

Background: miR-17/20a are oncogenic microRNAs in human cancers.
Results: A p53 kinase DAPK3 was identified as a key target for miR-17/20a to promote tumorigenesis.
Conclusion: miR-17/20a and DAPK3 form a positive feed-forward loop to amplify tumorigenic signals.
Significance: Data suggest a key microRNA-mediated tumorigenic pathway.
Keywords: DNA damage, microRNA (miRNA), oncogene, p53, tumor
Abstract
MicroRNAs (miRs) are a class of small regulatory RNAs that have been implicated in diverse biological pathways, including cancer. miR-17/20a encoded by the c13orf25 locus is among the first miRs discovered to have oncogenic functions. The E2F family members have been established as the targets for these oncomiRs, which form a negative feedback loop to control cell cycle progression. However, this pathway does not seem to be sufficient to account for elevated expression of these oncomiRs in cancer cells to promote tumorigenesis. Here we report that miR-17/20a targets a p53 activating kinase DAPK3, leading to p53-dependent transcriptional de-repression of the oncomiRs. We demonstrate that DAPK3 plays a central role in preventing miR-17/20a depletion-induced genome instability and in miR-17/20a overexpression-triggered tumor formation. This newly identified tumorigenic pathway may thus contribute to miR-17/20a amplification and tumor growth in diverse human cancers.
Introduction
miRs2 are a class of small RNAs involved in the regulation of gene expression at a post-transcription level in diverse biological pathways (1). However, in most cases their full biological functions are only partially understood based on characterization of limited targets. For example, the miR-17-92a cluster is frequently overexpressed in multiple human cancers (2–4). Among six miRs expressed from this cluster, miR-17 and 20a are particularly oncogenic (5, 6). These two oncomiRs differ by only the 1st and the 12th bases. Because neither is involved in miR targeting according to the Ago2-miR-20a crystal structure (7), the two miRs are thought to essentially share similar biological functions.
c-Myc and E2F1 are long known to stimulate one another at the transcription level, thus forming a positive feed-forward loop (8). However, as uncontrolled expression of E2F1 is known to trigger apoptosis, this transcription activator has to be tightly regulated, which is accomplished at least in part by a negative feedback loop between miR-17/20a and E2F family members (6). As a transcription activator of the miR-17-92a locus, such an E2F dampening mechanism would also prevent elevated miR-17/20a expression, a hallmark of lymphomas and other cancer types in humans (2–4). Overexpressed miR-17/20a has been shown to promote tumorigenesis by reducing the cyclin-dependent inhibitor p21 to stimulate cell proliferation and by limiting the pro-apoptotic factor BCL2L11 to inhibit apoptosis of tumor cells (3, 9).
The tumor suppressor p53 has also been implicated in mediating transcriptional repression of the miR-17-92a cluster through binding the TATA box of the miR gene promoter, suggesting a key contribution of p53 to elevated miR-17/20a expression in human cancers (10). However, it has been unclear how this pathway might be integrated into the tumorigenic program elicited by these oncomiRs.
Our current study elucidates death-associated protein kinase 3 (DAPK3), a p53 activating kinase, as a key target for miR-17/20a and establishes a positive feed-forward loop in which miR-17/20a inhibits DAPK3 and reduced DAPK3 in turn restricts the p53 function in repressing the miR gene expression, thus leading to transcriptional de-repression of these oncomiRs. This newly elucidated pathway is likely coupled with the E2F pathway and other induced tumorigenic events to orchestrate elevated expression of these oncomiRs in human cancers. We also provide a series of functional evidence for a central function of DAPK3 in miR-17/20a-dependent regulation of genome stability and tumorigenesis on both cellular and animal models. By limiting p53 activation, this pathway may also contribute to tumorigenic events in wild type p53 cells.
Experimental Procedures
Cell Culture, RNA Oligonucleotide, and Antibodies
HeLa cells, HEK293T cells, and mouse breast cancer 4T1 cells were maintained in DMEM medium (Gibco) supplemented with 10% FBS (Hyclone).
The RNA oligonucleotides used in this study were purchased from Genepharma (Shanghai, China), including siRNA, miR mimics, and antagomirs (miR-17 mimic, 5′-caaagugcuuacagugcagguag-3′; miR-17 mimic mutant, 5′-CAAACUGUUUUCAAUGGAGAUAA-3′; miR-20a mimic, 5′-uaaagugcuuauagugcagguag-3′; miR-20a mimic mutant, 5′-UAAACUGUUUUUAAUGGAGAUAA-3′; miR-17 antagomir, 5′-CUACCUGCACUGUAAGCACUUUG-3′; miR-20a antagomir, 5′-CUACCUGCACUAUAAGCACUUUA-3′; siDAPK1-1, 5′-CAAGAAACGUUAGCAAAUGTT-3′; siDAPK1-2, 5′-GGUCAAGGAUCCAAAGAAGTT-3′; siDAPK2-1: 5′-GGGACGCCGGAATTTGTTGCTTT-3′; siDAPK2-2; 5′-GAGCCCCTGGGTCTGGAGGCTTT-3′; siDAPK2-3, 5′-TCTGGAGGCTGACATGTGGAGTT-3′; siDAPK3-1, 5′-GCACTCTAAGCGCATCGCACACTTT-3′; siDAPK3-2, 5′-GGGAACGAGTTCAAGAACATCTTCG-3′; siDAPK3-3, 5′-CCAGAGCCTGGAACATTCCTGGATT-3′; siE2F1-1, 5′-CCGTGGACTCTTCGGAGAACTTTCATT-3′; siE2F1-2, 5′-CGGAGAACTTTCAGATCTCCCTTAATT-3′; siE2F1–3, 5′-GAGAACTTTCAGATCTCCCTTAAGATT-3′; negative control (NC) small RNAs si-NC (5′-UUCUCCGAACGUGUCACGUTT-3′), miR-NC, (5′-UUCUCCGAACGUGUCACGUTT-3′), and inhibitor-NC (5′-CAGUACUUUUGUGUAGUACAA-3′)).
The antibodies used in this study were from different vendors, including anti-DAPK1 (Abcam), anti-DAPK2 (Abgent), anti-DAPK3 (Thermal scientific), anti-ACTIN (PTGlab), anti-E2F1 (Abcam), anti-ATM (Millipore), anti-ATM(Ser-1981) (Millipore), anti-p53BP1 (BD Transduction LaboratoriesTM), anti-p53BP1(Ser-25/29) (Bioss), anti-H2AX (PTGlab), anti-γ-H2AX (BioLegend), anti-p53 (Cell Signaling Technology), anti-p53(Ser-20) (Thermal scientific), anti-FLAG (PTGlab), and anti-Ago2 (Abnova).
Biotin Pulldown Assays
HeLa cells were seeded at 50% confluency in 10-cm dishes and transfected with 50 nm biotin-labeled miR-17/20a with RNAiMAX (Life Technologies) according to the manufacturer's instructions. After transfection for 48 h, cells were harvested, treated with RNase-free radioimmune precipitation lysis buffer, and incubated on ice for 30 min with shaking 6 times at the 5-min interval. Cell lysates were clarified by centrifugation at 12,000 rpm for 20 min at 4 °C. The supernatants were transferred into a fresh RNase-free tube, 300 μl of New England BioLabs streptavidin beads were added (pre-washed and resuspended in RNase-free radioimmune precipitation lysis buffer), and the supernatants were incubated on a roller for 1 h at 4 °C. The beads were washed three times with RNase-free radioimmune precipitation lysis buffer. RNA was extracted with TRIzol (Life Technologies) according to the manufacturer's instruction.
All biotinylated oligonucleotides carry C6-biotin at the 3′ terminus including the following: Bio-miR-17 (sense),5′-CAAAGUGCUUAUAGUGCAGGUAG-3′; Bio-miR-17 (antisense), 5′-CUACCUGCACUGUAAGCACUUUG-3′; Bio-miR-20a (sense), 5′-UAAAGUGCUUAUAGUGCAGGUAG-3′; Bio-miR-20a (antisense), 5′-CUACCUGCACUAUAAGCACUUUA-3′; Bio-siGFP (sense), 5′-UGAAUUAGAUGGCGAUGUUdTdT-3′; Bio-siGFP (antisense), 5′-AACAUCGCCAUCUAAUUCAdTdT-3′.
RT-PCR and RT-qPCR
Total RNA was extracted by TRIzol (Life Technologies) and treated with RQ1 DNase I and ImpromII (Promega). For RT reactions, 1 μg of total RNA was used in a 30-μl RT reaction containing 10 pmol of the RT primer. After the RT reaction, 1 μl of cDNA was used for the PCR reaction along with the Tiangen SYBR mix according to manufacturer's instruction. Levels of GAPDH and GFP mRNAs were measured for normalization. The primers used are as follows: DAPK1 forward (F), 5′-AATCCTAGACGTGGTCCGGTAT-3′; DAPK1 reverse (R), 5′-GAGTCCTCGGTGCGTATCCTT-3′; DAPK2 F, 5′-GTGAGGGACCAATCCTGTGAC-3′; DAPK2 R, 5′-AGAAGCCAAGCATCAGGTAAA-3′; DAPK3 F, 5′-GTGCCCAACCCACGAATCAA-3′; DAPK3 R, 5′-AGCGGCTCATAGTTCACAATCTC-3′; GAPDH F, 5′-CTCACTGCTGGGGAGTCCCT-3′; GAPDH R, 5′-GGTCTACATGGCAACTGTG-3′; primary miR-17-92 F, 5′-CAGTAAAGGTAAGGAGAGCTCAATCTG-3′; primary miR-17-92 R, 5′-CATACAACCACTAAGCTAAAGAATAATCTGA-3′; miR-17 RT, 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCTACCTGCACTG-3′; miR-18a RT, 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCTATCTGCACTA-3′; miR-19a RT, 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACTCAGTTTTGCATA-3′; miR-20a RT, 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCTACCTGCACTA-3′; miR-19b RT, 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACTCAGTTTTGCATG-3′; miR-92a RT, 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACACAGGC-3′; miR-17-92 universal R, 5′-CAGTGCAGGGTCCGAGG-3′; miR-17 F, 5′-CGGCGGCAAAGTGCTTAC-3′; miR-18a F, 5′-CGGCGGTAAGGTGCATCT-3′; miR-19a F, 5′-CGGCGGTGTGCAAATCT-3′; miR-20a F, 5′-CGGCGGTAAAGTGCTTAT-3′; miR-19b F, 5′-CGGCGGTGTGCAAATCC-3′; miR-92a F, 5′-CGGCGGTATTGCACTTGT-3′; qIRES-FLAG-DAPK3 F, 5′-AACCGTCAGATCCGCTAGCGCTA-3′; qIRES-FLAG-DAPK3 R, 5′-CAGCTCCTCCCCCATCTCAT-3′; IRES FLAG eGFP F, 5′-AGCAGAAGAACGGCATCAAGGT-3′; IRES FLAG eGFP F, 5′-GGACTGGGTGCTCAGGTAGTGG-3′; let-7b primer set was purchased from RiboBio.
Immunostaining
Cells were seeded in 24-well plates to reach ∼30% confluency and then transfected with Lipofectamine RNAiMAX (Life Technologies) with 50 nm miR-17/20a mimics or antagomirs with or without 50 nm siRNA against DAPK1, DAPK2, DAPK3, or E2F1. After incubation for 24 h, cells were further transfected with 0.8 μg of expression plasmids, and 48 h after transfection cells were fixed with 4% paraformaldehyde, permeabilized with 1% Triton X-100, and incubated overnight with specific antibodies against various double-strand DNA (dsDNA) damage markers. Cells were washed with PBS, incubated with fluorescence-labeled secondary antibody for 1 h, and mounted in DAPI solution for confocal microscopy.
Target Site Validation and Western Blotting
The DAPK3 coding region along with a FLAG tag was cloned in the IRES-eGFP vector, and a synonymous mutation was introduced in the miR-17/20a target site. The following primers were used for validating the target site: IRES-GFP-FLAG-DAPK3 forward (F), 5′-CCGGAATTCGACTACAAGGACGATGATGACAAGTCCACGTTCAGGCAGGAGGA-3′; IRES-GFP-FLAG-DAPK3 reverse (R), 5′-CGCGGATCCATGCTAGCGCAGCCCGCACTCCA-3′; IRES-GFP-FLAG-DAPK3 mutant F, 5′-CCTGGACGGCGTTCATTATCTCCATTCAAAACGGATCGCACACTTTGACCTGAAGC-3′; IRES-GFP-FLAG-DAPK3Mut R, 5′-GCTTCAGGTCAAAGTGTGCGATCCGTTTTGAATGGAGATAATGAACGCCGTCCAGG-3′.
For Western blotting, 48 h after transfection, cells were washed twice with PBS and lysed with 1× SDS loading buffer. The cell lysates were resolved in 10% SDS-PAGE followed by transfer to PVDF membrane. The membrane was washed and blocked by TBS-Tween with 5% skim milk and incubated with specific primary antibody overnight. The membrane was then washed and incubated with secondary antibody, and signals were detected by chemiluminescence.
Stable Cell Lines and the Assay for Tumor Formation
miR-17/20a with or without DAPK3 and DAPK3 mutant were cloned in the pLenti-CMV-GFP-puro vector. The primers used for contrasting the lentiviral expression system are as follows: pLenti-miR-17/20a forward (F), 5′-CGCGGATCCATACGTGTCTAAATGGACCTCATAT-3′; pLenti-miR-17/20a reverse (R), 5′-ACGCGTCGACATATCCTGGAATAACACTAACTCCA-3′; miR-17/20a of pLenti-miR-17/20 plus DAPK3 F, 5′-CCCAAGCTTATACGTGTCTAAATGGACCTCATAT-3′; miR17–20a of pLenti-miR-17/20a plus DAPK3 R, 5′-ACGCGTCGACATATCCTGGAATAACACTAACTCCA-3′: DAPK3 of pLenti miR-17/20a plus DAPK3 F, 5′-CGCGGATCCATGTCCACGTTCAGGCAGGAGG-3′; DAPK3 of pLenti-miR-17/20a plus DAPK3 R, 5′-CCCAAGCTTCTAGCGCAGCCCGCACTCCACGC-3′.
4T1 cells were infected and selected according to the lentiviral expression and packaging protocol from the Addgene website. Briefly, HEK293T cells were seeded at 40% confluency in 10-cm dishes, and 4 μg of pLenti plus 3 μg of pxPAX2 and 1 μg of pMD2.G were co-transfected when cells reached 90% confluency with Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions. After 48 h the lentiviral particle-containing medium was clarified by centrifugation at 1000 rpm for 5 min at 4 °C. The supernatant was transferred to a clean tube followed by the addition of Polybrene to 8 μg/ml. The packaged virus was used to infect 4T1 cells at 80% confluency for 24 h, and infected cells were selected with puromycin at 1 μg/ml.
Female Balb/C nude mice at 5 weeks of age were subcutaneously injected with 5 × 105 4T1 cells infected with various miRs and/or expression plasmids as indicated in the text. Tumor size and weight were quantified 2 weeks after injection.
Results
Identification of the p53 Activating Kinase DAPK3 as a Key Target for miR-17/20a
To fully comprehend the regulatory network of miR-17/20a in cancer, it has been suggested that identification and characterization of additional targets for these oncomiRs will be necessary (11). Toward this goal, we applied an experimental strategy to identify miR targets by primer extension from specific miRs in immunoprecipitated Ago2-containing RNA-induced silencing complex (12). We identified a p53-Ser-20 kinase characterized earlier as DAPK3 to be a target for miR-17/20a (Fig. 1A) (13–16). Interestingly, the newly identified target site resides within the second exon of the DAPK3 gene, which is evolutionarily conserved among mammals (Fig. 1A). We also noted multiple Ago2 binding events on the site from several independent genome-wide Ago2-RNA interaction datasets (17–19), together suggesting that this target site is a bona fide site for miR-17/20a in the human genome.
FIGURE 1.

miR-17/20a targets DAPK3 in the coding region. A, an experimentally identified miR-20a target site resides in a conserved region in the second exon of the DAPK3 gene. B, biotinylated miR-17/20a specifically captured DAPK3 mRNA in a biotin pulldown assay. DAPK1 and -2 served as negative controls. C, base-pairing interactions between miR-17/20a and DAPK3 (upper panel) and Western blotting analysis of FLAG-tagged wild type and mutant DAPK3 in response to transfected miR-17/20a in HeLa cells (lower panel). P cmv, CMV promoter. D, base-pairing potential between a mutant miR-17/20a mimic and DAPK3 and restored base-pairing with a mutant DAPK3 (upper panel) and Western blotting analysis of FLAG-tagged wild type and mutant DAPK3 in HeLa cells transfected with the wild type and mutant miR-17/20a (lower panel). E, unaltered mRNA levels of FLAG-tagged wild type and mutant DAPK3 in response to transfected miR-17/20a mimics.
The miR-17/20a target site is unique to DAPK3, as the corresponding sequences in related DAPK1 and DAPK2 are highly diversified (Fig. 1A). To establish the targeting specificity, we used biotinylated miR-17/20a transfected in HeLa cells to capture DAPK3, but not DAPK1 or DAPK2 under the same experimental conditions (Fig. 1B). These data suggest specific binding of miR-17/20a with the DAPK3 mRNA.
Although miRs typically target 3′-UTRs, there are several reports that document the capacity of miRs to target protein-coding sequences (CDS) (17, 18, 20, 21). We have also taken this opportunity to further determine the mechanism for CDS-targeted miRs, such as the effect of transfected miR-17/20a on endogenous DAPK3 (see below in Fig. 5A). To determine the requirement for deduced base-pairing between miR-17/20a and the identified target site in DAPK3 mRNA, we cloned the DAPK3 cDNA as a fusion protein with a FLAG tag in the N terminus (Fig. 1C). For comparison, we introduced synonymous mutations in the DAPK3 target site to disrupt miR-17/20a binding (Fig. 1D). In response to miR-17/20a overexpression in transfected HeLa cells, the level of FLAG-DAPK3 was greatly reduced, whereas the GFP signal expressed from the IRES-eGFP unit in the same expression plasmid was unaffected (Fig. 1C, lower panel). The mutations in the miR-17/20a targeting site prevented the effect of transfected miR-17/20a and, instead, responded to a mutant miR mimic designed to restore the base-pairing potential with the mutant reporter (Fig. 1D, lower panel). We detected no change in the mRNA level with either wild type or mutant reporters under these experimental conditions (Fig. 1E). These data suggest that miR-17/20a targets DAPK3 in the CDS region to suppress its translation.
FIGURE 5.

DAPK3 contributes to miR-17/20a-enhanced tumorigenesis. A, Western blotting analysis to p21, c-Myc, and DAPK3 levels in mouse breast cancer 4T1 cells stably expressing miR-17/20a or in combination with the expression plasmids for wild type DAPK3 or a miR-17/20a-resistent DAPK3. B, tumor size of mice subcutaneously injected with 4T1 cells from A. C, quantification of tumor weight from B. *, p < 0.05; ***, p < 0.001. D, DAPK3 mRNA levels in c-Myc transgenic tissues and control tissues (from Ref. 6). E, an extended gene network regulated by miR-17/20a in tumorigenesis. miR-17/20a inhibits E2F1 and E2F1 promotes transcription from the miR-17-92 gene cluster. E2F1 and c-Myc enhance each other at the level of transcription, and c-myc also positively regulates miR-17/20a transcription. Blue shading highlights the new branch in this network, illustrating that miR-17/20a translationally inhibits DAPK3, which is responsible for phosphorylating p53 at Ser-20 (and perhaps other sites), and that activated p53 transcriptionally represses miR-17/20a, thus forming a positive feed-forward loop to elevate miR-17/20a in tumor cells. miR-17/20a is also known to inhibit BCL2L11 (BIM) to impair the apoptotic program and to inhibit p21 to promote cell cycle. Together, these events promote tumorigenesis with a central role of miR-17/20a in the regulation of this gene network.
A Key Role of DAPK3 in miR-17/20a-dependent Regulation of Genome Stability
miR-17/20a is known to provide a critical function in protecting the genome against DNA damage, as blockage of these oncomiRs has been shown to induce dsDNA breaks and cell cycle arrest (22). We first confirmed the induction of dsDNA breaks in HeLa cells transfected with antagomirs against miR-17/20a, as indicated by immunostaining analysis for dsDNA breaks markers, such as ATM(Ser-1981), p53BP1(Ser-25/29), and γ-H2AX (Fig. 2A, first column). These effects could also be validated by Western blotting with specific antibodies to detect elevated ATM(Ser-1981), and p53BP1(Ser-25/29), and γ-H2AX relative to the consistent total protein level in each case in response to transfected antagomirs against miR-17, miR-20a, or both (Fig. 2C). The elevation of DAPK3 expression was also confirmed under the same conditions.
FIGURE 2.

DAPK3 is critical for miR-17/20a depletion-induced dsDNA breaks. A, confocal microscopic analysis of dsDNA breaks by staining for ATM(Ser-1981), p53BP1(Ser-25/29), and γH2AX in HeLa cells transfected with miR-17/20a antagomirs or the pcDNA3-based plasmid to overexpress FLAG-DAPK3 or in combination with siDAPK1, siDAPK2, siDAPK3, or siE2F1 as individually marked on the top. B, Western blotting analysis to verify DAPK3 overexpression and the effects of multiple siRNAs against DAPK1, DAPK2, DAPK3, and E2F1. NC, negative control. C, Western blotting analysis of dsDNA breaks by detecting levels of ATM(Ser-1981), p53BP1(Ser-25/29), and γH2AX as well as DAPK3 in HeLa cells transfected with miR-17, miR-20a, and miR-17/20a. D, Western blotting analysis of dsDNA breaks by ATM(Ser-1981), p53BP1(Ser-25/29), and γH2AX in HeLa cells in response to DAPK3 overexpression.
Similarly, DAPK3 overexpression (Fig. 2B) exhibited the same effect at both immunostaining and Western blotting levels (Fig. 2, A and D). We next determined whether miR-17/20a depletion-induced genome instability is DAPK3-dependent. By treating the cell with siRNA against DAPK3 (Fig. 2B), we found that the treatment effectively prevented miR-17/20a antagomir-induced genome instability (Fig. 2A, third column). In contrast, we detected little effect after siRNA-mediated depletion of either DAPK1 or DAPK2 (Fig. 2A, fourth and fifth column), whose efficient knockdown was confirmed by Western blotting (Fig. 2B).
A previous study assigned a role of E2F1 in mediating miR-17/20a depletion-induced genome instability (22). However, in our hands we still detected significant signals for induced dsDNA breaks in HeLa cells co-transfected with miR-17/20a antagomirs in combination with siRNA against E2F1 (Fig. 2A, last column). Taken together, these data suggest that DAPK3 is a major mediator in miR-17/20a depletion-induced genome instability. Defects in this pathway may thus increase the mutation rate to propel tumorigenesis (23).
A Positive Feed-forward Loop to Amplify miR-17/20a
To further understand the potential functional interplay between DAPK3 and miR-17/20a expression, we found that DAPK3 overexpression repressed both the primary and mature miRs from the miR-17-92 cluster in HeLa cells (Fig. 3A). Given p53 has been implicated as a transcriptional repressor for the miR-17-92a cluster (10), we extended the analysis on a pair of isogenic HCT116 and HCT116 p53−/− cell lines. We observed with let-7 as an internal negative control that the steady state levels of miRs from the miR-17-92a cluster were all lower in wild type HCT116 cells relative to HCT116 p53−/− cells (Fig. 3B).
FIGURE 3.

DAPK3 mediates a feed forward loop between miR-17/20a and p53. A, RT-qPCR analysis of primary and mature miR-17/20a in HeLa cells transfected with a DAPK3 overexpression plasmid. B, RT-qPCR analysis of primary and mature miR-17/20a in HCT116 cells and HCT116 p53−/− cells. C, Western blotting analysis of p53 Ser-20 in HCT116 cells in response to DAPK3 overexpression. D, RT-qPCR analysis of primary and mature miR-17/20a in HCT116 cells in response to DAPK3 overexpression. E, RT-qPCR analysis of primary and mature miR-17/20a in HCT116 p53−/− cells in response to DAPK3 overexpression. *, p < 0.05; **, p < 0.01; ***, p < 0.001 based on triplicate experiments.
DAPK3 has been established as a key enzyme for catalyzing Ser-20 phosphorylation in the transactivation domain of p53, which is critical for the function of p53 in suppressing B-cell tumors (24). By overexpressing DAPK3 in wild type HCT116 cells, we confirmed elevated p53 phosphorylation at Ser-20 (Fig. 3C). This was again correlated with the repressive effect of DAPK3 overexpression on the expression of both primary and mature miRs from the miR-17-92a locus in wild type HCT116 cells (Fig. 3D). We next repeated the experiment on HCT116 p53−/− cells, finding that inactivation of p53 abolished the effect of overexpressed DAPK3 on miR-17-92a expression (Fig. 3E).
Taken together, these data suggest that DAPK3 is a key kinase for p53-Ser-20 and represses miR-17/20a transcription in a p53-dependent manner. These findings imply that elevated miR-17/20a would diminish DPAK3, which would in turn attenuate p53 activation, thus leading to transcriptional de-repression of the miR-17/20a locus. This positive feed-forward loop may thus contribute to miR-17/20a amplification in human cancers (see “Discussion”).
Contribution of DAPK3 to Genome Instability in a p53-independent Manner
Given p53 is involved in regulated expression of miR-17/20a, which in turn controls the level of DAPK3, and the well known involvement of p53 in dsDNA breaks, we wondered whether DAPK3 overexpression-induced genome instability might also be influenced by the p53 status. We transfected miR-17/20a antagomirs in HCT116 or HCT116 p53−/− cells, finding elevated γH2AX in both cell types (Fig. 4, A and B). Similarly, DAPK3 overexpression in both cell types also induced dsDNA breaks, as indicated by elevated γH2AX (Fig. 4, C and D). These observations suggest that another pathway(s), rather than p53, is responsible for induced dsDNA breaks in response to antagomir-mediated depletion of miR-17/20a and DAPK3 overexpression.
FIGURE 4.
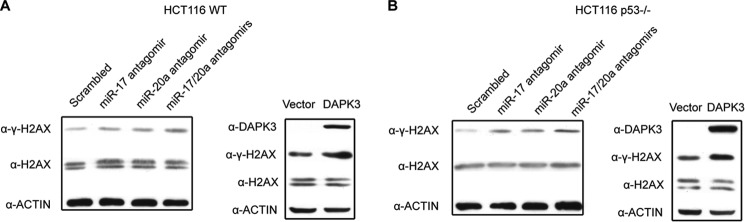
Genome instability in response to depletion of miR-17/20a or DAPK3 overexpression is independent of p53. A, analysis of dsDNA breaks by Western blotting for γH2AX in HCT116 cells transfected with miR-17 antagomir, miR-20a antagomir, or both (left panel) or in response to DAPK3 overexpression plasmid (right panel). B, analysis of dsDNA breaks by Western blotting for γH2AX in HCT116 p53−/− cells under the same conditions as A.
A Key Role of DAPK3 in miR-17/20a-dependent Tumorigenic Pathway
We next wished to establish a direct contribution of DAPK3 to miR-17/20a oncogenic functions in an animal model. For this purpose, we utilized a breast cancer 4T1 cell line in which we stably augmented the miR-17/20a level by lentivirus infection. Western blotting showed that this led to diminished expression of the endogenous DAPK3 (Fig. 5A). For comparison, we also engineered co-expression of wild type DAPK3 or a variant DAPK3 that carries synonymous mutations in the miR-17/20a target site, and as expected, we detected elevated DAPK3 when the miR-17/20a target was mutated (Fig. 5A). To connect DAPK3 to other key components in the oncogenic miR-17/20a pathway, we examined the levels of p21 and c-Myc, showing dramatic elevation of both in response to DAPK3 overexpression. These data suggest that DAPK3 plays a central role in controlling the miR-17/20a-regulated oncogenic pathway.
We subcutaneously injected these cells in Balb/C nude mice and examined the tumor size 2 weeks later. We found that augmented expression of miR-17/20a indeed enhanced tumor growth, as evidenced by the larger tumor mass (Fig. 5B). In contrast, restored expression of wild type DAPK3 attenuated the effect, and the expression of a miR-17/20a-resistant DAPK3 was more effective in attenuating the tumorigenic potential (Fig. 5B). The data were quantified based on tumor weight, which is consistent with the observed tumor size (Fig. 5C). Taken together, these data suggest that DAPK3 down-regulation contributes to the oncogenic functions of miR-17/20a in tumor development.
Discussion
By applying an experimental strategy for identifying miR targets, we unveiled DAPK3 as a new target for oncogenic miR-17/20a. The target site resides in the highly conserved second exon of the DAPK3 mRNA. We demonstrate that miR-17/20a was highly effective in regulating the expression of both endogenous and exogenous DAPK3. These findings provide a further example for the functionality of CDS-targeted miRs.
Importantly, we provide evidence that regulated DAPK3 expression is critical for miR-17/20a-dependent protection of genome stability, which was previously attributed to E2F1 (22). However, our data suggest that the major role in this pathway is provided by DAPK3. Interestingly, overexpression of DAPK3 in both HeLa and HCT116 cells induced dsDNA breaks regardless of the p53 status. These results indicate that elevated DAPK3 induces genome instability via a p53-independent mechanism, which warrants future investigation.
As miR-17/20a overexpression has been observed in diverse human cancers, it remains unclear how overexpression of these oncomiRs is maintained and/or regulated in cancers. Although E2F family members have been well characterized as the targets of these oncomiRs, the negative feedback loop formed between E2Fs and miR-17/20a seems to be sufficient to account for homeostatic expression of these oncomiRs but insufficient to explain their overexpression in human cancers. We now elucidate a new pathway that involves the p53 activating kinase DAPK3. By integrating the existing information, we are able to construct a highly intertwined regulatory network as follows.
c-Myc has been well established to transcriptionally activate the miR-17-92a locus (6), which is predicted to diminish DAPK3 expression according to the newly identify pathway in the current study. We examined the existing data from several c-Myc overexpression cell lines (6). Compared to isogenic control lines, we indeed noted lower DAPK3 mRNA levels in multiple c-Myc overexpressing lines (Fig. 5D). More importantly, our data also suggest a positive feed-forward loop, as highlighted in blue shade (Fig. 5E). In this loop, initially elevated miR-17/20a, which may be induced by increased c-Myc followed by the homeostatic regulation via E2Fs, may diminish DAPK3. This would limit the p53 activation potential, thus suppressing its tumor suppressor function. Such attenuated p53 function would also cause transcriptional de-repression of miR-17/20a, allowing the maintenance of these elevated oncomiRs in tumor cells.
Moreover, such reduced p53 would also be less effective in activating p21. In this regard a previous study suggests that DAPK3 may also function as a key kinase for phosphorylating p21 to increase its half-life (25). Consistently, we detected a dramatic increase in p21 expression in DAPK3 overexpression cells (Fig. 5A). Via these combined mechanisms, diminished DAPK3 may thus promote tumorigenesis by reducing p21 expression and its protein stability, thereby enhancing tumor cell proliferation.
The regulatory network elucidated in the present study provides critical insights into the function of DAPK3 in both healthy and disease states. In healthy cells and tissues, a balanced DAPK3 level may be critical to ensure genome stability, which likely functions in conjunction with E2F family members to keep miR-17/20a at a hemostatic level. When cells undergo oncogenic transformation, such as those induced by miR-17/20a overexpression in response to c-Myc amplification or other oncogenic events, the cellular defense system mediated by DAPK3 is diminished. The data presented in this report demonstrate that this pathway plays a central role in miR-17/20a-induced tumorigenesis.
Author Contributions
Z. C. and X.-D. F. designed the experiments. Z. C., R. C., K. Z., Y. X., and C. Z. performed experiments, J. Z., Y. Z., and H. S. analyzed the data, and Z. C., K. Z., J. Z., and X. D. F. wrote the paper.
This work was supported, in whole or in part, by National Institutes of Health Grants GM049369 and GM052872. This work was also supported by the China 973 program (2011CB811300 and 2012CB910800) and Chinese 111 program Grant B06018. The authors declare that they have no conflicts of interest with the contents of this article.
- miR
- microRNA
- qPCR
- quantitative PCR
- DAPK3
- death-associated protein kinase 3.
REFERENCES
- 1. He L., Hannon G. J. (2004) MicroRNAs: small RNAs with a big role in gene regulation. Nat. Rev. Genet. 5, 522–531 [DOI] [PubMed] [Google Scholar]
- 2. Ota A., Tagawa H., Karnan S., Tsuzuki S., Karpas A., Kira S., Yoshida Y., Seto M. (2004) Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res. 64, 3087–3095 [DOI] [PubMed] [Google Scholar]
- 3. Petrocca F., Vecchione A., Croce C. M. (2008) Emerging role of miR-106b-25/miR-17-92 clusters in the control of transforming growth factor beta signaling. Cancer Res. 68, 8191–8194 [DOI] [PubMed] [Google Scholar]
- 4. Volinia S., Calin G. A., Liu C. G., Ambs S., Cimmino A., Petrocca F., Visone R., Iorio M., Roldo C., Ferracin M., Prueitt R. L., Yanaihara N., Lanza G., Scarpa A., Vecchione A., Negrini M., Harris C. C., Croce C. M. (2006) A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. U.S.A. 103, 2257–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. He L., Thomson J. M., Hemann M. T., Hernando-Monge E., Mu D., Goodson S., Powers S., Cordon-Cardo C., Lowe S. W., Hannon G. J., Hammond S. M. (2005) A microRNA polycistron as a potential human oncogene. Nature 435, 828–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. O'Donnell K. A., Wentzel E. A., Zeller K. I., Dang C. V., Mendell J. T. (2005) c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435, 839–843 [DOI] [PubMed] [Google Scholar]
- 7. Elkayam E., Kuhn C. D., Tocilj A., Haase A. D., Greene E. M., Hannon G. J., Joshua-Tor L. (2012) The structure of human argonaute-2 in complex with miR-20a. Cell 150, 100–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sylvestre Y., De Guire V., Querido E., Mukhopadhyay U. K., Bourdeau V., Major F., Ferbeyre G., Chartrand P. (2007) An E2F/miR-20a autoregulatory feedback loop. J. Biol. Chem. 282, 2135–2143 [DOI] [PubMed] [Google Scholar]
- 9. Ivanovska I., Ball A. S., Diaz R. L., Magnus J. F., Kibukawa M., Schelter J. M., Kobayashi S. V., Lim L., Burchard J., Jackson A. L., Linsley P. S., Cleary M. A. (2008) MicroRNAs in the miR-106b family regulate p21/CDKN1A and promote cell cycle progression. Mol. Cell. Biol. 28, 2167–2174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yan H. L., Xue G., Mei Q., Wang Y. Z., Ding F. X., Liu M. F., Lu M. H., Tang Y., Yu H. Y., Sun S. H. (2009) Repression of the miR-17-92 cluster by p53 has an important function in hypoxia-induced apoptosis. EMBO J. 28, 2719–2732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chang T. C., Yu D., Lee Y. S., Wentzel E. A., Arking D. E., West K. M., Dang C. V., Thomas-Tikhonenko A., Mendell J. T. (2008) Widespread microRNA repression by Myc contributes to tumorigenesis. Nat. Genet. 40, 43–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang X., Zuo X., Yang B., Li Z., Xue Y., Zhou Y., Huang J., Zhao X., Zhou J., Yan Y., Zhang H., Guo P., Sun H., Guo L., Zhang Y., Fu X. D. (2014) MicroRNA directly enhances mitochondrial translation during muscle differentiation. Cell 158, 607–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gozuacik D., Kimchi A. (2006) DAPk protein family and cancer. Autophagy 2, 74–79 [DOI] [PubMed] [Google Scholar]
- 14. Bi J., Lau S. H., Hu L., Rao H. L., Liu H. B., Zhan W. H., Chen G., Wen J. M., Wang Q., Li B., Guan X. Y. (2009) Downregulation of ZIP kinase is associated with tumor invasion, metastasis and poor prognosis in gastric cancer. Int. J. Cancer 124, 1587–1593 [DOI] [PubMed] [Google Scholar]
- 15. Kawai T., Matsumoto M., Takeda K., Sanjo H., Akira S. (1998) ZIP kinase, a novel serine/threonine kinase which mediates apoptosis. Mol. Cell. Biol. 18, 1642–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kögel D., Plöttner O., Landsberg G., Christian S., Scheidtmann K. H. (1998) Cloning and characterization of Dlk, a novel serine/threonine kinase that is tightly associated with chromatin and phosphorylates core histones. Oncogene 17, 2645–2654 [DOI] [PubMed] [Google Scholar]
- 17. Chi S. W., Zang J. B., Mele A., Darnell R. B. (2009) Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 460, 479–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hafner M., Lianoglou S., Tuschl T., Betel D. (2012) Genome-wide identification of miRNA targets by PAR-CLIP. Methods 58, 94–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xue Y., Ouyang K., Huang J., Zhou Y., Ouyang H., Li H., Wang G., Wu Q., Wei C., Bi Y., Jiang L., Cai Z., Sun H., Zhang K., Zhang Y., Chen J., Fu X. D. (2013) Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated microRNA circuits. Cell 152, 82–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Miranda K. C., Huynh T., Tay Y., Ang Y. S., Tam W. L., Thomson A. M., Lim B., Rigoutsos I. (2006) A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell 126, 1203–1217 [DOI] [PubMed] [Google Scholar]
- 21. Tay Y., Zhang J., Thomson A. M., Lim B., Rigoutsos I. (2008) MicroRNAs to Nanog, Oct4, and Sox2 coding regions modulate embryonic stem cell differentiation. Nature 455, 1124–1128 [DOI] [PubMed] [Google Scholar]
- 22. Pickering M. T., Stadler B. M., Kowalik T. F. (2009) miR-17 and miR-20a temper an E2F1-induced G1 checkpoint to regulate cell cycle progression. Oncogene 28, 140–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang L., Huang J., Yang N., Greshock J., Megraw M. S., Giannakakis A., Liang S., Naylor T. L., Barchetti A., Ward M. R., Yao G., Medina A., O'brien-Jenkins A., Katsaros D., Hatzigeorgiou A., Gimotty P. A., Weber B. L., Coukos G. (2006) microRNAs exhibit high frequency genomic alterations in human cancer. Proc. Natl. Acad. Sci. U.S.A. 103, 9136–9141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Craig A. L., Chrystal J. A., Fraser J. A., Sphyris N., Lin Y., Harrison B. J., Scott M. T., Dornreiter I., Hupp T. R. (2007) The MDM2 ubiquitination signal in the DNA-binding domain of p53 forms a docking site for calcium calmodulin kinase superfamily members. Mol. Cell. Biol. 27, 3542–3555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burch L. R., Scott M., Pohler E., Meek D., Hupp T. (2004) Phage-peptide display identifies the interferon-responsive, death-activated protein kinase family as a novel modifier of MDM2 and p21WAF1. J. Mol. Biol. 337, 115–128 [DOI] [PubMed] [Google Scholar]