were quantified, and their ratios (%) relative to total DNA are graphed on the right side or below. Values represent the means of three independent experiments, and error bars indicate S.E. A, inset is Kd of 9ΔC-1-1, which was estimated as 2.2 × 10−7 from the quantified shifted bands, although the values of the shifted bands of 9-1-1 were too low to estimate the precise Kd value from this study.
Properties of DNA Binding by 9ΔC-1-1
We studied the binding using four structured DNAs as follows: primer-template duplex with recessed 5′ end, primer-template duplex with recessed 3′ end (3′ end), 90-mer ssDNA, and 90-mer dsDNA (Fig. 1C). As shown in this result, the binding did not have any obvious preference for these DNA structures. When we tested DNA binding to ssDNA and dsDNA ranging in size from 30- to 90-mer (Fig. 1D), the binding efficiency increased as a function of length; furthermore, for DNAs shorter than 50-mers, 9ΔC-1-1 exhibited a binding preference for dsDNA relative to ssDNA. The same effect of DNA length for the DNA binding of 9ΔC-1-1 was observed in a previous study, in which DNA binding was studied using a primer-template structure (31). For 49-mers, we observed a pronounced preference for dsDNA relative to ssDNA (Fig. 1E); in that experiment, we used 9ΔC-1-1 that had lower specific activity than in other experiments. In the same experiment, we tested primer-template- and fork-structured DNAs along with dsDNA; the levels of binding to these DNA structures were similar to those of dsDNA, except that binding to the fork structure was slightly elevated. Collectively, these findings indicated that 9ΔC-1-1 bound to DNA with a preference for double strand structure relative to single strand structure; longer DNA chains and certain secondary DNA structures, such as branches, stabilized the binding.
C-tail Bound to CRS
Suppression of 9-1-1 DNA binding by the C-tail implied that the C-tail interacts with the CRS. To address this question, we studied direct binding of GST/FLAG-tagged C-tail fragments (aa 272–391) to CRS. Glutathione beads pre-bound with GST alone or GST/FLAG-tagged C-tail expressed in E. coli were mixed with purified 9-1-1 or 9ΔC-1-1 (Fig. 2A). The amount of 9ΔC-1-1 detected in the C-tail bound fraction was higher than 5% of the input, whereas the amount of 9-1-1 was much lower. Thus, the C-tail could indeed bind efficiently to CRS, but the binding was suppressed when CRS had a C-tail, as in 9-1-1.
FIGURE 2.
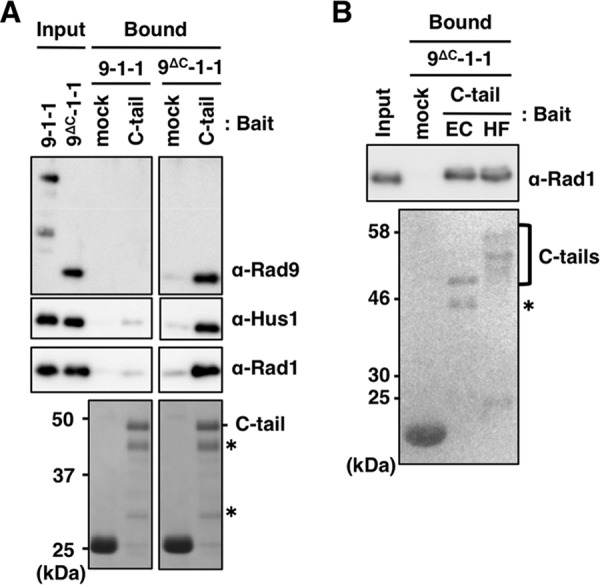
Binding of C-tail with 9-1-1 and 9ΔC-1-1. A and B, glutathione-Sepharose beads pre-bound with GST only or GST/FLAG-tagged C-tails (from E. coli cells in A, and EC and HF in B from E. coli and insect cells, respectively) were mixed with 10 pmol each of purified 9-1-1 or 9ΔC-1-1 at 4 °C for 1 h. After washing the beads, input proteins (1%) and bound proteins (20%) were analyzed by immunoblotting using the indicated antibodies for 9-1-1 subunits (upper panel), and staining with Coomassie Brilliant Blue (A) or Ponceau S (B) for C-tail (lower panels). Asterisks indicate degraded C-tail fragments.
As described above, the C-tail is phosphorylated at multiple sites, and some of them (e.g. Ser-341 and Ser-387) are involved in protein/protein interactions and are necessary for binding with TopBP1. Sohn and Cho (31) suggested that phosphorylation of the C-tail might induce a movement of the C-tail that opens the DNA-binding site of CRS. We studied the importance of phosphorylation in the C-tail for binding to CRS by comparing binding between highly phosphorylated C-tails produced in High Five insect cells (HF) and unphosphorylated C-tails expressed in E. coli (EC). The HF appeared as multiple bands that migrated more slowly than the EC (Fig. 2B, lower panel). However, we could not detect any significant differences between their binding activities (Fig. 2B, upper panel), indicating that their binding was not affected by phosphorylation, at least of the type that occurred in insect cells.
Binding of C-tail to CRS Requires a 15-aa Sequence
To determine which region of the C-tail is responsible for binding, we constructed several deletions of GST/FLAG-tagged C-tail (Fig. 3A). These deletions were pre-bound to glutathione beads and mixed with purified 9ΔC-1-1. Comparable amounts of C-tail fragments bound to the beads, as demonstrated by Ponceau S staining (Fig. 3B, lower panels). The bound 9ΔC-1-1 was detected by immunoblotting to detect Rad1. First, we separated the C-tail into its N- and C-terminal halves and prepared C-N and C-C, respectively. As demonstrated in Fig. 3B, C-C (but not C-N) bound 9ΔC-1-1, although the binding efficiency was 30% lower than that of the C-tail. This observation suggested that C-C contained a structure essential for binding but that some additional structure in C-N might stabilize the interaction. Thus, we searched C-C for the sequence required for binding. To this end, we prepared seven deletions, Δ336, Δ341, Δ351d5, Δ351, Δ356, Δ361, and Δ371, in which 5 or 10 aa were deleted from C-C. Analyses of the binding of the deletions to 9ΔC-1-1 demonstrated that any deletions from C-C decreased the binding efficiency. Several of the constructs, Δ336, Δ341, Δ351d5, and Δ371, bound about 30% as efficiently as C-C, whereas Δ351, Δ356, and Δ361 did not bind at all (Fig. 3B, left panel). The region remaining in the former four and eliminated from the latter three was the 15-aa stretch (aa 356–370), which could be considered essential for the CRS binding. The weak binding property of the four binding-positive fragments indicated that the sequences surrounding the 15-aa stretch would facilitate its binding with CRS. To support this notion, we prepared mutant C-tail fragments, C-tailΔ351d5 and C-tailΔ351, which had 5- and 10-aa deletions of Δ351d5 and Δ351, respectively, as representatives of binding-positive and -negative structures (Fig. 3A). As expected, the C-tailΔ351d5 bound to 9ΔC-1-1 as efficiently as the C-tail, whereas the C-tailΔ351 did not (Fig. 3B, right panel). Thus, the essential sequence for C-tail binding with CRS exists in the 15-aa stretch (aa 356–370), although its flanking sequences play a role in facilitating or stabilizing binding.
FIGURE 3.
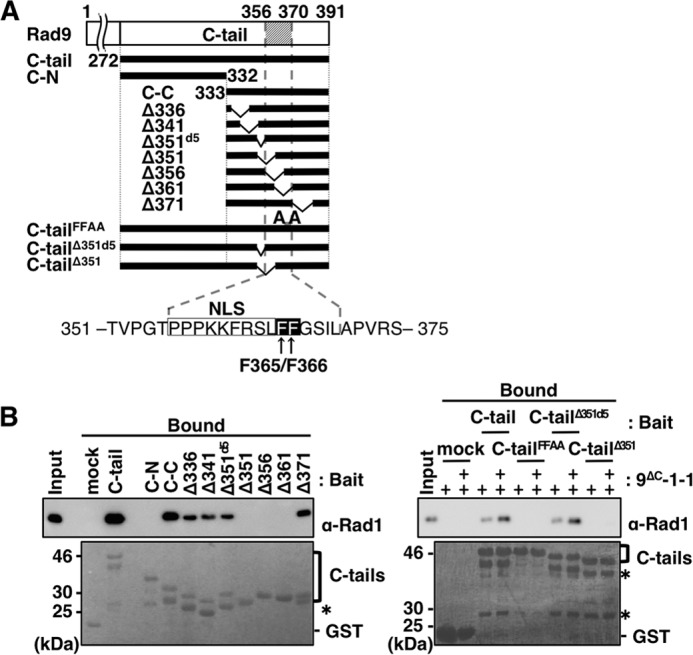
Analyses of amino acid sequence of C-tail necessary for its binding to 9ΔC-1-1. A, schematic illustration of human C-tail and its deletions used in this study. C-tail, C-N, and C-C contain aa 272–391, aa 272–332, and aa 333–391, respectively. Constructs Δ336, Δ341, Δ351d5, Δ351, Δ356, Δ361, and Δ371 harbor internal deletions of aa 336–345, aa 341–350, aa 351–355, aa 351–360, aa 356–365, aa 361–370, and aa 371–380 from C-C, respectively; C-tailΔ351d5 and C-tailΔ351 are the same deletions as Δ351d5 and Δ351, respectively, from the C-tail. C-tailFFAA harbors Ala substitutions at both Phe-365 and Phe-366, which were expressed as fusions with GST/FLAG tags at their N-terminal portion. Below is the sequence of the C-tail, including the 15-aa stretch (aa 351–375) required for interaction with CRS. The NLS and FF motif (Phe-365/Phe-366) are boxed in white and black, respectively. B, glutathione-Sepharose beads pre-bound with GST only (mock), GST/FLAG-tagged C-tail (C-tail), C-tail mutant (C-tailFFAA), or its deletions (C-N, C-C, Δ336, Δ341, Δ351d5, Δ351, Δ356, Δ361, Δ371, C-tailΔ351d5, and C-tailΔ351) were mixed with 10 pmol alone (left panel) or 5 and 10 pmol (+, and ++, respectively) (right panel) of purified 9ΔC-1-1, and incubated at 4 °C for 1 h. After washing the beads, 50 fmol (left panel) and 100 fmol (right panel) of purified 9ΔC-1-1 and 10% (left panel) or 20% (right panel) of the bound fractions were analyzed by immunoblotting using anti-Rad1 antibody (upper panels) and staining with Ponceau S for the C-tail and its deletions (lower panels). Asterisks indicate degraded C-tail fragments.
The amino acid sequence of the 15-aa stretch involved in the interaction of the C-tail with CRS is highly conserved from human to chicken, and it contains several functional motifs, including a nuclear localization signal (NLS; 356PPPKKFRSL364) (33). Immediately adjacent to this NLS are two highly conserved consecutive phenylalanine residues (FF), Phe-365 and Phe-366. The FF sequence is often crucial for protein/protein interactions, e.g. for the binding of the polymerase δ p66 subunit C terminus to PCNA (40) and the binding of the internal sequence of polymerase κ to REV1 (41). We constructed the Ala substitution mutant of the FF sequence in the C-tail (C-tailFFAA), and investigated its binding to 9ΔC-1-1. The results revealed that the FF motif was essential for the binding (Fig. 3B, right panel).
Binding of C-tail to CRS Interferes with DNA Binding
Based on a prediction that the intramolecular binding of the C-tail to CRS would suppress DNA binding, we demonstrated that the C-tail bound to 9ΔC-1-1. Therefore, we next investigated whether the suppression was mediated by intramolecular binding. Prior to the experiment, we first tested whether the C-tail added in trans would suppress DNA binding. To this end, we added purified GST/FLAG-tagged C-tail to EMSA with ssDNA and 9ΔC-1-1 (Fig. 4A, left panel). DNA binding was suppressed by C-tail in a dose-dependent manner and reached 50% inhibition when a 10-fold molar excess of C-tail was added (Fig. 4A, right panel). When the same experiment was performed with C-tailFFAA, we observed no inhibition in the same protein concentration range. This result clearly demonstrated that the C-tail exerted its suppressive function on DNA binding through an interaction with CRS in trans.
FIGURE 4.
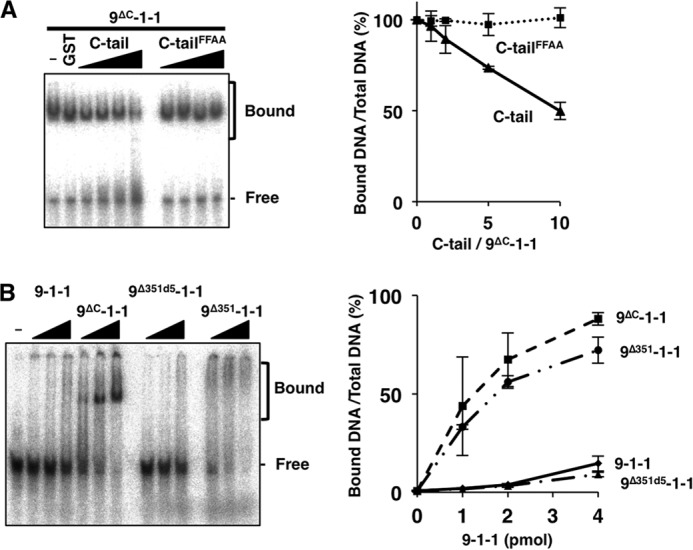
Effects of intramolecular binding on DNA binding by 9-1-1. A, increasing amounts of C-tail or C-tailFFAA (2.5, 5, 10, or 20 pmol) were added to a DNA-binding assay containing 9ΔC-1-1 (2 pmol) and ssDNA (90-mer, 5 fmol) and analyzed as in Fig. 1 (left panel). The ratios of bound DNA (% of total DNA) obtained as the means of two independent experiments as in Fig. 1D are plotted in the right graph; error bars indicate S.E. B, increasing amounts of 9-1-1 and 9-1-1 mutant (9ΔC-1-1, 9Δ351d5-1-1, 9Δ351-1-1) (1, 2, or 4 pmol each) were incubated with ssDNA, analyzed (left panel), and quantitated (right panel) as in A.
Next, we investigated whether C-tail in cis could suppress DNA binding. For this purpose, we prepared purified 9-1-1 containing wild-type or mutant Rad9 (9-1-1, 9ΔC-1-1, 9Δ351d5-1-1, and 9Δ351-1-1), and we investigated the binding to 90-mer ssDNA by EMSA. The mobility-shifted bands were more intense in the presence of 9ΔC-1-1 than 9-1-1, similar to the results obtained with 90-mer 5′ end DNA (Fig. 1A). 9Δ351-1-1, which contained a C-tail that is unable to bind to CRS, bound DNA almost as efficiently as 9ΔC-1-1, although the shifted band profiles were different (Fig. 4B). This difference may be due to differences arising in protein structures in the presence or absence of the extended C-tail. By contrast, 9Δ351d5-1-1, containing a C-tail harboring a 5-aa deletion, was able to bind to CRS but exhibited weak DNA binding, as in the case of 9-1-1. Thus, the CRS binding ability of C-tails and the DNA binding ability of the 9-1-1 complex containing them were inversely related.
C-tail Sequences Necessary for TopBP1 Binding Partly Overlap with the 15-aa Stretch Required for CRS Binding
The C-tail in free 9-1-1 should fold back to its target site within CRS, raising questions regarding how and when the C-tail can unfold from the CRS. Two possibilities should be considered, as follows. First, DNA binding itself could release the C-tail and make it competent to interact with other proteins. Second, binding of other proteins to the C-tail could make 9-1-1 competent for DNA binding. In either case, C-tail or CRS could associate with other protein(s) through their contact structures. The most plausible target is TopBP1, because it bound to the C-tail through Ser-341 and Ser-387, which are phosphorylated by CK2. These residues are located within the C-C fragment and proximal to the 15-aa stretch. To identify the region responsible for interaction with TopBP1, in addition to the two Ser residues, we used GST/FLAG-tagged mutant C-tail fragments pre-bound to glutathione beads, followed by CK2 phosphorylation (Fig. 5A). Because CK2 phosphorylation contributes only slightly to the C-tail migration in SDS-PAGE (Figs. 5, B and C, and 6), we used phospho-specific antibody (24) to confirm that their target Ser residues were indeed phosphorylated (Fig. 5B). Lysate of insect cells expressing His-TopBP1 was added to these beads, and the bound TopBP1 was detected by Coomassie Brilliant Blue staining. As we have shown previously (24), the TopBP1 bound to the C-tail, in a manner-dependent on CK2-mediated phosphorylation of C-tail (Fig. 5C). This phospho-dependent TopBP1 binding was also confirmed by using the 2A mutant, the Ala substitution mutant of the CK2 sites, Ser-341 and Ser-387 (Fig. 5D, left panel); TopBP1 bound to C-C as C-tail but not to C-C2A. We also carried out the same TopBP1 binding assay on five deletions (Δ351d5, Δ351, Δ356, Δ361, and Δ371). Δ351d5, Δ351, and Δ371 significantly bound to TopBP1 (more than 10% of input), but Δ356 and Δ361 bound at the background level (less than 3% of input) (Fig. 5D, left and middle panels). Thus, the 10-aa sequence (aa 361–370) of Rad9 is necessary for TopBP1 binding. The C-tailFFAA carrying Ala substitutions of the FF sequence crucial for CRS binding bound to TopBP1 only at the background level (Fig. 5D, right panel), indicating that the C-tail may bind to CRS and TopBP1 via similar mechanisms and that binding of TopBP1 to the C-tail occurs via the interaction with the 10-aa sequence in addition to the two phospho-Ser residues at Ser-341 and Ser-387. Because the C-tail sequences necessary for CRS and TopBP1 binding were partly coincident and included common crucial hydrophobic residues, it is likely that the binding of C-tail to CRS and TopBP1 is competitive.
FIGURE 5.

Analyses of the interaction of TopBP1 with C-tail, its Ala substitution mutant, and its deletions. A, C-tail fragments used for this analysis as indicated in Fig. 3A, except for C-C2A, which contains Ala substitutions at both Ser-341 and Ser-387. B, glutathione-Sepharose beads pre-bound with GST only (mock), GST/FLAG-tagged C-tail (C-tail), and its derivatives were prepared as Fig. 3, except for treatment with CK2 for 1 h at 30 °C, followed by three washes. A portion (20%) of the bound fractions was used for Coomassie Brilliant Blue (CBB) staining (upper left panel), and 0.5% was used for immunoblotting with the indicated antibodies (remaining panels). C and D, prepared beads as B with (C and D) or without (C) CK2 treatment were mixed with 5 or 10 μl of lysates of insect cells expressing TopBP1; 1 μl of the lysates and 25% of the bound fractions were analyzed by Coomassie Brilliant Blue staining. Asterisks indicate degraded C-tail fragments.
FIGURE 6.
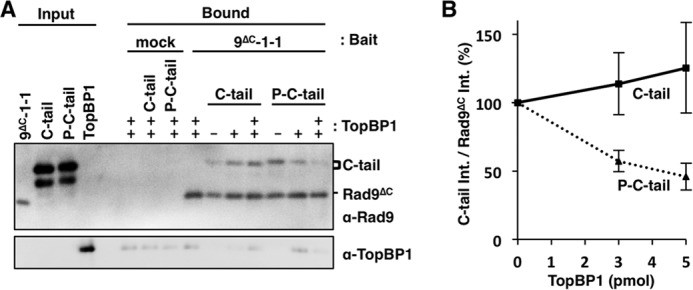
TopBP1 and CRS compete each other for C-tail. A, anti-FLAG beads were pre-bound with 10 pmol of purified FLAG-tagged 9ΔC-1-1 and incubated with GST-tagged C-tail with or without CK2 phosphorylation (P-C-tail or C-tail) at 4 °C for 1 h. After washing the beads, 0, 3, or 5 pmol (−, +, or ++, respectively) of purified TopBP1 were added and further incubated for 1 h at 4 °C. One percent of the input fractions and 30% of the bound fractions were analyzed by immunoblotting using indicated antibodies. B, band intensities of C-tail in the bound fractions were divided by that of Rad9ΔC and the values relative to TopBP1 minus experiments as 100% were plotted as means ± S.E. for four independent experiments.
CRS and TopBP1 Compete for Binding to C-tail
To see whether CRS and TopBP1 compete with each other for the binding to the C-tail, we performed competition assay using purified FLAG-tagged 9ΔC-1-1, GST-tagged C-tail, and His-tagged TopBP1. We immobilized the FLAG-tagged 9ΔC-1-1 onto anti-FLAG beads and added phosphorylated or unphosphorylated GST-tagged C-tail to the beads. After removing the unbound C-tail, we added TopBP1 to the suspension. As shown in Fig. 6, the phosphorylated C-tail was released from the 9ΔC-1-1 beads by adding TopBP1, although the unphosphorylated C-tail was not. Thus, we demonstrated the evidence that CRS and TopBP1 compete for the binding to the C-tail in a manner dependent on its phosphorylation by CK2.
Discussion
We confirmed that human 9-1-1 has an intrinsic DNA binding activity that is masked by the intramolecular binding of C-tail. Sohn and Cho (31) suggested that the binding might occur through entrapment of duplex DNA at the central hole of CRS at a tilted angle, as observed for E. coli β-clamp/DNA binding (42). Regarding this binding mode, they demonstrated some duplex-length dependence of the binding; specifically, the complex could bind duplexes longer than 15 bp, implying that the duplex must be above some critical length to be trapped stably at the hole. We observed that the complex bound both ssDNA and dsDNA longer than 50 nucleotides with similar efficiencies, but it bound less efficiently to those shorter than 50 nucleotides (Fig. 1D). The effect of DNA length on binding supported a model involving DNA entrapment at the central hole. Furthermore, ssDNA could be entrapped as efficiently as dsDNA, but only when the length was greater than 50 nucleotides. This result may reflect a larger spatial occupation of dsDNA than ssDNA that makes the DNA be entrapped stably at the inside of hole. This prediction is supported by the higher binding efficiency of fork-structured DNA, in which the branched end will prevent release of the DNA from the hole (Fig. 1E).
The predicted DNA-binding mode of 9ΔC-1-1 implied that DNA binding plays a positive role in loading of 9-1-1 by Rad17-RFC. In relation to this, Ddc1-Mec3-Rad17 of Saccharomyces cerevisiae, the counterpart of human 9-1-1, is loaded by its loader with the same efficiency irrespective of the presence or the absence of its C-tail structure (43). Thus, the DNA binding of 9-1-1 may not have any active role in its loading.
Sohn and Cho (31) suggested two possible roles of the C-tail in DNA binding. First, the C-tail could block the central hole of 9-1-1, preventing DNA from passing through the hole. Second, binding to DNA by 9-1-1 could require that the DNA be tilted relative to the plane of the ring, as in the β-clamp (42); however, the C-tail would force the DNA angle to be nearly perpendicular, allowing 9-1-1 to slide off the DNA. Our result showing that C-tail added in trans inhibited binding (Fig. 4A) excludes the second possibility. Furthermore, contrary to their suggestion, we demonstrated that the C-tail could bind to CRS regardless of whether it was phosphorylated (Fig. 2B). Thus, DNA binding by 9-1-1 occurred independently of the C-tail's phosphorylation state.
How does C-tail fold back on CRS to interfere with the passage of DNA through the ring? To address this issue, we mapped the 15-aa stretch harboring the FF sequence necessary for binding. Similar hydrophobic sequences exist in other regions involved in protein/protein interaction, including the PCNA interaction protein (PIP) box, mentioned above. In the case of PCNA, the PIP peptide interacts with the hydrophobic groove formed by the interdomain connecting loop (44, 45). The three subunits of 9-1-1 have individual hydrophobic grooves with interdomain connecting loops, similar to those in PCNA. Thus, either of the hydrophobic grooves in 9-1-1 represent potential targets. These structures have also been proposed as targets of various clamp-binding proteins (30, 31). If the target structure for C-tail is shared with other binding proteins, the unfolding of C-tail from CRS may be involved not only with the binding but also with interaction of 9-1-1-binding protein(s). These predictions suggest that C-tail/CRS binding functions as a molecular switch between 9-1-1 and its interacting partners. In future studies, this model should be tested by detailed analyses of the contact structures of 9-1-1 and its partners.
TopBP1 binding to the C-tail required 10 aa (aa 361–370), including the FF sequence, which coincides partly with the 15 aa required for CRS binding (Figs. 3B, right panel, and 5D). Thus, the binding of CRS and TopBP1 to C-tail should be competitive. This prediction could be confirmed as we observed that C-tail prebound to 9ΔC-1-1 was released from it in accordance with the amount of added TopBP1 only when C-tail was phosphorylated by CK2 (Fig. 6). These data also demonstrated that the C-tail did not likely bind to CRS and TopBP1 simultaneously. Judging from the low DNA binding activity of 9-1-1, it is likely that under ordinary conditions the C-tail folds against CRS. In this situation, TopBP1 is able to unfold C-tail from CRS, because added TopBP1 can bind efficiently to 9-1-1 (24). It is also possible that the binding of TopBP1 to the C-tail may promote the binding of 9-1-1 to DNA. We tried to test this possibility. However, TopBP1 alone binds to both ssDNA and dsDNA, whose activity makes it difficult to test the effect of TopBP1 on DNA binding of 9-1-1 using EMSA. Because the presence or absence of C-tails in 9-1-1 clamps does not affect DNA loading efficiency in budding yeast (43), the DNA-binding potential of 9-1-1 mediated by C-tail-binding protein(s), such as TopBP1, should not directly affect loading of 9-1-1 onto DNA.
Length and secondary structures affected the efficiency of DNA binding by 9-1-1, as demonstrated here and by Sohn and Cho (31), implying that both ssDNA and dsDNA pass through the central hole and are retained relatively stably within this structure. If the release of the C-tail from CRS induces DNA binding independently of the Rad17-RFC loader, how does it work? For 9-1-1 to capture DNA without Rad17-RFC, newly produced DNA ends must be present. Indeed, 9-1-1 has been implicated in DSBs repair and HR (46–50). Thus, the interaction of 9-1-1 with several DSBs repair or HR factors may release C-tail from CRS, induce binding of 9-1-1 to the DNA ends, and provide a platform for other DNA repair proteins, DNA polymerases, or checkpoint factors (51–55).
Alternatively, because DSBs are a minor cause of activation of the ATR/ATRIP pathway (11, 12), we propose that the DNA binding observed here represents the consequence of Rad17-RFC-dependent 9-1-1 loading at a damaged DNA site, instead of functional activity of 9-1-1. If so, what is the functional significance of the link between DNA binding and unfolding? We demonstrated that the structures in the C-tail necessary for intramolecular binding and the TopBP1 interaction are mostly coincident. Thus, the unfolded C-tail, fully competent for the TopBP1 interaction, should be produced by loading of 9-1-1 by Rad17-RFC. Mec1 (human ATR) in budding yeast interacts with Ddc1-Mec3-Rad17 (human 9-1-1) (43). We further propose that 9-1-1 with an unfolded C-tail is competent for ATR/ATRIP binding, as well as binding of TopBP1, and that that mechanism ensures the recruitment of multiple checkpoint components at the same damaged site. The canonical clamp PCNA functions as a platform for multiple replication proteins on target DNA (56, 57). Our model explains the parallel roles of 9-1-1 as a clamp and (once it is loaded on a damaged DNA) a functional platform for multiple checkpoint proteins on target DNAs.
This model remains mostly unproven, but it plausibly explains the molecular connection to activation of ATR/ATRIP-mediated checkpoint signaling. To test this model, it will be necessary to reconstitute the assembly of these proteins at a DNA·9-1-1 complex and monitor the activation of ATR/ATRIP by the reconstituted complex.
Author Contributions
Y. T. and E. O. designed, performed, and analyzed most of the experiments and wrote the paper. R. I.-O. partly performed and analyzed experiments in Fig. 5. T. T. coordinated the study and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We thank Dr. Y. Ishino (Kyushu University, Japan) for the gracious gift of substrate oligo DNAs.
This work was supported by Grant-in-aid for Scientific Research (KAKENHI) 25131714 (to T. T.) and 22770172 (to E. O.), Uehara Memorial Foundation Grant 103-2012 (to E. O.), and Grant-in-aid for Research Fellows of the Japan Society for the Promotion of Science 11J04189 (to Y. T.). The authors declare that they have no conflicts of interest with the contents of this article.
- ATM
- ataxia telangiectasia mutated
- 9-1-1
- Rad9-Hus1-Rad1
- C-tail
- Rad9 C terminus
- aa
- amino acid
- CRS
- core ring structure of 9-1-1
- ATR
- ATM and Rad3-related
- DSB
- double strand break
- HR
- homologous recombination
- ATRIP
- ATR-interacting protein
- ss
- single-stranded
- RFC
- replication factor C
- TopBP1
- topoisomerase IIβ-binding protein 1
- CK2
- casein kinase 2
- PCNA
- proliferating cell nuclear antigen
- NLS
- nuclear localization signal
- HF
- phosphorylated C-tail expressed in High Five insect cells
- EC
- unphosphorylated C-tail expressed in Escherichia coli.
REFERENCES
- 1. Weinert T. A., Hartwell L. H. (1988) The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science 241, 317–322 [DOI] [PubMed] [Google Scholar]
- 2. Hartwell L. H., Weinert T. A. (1989) Checkpoints: controls that ensure the order of cell cycle events. Science 246, 629–634 [DOI] [PubMed] [Google Scholar]
- 3. Paterson M. C., Smith B. P., Lohman P. H., Anderson A. K., Fishman L. (1976) Defective excision repair of γ-ray-damaged DNA in human (ataxia telangiectasia) fibroblasts. Nature 260, 444–447 [DOI] [PubMed] [Google Scholar]
- 4. Painter R. B., Young B. R. (1980) Radiosensitivity in ataxia-telangiectasia: a new explanation. Proc. Natl. Acad. Sci. U.S.A. 77, 7315–7317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bentley N. J., Holtzman D. A., Flaggs G., Keegan K. S., DeMaggio A., Ford J. C., Hoekstra M., Carr A. M. (1996) The Schizosaccharomyces pombe rad3 checkpoint gene. EMBO J. 15, 6641–6651 [PMC free article] [PubMed] [Google Scholar]
- 6. Cliby W. A., Roberts C. J., Cimprich K. A., Stringer C. M., Lamb J. R., Schreiber S. L., Friend S. H. (1998) Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 17, 159–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kastan M. B., Zhan Q., el-Deiry W. S., Carrier F., Jacks T., Walsh W. V., Plunkett B. S., Vogelstein B., Fornace A. J., Jr. (1992) A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 71, 587–597 [DOI] [PubMed] [Google Scholar]
- 8. Cortez D., Wang Y., Qin J., Elledge S. J. (1999) Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science 286, 1162–1166 [DOI] [PubMed] [Google Scholar]
- 9. Morrison C., Sonoda E., Takao N., Shinohara A., Yamamoto K., Takeda S. (2000) The controlling role of ATM in homologous recombinational repair of DNA damage. EMBO J. 19, 463–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sekiguchi J., Ferguson D. O., Chen H. T., Yang E. M., Earle J., Frank K., Whitlow S., Gu Y., Xu Y., Nussenzweig A., Alt F. W. (2001) Genetic interactions between ATM and the nonhomologous end-joining factors in genomic stability and development. Proc. Natl. Acad. Sci. U.S.A. 98, 3243–3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cortez D., Guntuku S., Qin J., Elledge S. J. (2001) ATR and ATRIP: partners in checkpoint signaling. Science 294, 1713–1716 [DOI] [PubMed] [Google Scholar]
- 12. Liu Q., Guntuku S., Cui X. S., Matsuoka S., Cortez D., Tamai K., Luo G., Carattini-Rivera S., DeMayo F., Bradley A., Donehower L. A., Elledge S. J. (2000) Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 14, 1448–1459 [PMC free article] [PubMed] [Google Scholar]
- 13. Gatei M., Zhou B. B., Hobson K., Scott S., Young D., Khanna K. K. (2001) Ataxia telangiectasia mutated (ATM) kinase and ATM and Rad3 related kinase mediate phosphorylation of Brca1 at distinct and overlapping sites. In vivo assessment using phospho-specific antibodies. J. Biol. Chem. 276, 17276–17280 [DOI] [PubMed] [Google Scholar]
- 14. Wold M. S., Kelly T. (1988) Purification and characterization of replication protein A, a cellular protein required for in vitro replication of simian virus 40 DNA. Proc. Natl. Acad. Sci. U.S.A. 85, 2523–2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zou L., Elledge S. J. (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542–1548 [DOI] [PubMed] [Google Scholar]
- 16. Zou L., Liu D., Elledge S. J. (2003) Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc. Natl. Acad. Sci. U.S.A. 100, 13827–13832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lindsey-Boltz L. A., Bermudez V. P., Hurwitz J., Sancar A. (2001) Purification and characterization of human DNA damage checkpoint Rad complexes. Proc. Natl. Acad. Sci. U.S.A. 98, 11236–11241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Burtelow M. A., Roos-Mattjus P. M., Rauen M., Babendure J. R., Karnitz L. M. (2001) Reconstitution and molecular analysis of the hRad9-hHus1-hRad1 (9-1-1) DNA damage responsive checkpoint complex. J. Biol. Chem. 276, 25903–25909 [DOI] [PubMed] [Google Scholar]
- 19. Bermudez V. P., Lindsey-Boltz L. A., Cesare A. J., Maniwa Y., Griffith J. D., Hurwitz J., Sancar A. (2003) Loading of the human 9-1-1 checkpoint complex onto DNA by the checkpoint clamp loader hRad17-replication factor C complex in vitro. Proc. Natl. Acad. Sci. U.S.A. 100, 1633–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ellison V., Stillman B. (2003) Biochemical characterization of DNA damage checkpoint complexes: clamp loader and clamp complexes with specificity for 5′ recessed DNA. PLoS Biol. 1, E33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Majka J., Binz S. K., Wold M. S., Burgers P. M. (2006) Replication protein A directs loading of the DNA damage checkpoint clamp to 5′-DNA junctions. J. Biol. Chem. 281, 27855–27861 [DOI] [PubMed] [Google Scholar]
- 22. Delacroix S., Wagner J. M., Kobayashi M., Yamamoto K., Karnitz L. M. (2007) The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 21, 1472–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee J., Kumagai A., Dunphy W. G. (2007) The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J. Biol. Chem. 282, 28036–28044 [DOI] [PubMed] [Google Scholar]
- 24. Takeishi Y., Ohashi E., Ogawa K., Masai H., Obuse C., Tsurimoto T. (2010) Casein kinase 2-dependent phosphorylation of human Rad9 mediates the interaction between human Rad9-Hus1-Rad1 complex and TopBP1. Genes Cells 15, 761–771 [DOI] [PubMed] [Google Scholar]
- 25. Ueda S., Takeishi Y., Ohashi E., Tsurimoto T. (2012) Two serine phosphorylation sites in the C terminus of Rad9 are critical for 9-1-1 binding to TopBP1 and activation of the DNA damage checkpoint response in HeLa cells. Genes Cells 17, 807–816 [DOI] [PubMed] [Google Scholar]
- 26. Ohashi E., Takeishi Y., Ueda S., Tsurimoto T. (2014) Interaction between Rad9-Hus1-Rad1 and TopBP1 activates ATR-ATRIP and promotes TopBP1 recruitment to sites of UV damage. DNA Repair 21, 1–11 [DOI] [PubMed] [Google Scholar]
- 27. Hashimoto Y., Tsujimura T., Sugino A., Takisawa H. (2006) The phosphorylated C-terminal domain of Xenopus Cut5 directly mediates ATR-dependent activation of Chk1. Genes Cells 11, 993–1007 [DOI] [PubMed] [Google Scholar]
- 28. Kumagai A., Lee J., Yoo H. Y., Dunphy W. G. (2006) TopBP1 activates the ATR-ATRIP complex. Cell 124, 943–955 [DOI] [PubMed] [Google Scholar]
- 29. Choi J. H., Lindsey-Boltz L. A., Sancar A. (2007) Reconstitution of a human ATR-mediated checkpoint response to damaged DNA. Proc. Natl. Acad. Sci. U.S.A. 104, 13301–13306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Doré A. S., Kilkenny M. L., Rzechorzek N. J., Pearl L. H. (2009) Crystal structure of the rad9-rad1-hus1 DNA damage checkpoint complex–implications for clamp loading and regulation. Mol. Cell 34, 735–745 [DOI] [PubMed] [Google Scholar]
- 31. Sohn S. Y., Cho Y. (2009) Crystal structure of the human rad9-hus1-rad1 clamp. J. Mol. Biol. 390, 490–502 [DOI] [PubMed] [Google Scholar]
- 32. Xu M., Bai L., Gong Y., Xie W., Hang H., Jiang T. (2009) Structure and functional implications of the human rad9-hus1-rad1 cell cycle checkpoint complex. J. Biol. Chem. 284, 20457–20461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hirai I., Wang H. G. (2002) A role of the C-terminal region of human Rad9 (hRad9) in nuclear transport of the hRad9 checkpoint complex. J. Biol. Chem. 277, 25722–25727 [DOI] [PubMed] [Google Scholar]
- 34. Roos-Mattjus P., Hopkins K. M., Oestreich A. J., Vroman B. T., Johnson K. L., Naylor S., Lieberman H. B., Karnitz L. M. (2003) Phosphorylation of human Rad9 is required for genotoxin-activated checkpoint signaling. J. Biol. Chem. 278, 24428–24437 [DOI] [PubMed] [Google Scholar]
- 35. St Onge R. P., Besley B. D., Pelley J. L., Davey S. (2003) A role for the phosphorylation of hRad9 in checkpoint signaling. J. Biol. Chem. 278, 26620–26628 [DOI] [PubMed] [Google Scholar]
- 36. Wu X., Shell S. M., Zou Y. (2005) Interaction and colocalization of Rad9/Rad1/Hus1 checkpoint complex with replication protein A in human cells. Oncogene 24, 4728–4735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu X., Vaithiyalingam S., Glick G. G., Mordes D. A., Chazin W. J., Cortez D. (2008) The basic cleft of RPA70N binds multiple checkpoint proteins, including RAD9, to regulate ATR signaling. Mol. Cell. Biol. 28, 7345–7353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yilmaz S., Sancar A., Kemp M. G. (2011) Multiple ATR-Chk1 pathway proteins preferentially associate with checkpoint-inducing DNA substrates. PLoS One 6, e22986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang Y., Ishino S., Yamagami T., Kumamaru T., Satoh H., Ishino Y. (2012) The OsGEN-L protein from Oryza sativa possesses Holliday junction resolvase activity as well as 5′-flap endonuclease activity. J. Biochem. 151, 317–327 [DOI] [PubMed] [Google Scholar]
- 40. Bruning J. B., Shamoo Y. (2004) Structural and thermodynamic analysis of human PCNA with peptides derived from DNA polymerase-δ p66 subunit and flap endonuclease-1. Structure 12, 2209–2219 [DOI] [PubMed] [Google Scholar]
- 41. Ohashi E., Hanafusa T., Kamei K., Song I., Tomida J., Hashimoto H., Vaziri C., Ohmori H. (2009) Identification of a novel REV1-interacting motif necessary for DNA polymerase κ function. Genes Cells 14, 101–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Georgescu R. E., Kim S. S., Yurieva O., Kuriyan J., Kong X. P., O'Donnell M. (2008) Structure of a sliding clamp on DNA. Cell 132, 43–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Navadgi-Patil V. M., Burgers P. M. (2009) The unstructured C-terminal tail of the 9-1-1 clamp subunit Ddc1 activates Mec1/ATR via two distinct mechanisms. Mol. Cell 36, 743–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gulbis J. M., Kelman Z., Hurwitz J., O'Donnell M., Kuriyan J. (1996) Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA. Cell 87, 297–306 [DOI] [PubMed] [Google Scholar]
- 45. Jónsson Z. O., Hindges R., Hübscher U. (1998) Regulation of DNA replication and repair proteins through interaction with the front side of proliferating cell nuclear antigen. EMBO J. 17, 2412–2425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brandt P. D., Helt C. E., Keng P. C., Bambara R. A. (2006) The Rad9 protein enhances survival and promotes DNA repair following exposure to ionizing radiation. Biochem. Biophys. Res. Commun. 347, 232–237 [DOI] [PubMed] [Google Scholar]
- 47. Wang X., Hu B., Weiss R. S., Wang Y. (2006) The effect of Hus1 on ionizing radiation sensitivity is associated with homologous recombination repair but is independent of nonhomologous end-joining. Oncogene 25, 1980–1983 [DOI] [PubMed] [Google Scholar]
- 48. Cotta-Ramusino C., McDonald E. R., 3rd, Hurov K., Sowa M. E., Harper J. W., Elledge S. J. (2011) A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science 332, 1313–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pandita R. K., Sharma G. G., Laszlo A., Hopkins K. M., Davey S., Chakhparonian M., Gupta A., Wellinger R. J., Zhang J., Powell S. N., Roti Roti J. L., Lieberman H. B., Pandita T. K. (2006) Mammalian Rad9 plays a role in telomere stability, S- and G2-phase-specific cell survival, and homologous recombinational repair. Mol. Cell. Biol. 26, 1850–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Canfield C., Rains J., De Benedetti A. (2009) TLK1B promotes repair of DSBs via its interaction with Rad9 and Asf1. BMC Mol. Biol. 10, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Toueille M., El-Andaloussi N., Frouin I., Freire R., Funk D., Shevelev I., Friedrich-Heineken E., Villani G., Hottiger M. O., Hübscher U. (2004) The human Rad9/Rad1/Hus1 damage sensor clamp interacts with DNA polymerase β and increases its DNA substrate utilisation efficiency: implications for DNA repair. Nucleic Acids Res. 32, 3316–3324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Friedrich-Heineken E., Toueille M., Tännler B., Bürki C., Ferrari E., Hottiger M. O., Hübscher U. (2005) The two DNA clamps Rad9/Rad1/Hus1 complex and proliferating cell nuclear antigen differentially regulate flap endonuclease 1 activity. J. Mol. Biol. 353, 980–989 [DOI] [PubMed] [Google Scholar]
- 53. Smirnova E., Toueille M., Markkanen E., Hübscher U. (2005) The human checkpoint sensor and alternative DNA clamp Rad9-Rad1-Hus1 modulates the activity of DNA ligase I, a component of the long-patch base excision repair machinery. Biochem. J. 389, 13–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shi G., Chang D. Y., Cheng C. C., Guan X., Venclovas C., Lu A. L. (2006) Physical and functional interactions between MutY glycosylase homologue (MYH) and checkpoint proteins Rad9-Rad1-Hus1. Biochem. J. 400, 53–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Park M. J., Park J. H., Hahm S. H., Ko S. I., Lee Y. R., Chung J. H., Sohn S. Y., Cho Y., Kang L. W., Han Y. S. (2009) Repair activities of human 8-oxoguanine DNA glycosylase are stimulated by the interaction with human checkpoint sensor Rad9-Rad1-Hus1 complex. DNA Repair 8, 1190–1200 [DOI] [PubMed] [Google Scholar]
- 56. Moldovan G. L., Pfander B., Jentsch S. (2007) PCNA, the maestro of the replication fork. Cell 129, 665–679 [DOI] [PubMed] [Google Scholar]
- 57. Mayanagi K., Kiyonari S., Saito M., Shirai T., Ishino Y., Morikawa K. (2009) Mechanism of replication machinery assembly as revealed by the DNA ligase-PCNA-DNA complex architecture. Proc. Natl. Acad. Sci. U.S.A. 106, 4647–4652 [DOI] [PMC free article] [PubMed] [Google Scholar]