

Background: The p63 isoforms ΔNp63α, ΔNp63β, ΔNp63γ, and Grainyhead-like 2 (GRHL2) play distinct roles in regulating the epithelial phenotype.
Results: p63 modulation leads to epithelial-mesenchymal transition in human keratinocytes, and GRHL2 binds directly to the p63 promoter.
Conclusion: GRHL2/p63 reciprocal regulation maintains the epithelial phenotype and plasticity.
Significance: The GHRL2/p63 model is crucial for understanding epithelial plasticity and metastasis, epithelial wound healing, and tissue regeneration.
Keywords: epithelial-mesenchymal transition (EMT), epithelium, gene regulation, keratinocyte, p63, GRHL2
Abstract
In this study, we investigated the effects of p63 modulation in epithelial plasticity in human keratinocytes. The p63 isoforms ΔNp63α, ΔNp63β, and ΔNp63γ were ectopically expressed in normal human epidermal keratinocytes (NHEKs). The epithelial or mesenchymal state was determined by morphological changes and altered expression of various markers, e.g. fibronectin, E-Cadherin, and keratin 14. Overexpression of ΔNp63α and ΔNp63β but not ΔNp63γ isoforms led to morphological changes consistent with epithelial-mesenchymal transition (EMT). However, only ΔNp63α overexpression was able to maintain the morphological changes and molecular phenotype consistent with EMT. Interestingly, knockdown of all p63 isoforms by transfection of p63 siRNA also led to the EMT phenotype, further confirming the role of p63 in regulating the epithelial phenotype in NHEKs. EMT in NHKs accompanied loss of Grainyhead-Like 2 (GHRL2) and miR-200 family gene expression, both of which play crucial roles in determining the epithelial phenotype. Modulation of GRHL2 in NHKs also led to congruent changes in p63 expression. ChIP revealed direct GRHL2 binding to the p63 promoter. GRHL2 knockdown in NHK led to impaired binding of GRHL2 and changes in the histone marks consistent with p63 gene silencing. These data indicate the presence of a reciprocal feedback regulation between p63 and GRHL2 in NHEKs to regulate epithelial plasticity.
Introduction
p63 is a transcription factor that regulates epithelial phenotype and keratinocyte proliferation and is an important marker of epidermal and stratified keratinocyte stem cells (4, 5). p63 is expressed as six isoforms. Three of these isoforms contain the N-terminal transactivation domain (TAp63α, TAp63β, and TAp63γ), whereas the remainder (ΔNp63α, ΔNp63β, and ΔNp63γ) do not (6). Our recent study has demonstrated that transduction of the ΔNp63α isoform and intact TGF-β signaling is necessary to induce epithelial-mesenchymal transition (EMT)3 in normal human epidermal keratinocytes (NHEKs) (4).
EMT is a process by which epithelial cells exhibit morphologic and molecular changes consistent with the mesenchymal phenotype and is involved in tissue and organ development and repair (7). Cells with the EMT phenotype exhibit loss of epithelial morphology and development of an elongated, spindled, mesenchymal morphology (8). EMT is facilitated by the loss of cell-cell adhesion mediated by E-Cadherin (E-Cad), and loss of E-Cad expression serves as a marker for EMT (9). Also, loss of keratin 14 (K14) and enhanced expression of fibronectin (FN) and vimentin serve as mesenchymal markers (9–11).
GRHL2 is a novel transcription factor involved with epithelial morphogenesis, cell proliferation, and differentiation (12). Our recent studies have shown that GRHL2 transcriptionally regulates a broad spectrum of target genes, including the human telomerase (hTERT) gene, proliferating cell nuclear antigen gene, and epidermal differentiation complex genes (12, 13). Phenotypically, GRHL2 promotes epithelial cell proliferation and inhibits keratinocyte differentiation in NHEKs (12). Other studies suggest its inhibitory role in EMT and apoptosis in breast cancer cell lines, demonstrating its involvement in cancer progression (14). Mechanistically, GRHL2 down-regulates zinc finger E-box binding protein 1 (ZEB1), which triggers EMT through suppressing E-Cad expression (15). Therefore, GRHL2 is a determinant of epithelial phenotype through a transcriptional network of its target genes.
Our previous study has demonstrated that ectopic ΔNp63α expression in NHEKs yields the EMT phenotype in a TGF-β-dependent manner (4). In this report, we show that the induced EMT phenotype is specific to ectopic overexpression of ΔNp63α among the p63 isoforms. Alternatively, knockdown of all isoforms of p63 also led to the EMT phenotype through loss of GRHL2 and miR-200 family genes. Last, we found that GRHL2 directly binds to the p63 promoter and that p63 and GRHL2 regulate their mutual expression. This reciprocal feedback loop between p63 and GRHL2 may be critical in determining epithelial plasticity.
Experimental Procedures
Cells and Cell Culture
Primary cultured single-cell suspensions of NHEKs were isolated from discarded foreskin epidermal tissues and grown in EpiLife medium supplemented with growth factors (Invitrogen), as described elsewhere (16). Normal human oral keratinocytes (NHOKs) isolated from separated keratinizing oral epithelial tissues were prepared similar to NHEKs. NHEKs were serially subcultured and passed at 70% confluence until they reached replicative senescence. Replication kinetics were recorded as described previously (16). Normal human oral fibroblasts (NHOFs) were obtained from discarded gingival connective tissue and cultured in DMEM (Invitrogen) supplemented with 10% FBS (Invitrogen). Bone marrow mesenchymal stromal cells and dental pulp stem cells were cultured in α-minimum essential medium (Invitrogen) supplemented with 10% FBS (Invitrogen) and 5 mg/ml gentamicin sulfate (Gemini Bio-Products, West Sacramento, CA). The SCC4 and SCC9 cancer cell lines were cultured in DMEM/F12 (Invitrogen) and supplemented with 10% FBS and 0.4 pg/ml hydrocortisone, as described elsewhere (17). Immortalized keratinocyte cell line HaCaT cells were grown in EpiLife medium supplemented with growth factors (Invitrogen). Cells were maintained at 37 °C at 5% CO2 in a humidified chamber.
Retroviral Vector Construction and Transduction of Cells
We constructed retroviral vectors expressing human wild-type ΔNp63α, ΔNp63β, and ΔNp63γ. Full-length cDNAs of ΔNp63 were cloned from the cDNA library obtained from primary human keratinocytes by PCR amplification. ΔNp63α, ΔNp63β, and ΔNp63γ cDNAs were subcloned into the pLXSN retroviral expression vector (Clontech, Mountainview, CA) at XhoI/BamHI restriction sites, and isoforms were confirmed by sequencing. Retroviral construction and infection were performed as described elsewhere (18), and the infected cells were selected with 200 μg/ml G418 (Sigma). G418-resistant cells were continually maintained by serial subculture as outlined above.
Transwell Migration Assay
Transwell chambers with polycarbonate membranes were used to measure cell migration (Corning Inc., Corning, NY), according to methods described previously (19). NHEK/LXSN, NHEK/ΔNp63α, NHEK/ΔNp63β, and NHEK/ΔNp63γ cells were cultured in EpiLife (Invitrogen) and seeded in the upper chamber of the transwell. Cells were allowed to migrate for 24 h, and the transwell was washed with 1× PBS and fixed in 10% formalin for 10 min. Cells were then stained with 1% crystal violet dissolved in 10% formalin for a duration of 1 h. Non-migratory cells were removed from the chamber, and the transwells were photographed.
Immunofluorescence Staining
Cells were fixed for 15 min in 3.7% formaldehyde prior to permeabilization with 0.25% Triton X-100 for 10 min. Cells were then blocked in 10% normal goat serum for 1 h. Antibodies against p63 (A4A), E-Cad, β-catenin, ZEB1 (Santa Cruz Biotechnology, Santa Cruz, CA), GRHL2 (Abnova, Taipei City, Taiwan), and anti-FN (Sigma) were used as primary antibodies, and Alexa Fluor 488 IgG (Invitrogen) was used as a secondary antibody. Cells were counterstained with DAPI, and an Olympus BH2-RFCA fluorescence microscope was used to obtain images.
siRNA Transient Transfection
NHEKs were transfected with nonspecific, scrambled siRNA (Si-SCR) or siRNA targeting human p63 (Si-p63) at a concentration of 10 nm using Lipofectamine reagents (Invitrogen) according to the instructions of the manufacturer. NHEKs were transfected with Si-p63 once every 3 days over the course of 9 days. NHEK growth medium was replaced with mesenchymal growth medium (α-minimum essential medium/10% FBS and 5 mg/ml gentamicin sulfate) upon morphological transition to the mesenchymal phenotype.
Western Blotting
Whole cell extracts were isolated from the cultured cells, fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and transferred to Immobilon membrane (Millipore, Billerica, MA). Antibodies against p63 (A4A), p63 (H129), E-Cad, K14, ZEB1, N-Cad, GAPDH (Santa Cruz Biotechnology), GRHL2 (Abnova), FN, and Snail (Sigma) were used. The chemiluminescence signal was detected using HyGLO chemiluminescent HRP antibody detection reagent (Denville Scientific, South Plainfield, NJ).
Real-time qRT-PCR
Total RNA was extracted from cultured cells using the RNeasy Plus mini kit (Qiagen, Valencia, CA). Reverse transcription was performed with 5 μg of RNA by using the method described elsewhere (20). qRT-PCR was performed for the relative mRNA expression of TAp63, ΔNp63, and GRHL2; relative microRNA expression of miR-200a, miR-200b, miR-200c, miR-141, and miR-429; and enrichment of GRHL2, trimethylated histone 3 at lysine 4, and trimethylated histone 3 at lysine 27 in triplicates for each sample with LC480 SYBR Green I Master Mix (Roche) using LightCycler 480 (Roche). The second derivative quantitation cycle (Cq) value was calculated after running a total of 50 cycles to compare the relative change in mRNA and microRNA expression. The primer sequences and the PCR conditions are available upon request.
F-Actin Staining and Stress Fiber Formation
Cells were fixed for 10 min in 3.7% formaldehyde prior to permeabilization with 0.1% Triton X-100 for 5 min. Cells were stained with Alexa Fluor 594 Phalloidin (Invitrogen) for 20 min at room temperature. Cells were counterstained with DAPI, and an Olympus BH2-RFCA fluorescence microscope was used to obtain images.
Lentiviral Vector Construction and Knockdown of Endogenous GRHL2
The lentiviral vector expressing shRNA against GRHL2 (Sh-GRHL2) and the control vector expressing enhanced GFP were constructed as described previously (21). Endogenous GRHL2 was knocked down with Sh-GRHL2. All lentiviral vectors used allow the identification of infected cells by a GFP signal under an epifluorescence microscope.
Dual-Luciferase Reporter Assay
The promoter regions of GRHL2 and p63 were cloned into the pGL3B-Luc reporter plasmid (Promega, Madison, WI) expressing firefly luciferase. The promoter-luciferase constructs were transfected into SCC4 or SCC9 using Lipofectin reagent (Invitrogen) along with pRL-SV40 containing Renilla luciferase cDNA under the control of the SV40 enhancer/promoter. 48 h post-transfection, cells were collected, and the lysates were prepared using the Dual-Luciferase reporter assay system (Promega). Firefly and Renilla luciferase activities were measured using a luminometer (Turner Designs, Sunnyvale, CA). Renilla luciferase activity was used to control for the varied transfected efficiency. Gene promoter activity, reflected by firefly luciferase activity, was determined as the mean of at least triplicates per experiment.
ChIP and ChIP-qPCR Assay
A ChIP assay was performed as described previously (12). Nuclear proteins were cross-linked to DNA by adding 10% formaldehyde for 10 min, and 0.15 m glycine was used to stop cross-linking. The cells were then collected in ice-cold PBS supplemented with a protease inhibitor mixture and lysed in buffer (1% SDS, 10 mm EDTA, protease inhibitors, and 50 mm Tris-HCl (pH 8.1)). Genomic DNA was sonicated to produce DNA fragments 300–1000 bp in length. Cellular lysates were diluted 1:10 in ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mm EDTA, 16.7 mm NaCl, protease inhibitors, and 16.7 mm Tris-HCl (pH 8.1)). Chromatin solutions were incubated with 5 μl of p63 or GRHL2 primary antibody (or 5 μl of mouse IgG) overnight at 4 °C with rotation. Immunocomplexes were precipitated with 30 μl of a protein G-agarose slurry (Upstate Biotechnology, Lake Placid, NY) and eluted in 500 μl of buffer (1% SDS and 100 mm NaHCO3). Precipitated DNA was recovered by phenol extraction and used for PCR amplification to check the enrichment of p63, GRHL2, trimethylated histone 3 at lysine 4, or trimethylated histone 3 at lysine 27 in the gene promoter regions. qPCR was performed with the purified DNA using LightCycler 480 (Roche). Samples pulled down with IgG were included as the negative control. The qPCR readout was normalized relative to the amount of amplification from the input. The promoter regions analyzed in this study for p63 or GRHL2 enrichment were near transcription start sites (available upon request) that contribute to core promoter activity, and our previous data have shown that GRHL2 binds to these regions to regulate gene expression (12).
Results
Overexpression of ΔNp63α in NHEKs Yields the EMT Morphology and Phenotype
Exogenous δNp63 isoforms were transduced in NHEK using retroviral vectors. Primary NHEK were infected with LXSN-δNp63α, LXSN-δNp63β, LXSN-δNp63γ, or LXSN control vector. Among the transduced cells, cells expressing the ΔNp63α and ΔNp63β isoforms showed morphological changes indicative of EMT, e.g. an elongated, flattened, and spindle-like morphology (Fig. 1A, left panel). However, we observed that, in NHEKs expressing the ΔNp63β isoforms, few mesenchymal-like cells were formed, and they were not able to be maintained in culture. A transwell migration assay also showed enhanced cellular motility, a hallmark of EMT (22) in NHEK/ΔNp63α cells, whereas NHEK/ΔNp63β, NHEK/ΔNp63γ, and NHEK/LXSN cells showed limited transwell migration (Fig. 1A, right panel).
FIGURE 1.

Overexpression of ΔNp63α in NHEK yields the EMT phenotype. A, NHEKs were stably transduced using retroviruses containing an empty vector (LXSN) or the ΔNp63 isoforms α, β, or γ. Morphological changes were observed in cells after stable transduction of ΔNp63 isoforms by phase contrast. The change in cell motility was assessed by transwell migration assay. B, NHEK stably transduced using retroviruses containing an empty vector (LXSN) or the ΔNp63 isoforms α, β, or γ were serially subcultured and assessed for change in protein expression of the ΔNp63α, ΔNp63β, and ΔNp63γ isoforms, GRHL2, E-Cad, and FN at two different PD levels (PD24 and PD28) by Western blotting. The asterisk indicates a high molecular weight band of p63. C, NHEK stably transduced using retroviruses containing an empty vector (LXSN) or ΔNp63α isoforms at PD22, PD24, and PD28 were assessed for change in protein expression of ΔNp63α (using the ΔNp63α-specific antibody H129), GRHL2, K14, E-Cad, and FN. NHOFs were used as a negative control. GAPDH was using as a loading control. The asterisk indicates a high molecular weight band of p63.
To further determine the molecular phenotype of EMT in NHEKs transduced with various ΔNp63 isoforms, we performed Western blotting for the markers of the epithelial or mesenchyme state. Cells transduced with ΔNp63α, ΔNp63β, and ΔNp63γ were harvested at two different population doubling (PD) levels at PD24 and PD28. The EMT phenotype occurred between these two PD levels (Fig. 1C). We used both a pan-p63 antibody (p63 A4A) and a ΔNp63α-specific antibody (p63 H129) to assess protein expression of the ΔNp63 isoforms. We observed specific bands corresponding to the α, β, and γ isoforms of ΔNp63 that were distinctly visible at 60, 50, and 40 kDa, respectively (Fig. 1, B and C). We also noted that the high molecular weight p63 band denoted by the asterisk disappeared with occurrence of EMT only in the culture infected with LXSN-ΔNp63α at PD28, concomitant with the appearance of the EMT phenotype. We also found that only prolonged subculture of NHEK/ΔNp63α led to reduced protein expression of the epithelial markers E-Cad and K14 and increased expression of the mesenchymal marker FN (Fig. 1, B and C). Prolonged subculture of NHEK/ΔNp63α also led to down-regulation of GRHL2, a transcriptional regulator and EMT inhibitor that regulates epithelial proliferation and differentiation (12). Loss of E-Cad and enhanced FN expression in NHEK/ΔNp63α cells were also confirmed in situ (Fig. 2). In this particular culture, both cell types exhibiting the mesenchymal or epithelial phenotype were visible. Cells with the mesenchymal morphology also demonstrated an enhanced FN and reduced E-Cad signal. Collectively, these data indicate that ectopic expression of ΔNp63α, but not ΔNp63β and ΔNp63γ, in NHEKs triggers the EMT phenotype.
FIGURE 2.

Overexpression of ΔNp63α in NHEKs results in an EMT molecular profile. Changes in the expression of E-Cad and FN were assessed in NHEKs transduced with ΔNp63α, ΔNp63β, or ΔNp63γ by means of fluorescence microscopy. The arrow indicates NHEK/ΔNp63α cells exhibiting the EMT phenotype in both differential interference contrast (DIC) and fluorescence microscopy images.
Knockdown of all p63 Isoforms in NHEKs by Transient Transfection of siRNA Results in the EMT Phenotype
Because EMT occurrence by ΔNp63α overexpression was accompanied by loss of the major p63 band (Fig. 1, B and C, asterisk), we asked whether the loss of all p63 isoforms would lead to EMT. NHEKs were transiently transfected with Si-SCR or Si-p63. To assess phenotypic and morphological changes upon p63 knockdown, we examined the changes in protein expression of p63, E-Cad, and FN 3, 6, and 10 days post-transfection with Si-SCR or Si-p63. 10 days post-transfection, NHEK/Si-p63 cells exhibited a complete loss of p63 and E-Cad protein expression, whereas FN expression was notably enhanced (Fig. 3A). These cells also exhibited a mesenchymal cell morphology after 17 days compared with NHEK/Si-SCR cells (Fig. 3B).
FIGURE 3.

Knockdown of p63 in NHEK by transient transfection with Si-p63 results in the EMT phenotype. A, Western blotting was performed using whole cell lysates of NHEKs transiently transfected using Si-SCR and NHEKs transiently transfected using Si-p63 to confirm loss of p63 and E-Cad protein expression and enhanced FN expression (arrow) consistent with EMT. NHOFs were used as a positive control for the mesenchymal phenotype. GAPDH was used as a loading control. B, morphological changes were observed in Si-SCR- and Si-p63-transfected NHEKs to determine the onset of the EMT phenotype. C, relative mRNA expression of TAp63, ΔNp63, and GRHL2 was assessed by qRT-PCR in NHOFs and NHEKs transiently transfected using Si-SCR or Si-p63. D, stress fiber formation in NHEK/Si-SCR and NHEK/Si-p63 cells was determined by F-actin staining using Alexa Fluor 594 phalloidin. E, changes in p63, GRHL2, ZEB1, FN, E-Cad, and β-catenin expression were determined in NHEK/Si-SCR and NHEK/Si-p63 cells by immunofluorescent staining.
We confirmed successful knockdown of p63 by qRT-PCR (Fig. 3C). In the control cells (Si-SCR), the levels of ΔNp63 and GRHL2 were substantially higher than those in NHOFs. Upon Si-p63 transfection, p63 was almost completely knocked down to a level similar to those of NHOFs. The TAp63 expression level was negligible in all tested samples. Concomitantly, we found that the GRHL2 level was also reduced drastically by p63 knockdown, indicating a link between p63 and GRHL2 in the epithelial phenotype.
We also determined mesenchymal cell surface markers (e.g. CD44, CD73, CD90, and CD105) in NHEK/Si-p63 cells with the EMT phenotype. For comparison, we included bone marrow mesenchymal stromal cells, dental pulp stem cells, and parental NHEKs. NHEK/Si-p63 cells with EMT the phenotype showed elevated CD44, CD90, and CD105 expression compared with the parental NHEKs, whereas the level of CD73 was reduced, consistent with other MSC cell types (Fig. 4), further supporting EMT in cells after loss of p63 expression.
FIGURE 4.

Modulation of p63 results in the acquisition of a stem-like molecular profile in NHEK. Expression of stem cell-associated cell surface markers CD44, CD73, CD90, and CD105 in parental NHEKs and NHEK/Si-p63 cells were evaluated by flow cytometry. Bone marrow mesenchymal stromal cells (BM-MSCs) and dental pulp stem cells (DPSCs) were used as positive controls.
EMT is a process by which epithelial cells lose cell-to-cell contact adhesion and associated protein expression and exhibit a mesenchymal phenotype, including stress fiber formation and migration (22). As additional evidence of EMT, NHEK/Si-p63 cells demonstrated stress fiber formation with organized F-actin (Fig. 3D). We also performed fluorescence microscopy to determine changes in EMT marker expression upon p63 knockdown in NHEKs. NHEK/Si-p63 cells showed loss of p63 and GRHL2 expression, reduced expression of E-Cad and β-catenin, and enhanced expression of ZEB1 and FN (Fig. 3E). We used NHEKs treated for 10 days with 5 ng/ml TGF-β as a positive control for EMT, which, similarly, showed reduced GRHL2, p63, K14, and β-catenin expression and enhanced FN expression by immunofluorescence staining (Fig. 5). Collectively, these data indicate that knockdown of all p63 isoforms in NHEKs led to EMT.
FIGURE 5.

TGF-β induces the EMT phenotype in NHEKs. NHEKs were cultured in 5 ng/ml TGF-β for 10 days, and the expression of GRHL2, p63, FN, K14, and β-catenin was assessed by immunofluorescent staining. Cells were counterstained using DAPI.
Knockdown of p63 in NHEKs Results in Loss of GRHL2 and miR-200 Family Gene Expression
In the next experiments, we investigated the mechanism underlying p63-associated EMT. In both NHEK/ΔNp63α and NHEK/Si-p63 cells, EMT occurred with enhanced expression of ZEB1 and N-Cad, which are linked with mesenchymal and stem cell characteristics (23) (Fig. 6A). We also found that both NHEK/ΔNp63α and NHEK/Si-p63 cells exhibiting the EMT phenotype lost GRHL2 and E-Cad expression. Because GRHL2 is a direct upstream regulator of E-Cad (24), our data suggest that p63 modulation could lead to EMT through loss of GRHL2.
FIGURE 6.

Modulation of p63 results in the loss of GRHL2 and miR-200 family expression. A, NHEKs ectopically expressing ΔNp63α and transiently transfected with p63 siRNA were harvested before and after the onset of EMT. Non-transduced/transfected NHEKs were used as an epithelial cell control, and NHOFs were used as a mesenchyme control. Whole cell lysates were collected, and Western blot analysis was performed for p63, ZEB1, GRHL2, E-Cad, and N-Cad. GAPDH was used as a loading control. B, the change in expression of miR-200a, miR-200b, miR-200c, miR-141, and miR-429 was assessed by qRT-PCR using QuantiMir cDNA. C, a ChIP assay and ChIP-qPCR were performed to examine binding of p63 to the miR-200a, miR-200b, miR-200c, miR-141, and miR-429 promoter regions in NHOFs, parental NHEKs, and NHEK/ΔNp63α and NHEK/Si-p63 cells showing the EMT phenotype.
Recent studies have shown that GRHL2 suppresses EMT by up-regulating miR-200 family genes (25), which then down-regulate ZEB1 (26). In this study, acquisition of the EMT phenotype in NHEK/ΔNp63α and NHEK/Si-p63 cells resulted in strong down-regulation of miR-200a, miR-200b, miR-200c, miR-141, and miR-429 microRNA expression (Fig. 6B). To test whether p63 directly binds to and regulates the expression of miR-200 family genes, we performed a ChIP assay. NHEK/ΔNp63α and NHEK/Si-p63 cells with the EMT phenotype showed a loss of p63 enrichment on the promoter regions of the miR-200a, miR-200b, miR-200c, and miR-429 loci compared with those of parental NHEKs (Fig. 6C). No change in p63 enrichment on miR-141 was observed between NHOFs, parental NHEKs, and NHEK/ΔNp63α and NHEK/Si-p63 cells. These data suggest that p63 may regulate the epithelial phenotype in keratinocytes by direct regulation of GRHL2 and miR-200 family genes, although miR-141 may be bound and regulated by other factors downstream of p63.
Modulation of GRHL2 Alters p63 Expression and EMT Induction
In another experiment, we induced EMT in SCC4 and HaCaT epithelial cell lines by exposure to TGF-β. In a time-dependent manner after TGF-β treatment, protein expression of p63 and GRHL2 was reduced, and the expression of FN, N-Cad, ZEB-1, and Snail was enhanced in SCC4 compared with the untreated cells (Fig. 7A). Similarly, HaCaT cells showed reduced expression of GRHL2, p63, and E-Cad and enhanced expression of FN and ZEB-1 upon TGF-β treatment in a time-dependent manner (Fig. 7B). These data further confirm the regulatory relationship between p63 and GRHL2 in EMT.
FIGURE 7.

Modulation of GRHL2 alters p63 expression and the EMT phenotype in human keratinocytes and epithelial cancer cell lines. A, SCC4 cells were treated with 10 ng/ml TGF-β for 0, 3, 5, and 10 days and harvested. Whole cell lysates were collected, and Western blotting was performed for p63, GRHL2, FN, N-Cad, ZEB1, and Snail. GAPDH was used as a loading control. B, HaCaT cells were treated with 10 ng/ml TGF-β for 0, 1, 3, 7, and 10 days and harvested. Whole cell lysates were collected, and Western blotting was performed for GRHL2, p63, FN, ZEB-1, and E-Cad. GAPDH was used as a loading control. C, GRHL2 was overexpressed in NHOFs, NHOKs, and SCC9 cells and knocked down in SCC4 cells using shRNA. Whole cell lysates were collected, and Western blotting was performed for GRHL2, p63, and E-Cad. GAPDH was used as a loading control. D, the change in p63, GRHL2, and β-catenin expression was determined in SCC9 cells overexpressing GRHL2 by immunofluorescence staining. E, the change in mRNA expression of p63 was assessed by qRT-PCR in SCC9 cells overexpressing GRHL2 and SCC4 cells after GRHL2 knockdown. F, GRHL2 and p63 were knocked down independently in SCC4 cells, and GRHL2 was overexpressed in SCC9 cells, and a Dual-Luciferase reporter assay was performed to assess promoter activity.
Because GRHL2 is a transcription regulator for diverse gene targets, including those in the epidermal differentiation complex (12), we tested whether p63 itself would be under the regulation of GRHL2. GRHL2 was ectopically expressed in NHOFs and NHOKs by using retroviral vectors. Overexpression of GRHL2 resulted in a weak but notable up-regulation of p63 and E-Cad in NHOFs and very strong up-regulation in NHOKs (Fig. 7C). Also, GRHL2 overexpression in SCC9 cells, which lack endogenous GRHL2 gene expression, led to a visible p63 band and enhanced mRNA expression. Knockdown of GRHL2 in SCC4 cells, which express high endogenous GRHL2, almost completely abolished p63 expression (Fig. 7, C and E). Furthermore, in situ staining for GRHL2, p63, and β-catenin revealed protein expression only in SCC9 cells transduced with GRHL2, and not in control cells (Fig. 7D). Notably, there was heterogeneity in the level of p63 staining in SCC9 cells expressing exogenous GRHL2, presumably because of the varied level of GRHL2 expression in cells.
Because our prior data indicated that p63 was necessary for GRHL2 expression (Fig. 3C), the above data indicate a reciprocity in the regulation between p63 and GRHL2 in determining the epithelial phenotype. We explored this possibility by performing a Dual-Luciferase reporter assay. Knockdown of GRHL2 in SCC4 cells resulted in diminished p63 promoter activity, whereas GRHL2 overexpression in SCC9 cells resulted in enhanced p63 promoter activity (Fig. 7F). Interestingly, knockdown of p63 in SCC4 cells also led to a notable reduction of GRHL2 promoter activity. Therefore, GRHL2 and p63 mutually regulate their promoter activity through a reciprocal feedback loop.
To examine whether GRHL2 binds directly to the promoter region of p63, a ChIP assay was performed with SCC4 and NHEK cells. In both cell types, GRHL2 pulldown coimmunoprecipitated the p63 promoter fragment near the transcription start site. Also, GRHL2 enrichment on the p63 promoter was altered according to the level of GRHL2 protein, i.e. GRHL2 knockdown in SCC4 cells or overexpression in NHEKs (Fig. 8, A and B). As a positive control, we included GRHL2 binding to the promoter region of the hTERT gene, which is a known transcriptional target of GRHL2 (21). ChIP revealed a concordant GRHL2 binding signal to the hTERT promoter according to the GRHL2 protein level (Fig. 8C). ChIP-qPCR quantitatively showed the altered GRHL2 binding to the p63 promoter in concordance with the GRHL2 protein levels in SCC4 cells and NHEKs (Fig. 8, D and E). Furthermore, ChIP-qPCR analysis revealed reduced enrichment of trimethylated histone 3 at lysine 4, indicative of active chromatin on the p63 promoter, when GRHL2 was knocked down in SCC4 cells (Fig. 8F), in keeping with the loss of p63 expression in SCC4 cells with GRHL2 silencing. On the contrary, the repressive mark (trimethylated histone 3 at lysine 27) was slightly elevated with GRHL2 knockdown. Because p63 modulation showed regulatory effects on GRHL2 expression, these data indicate a reciprocal regulation between GRHL2 and p63 to establish the epithelial phenotype.
FIGURE 8.

GRHL2 directly binds to and activates the p63 promoter in human keratinocytes and epithelial cancer cell lines. A and B, a ChIP assay was performed to examine binding of GRHL2 to the p63 promoter in SCC4 cells infected with Sh-GRHL2 (A) and in NHEK overexpressing GRHL2 (B). C, a ChIP assay was performed to examine binding of GRHL2 to the hTERT promoter in NHEKs with or without GRHL2 overexpression. A pulldown assay using α-IgG was used as a negative control. D and E, ChIP-qPCR was performed to quantify the enrichment of GRHL2 on the p63 promoter in SCC4 cells with GRHL2 knockdown (D) and in NHEK overexpressing GRHL2 (E). F and G, enrichment of trimethylated histone 3 at lysine 4 (F) and trimethylated histone 3 at lysine 27 (G) on the p63 promoter in SCC4 cells with GRHL2 knockdown was also assessed by ChIP-qPCR.
Discussion
Our laboratory has shown previously that overexpression of the ΔNp63α isoform conferred the EMT phenotype in NHEKs in a TGF-β-dependent manner (4). This study also shows that ΔNp63β and ΔNp63γ do not fully transform NHEKs to the mesenchymal state, although there was morphological transition by ΔNp63β expression in NHEKs. One of the speculations from our prior study was that the ΔNp63α isoforms abolished the expression of the high molecular weight (∼75 kDa, Fig. 1, B and C, asterisk) p63 protein with unknown identity, which invariably occurred during EMT (4). As an alternative approach, in this study we knocked down all p63 isoforms using pan-p63 siRNA. This transient knockdown of p63 isoforms led to the EMT phenotype 10 days post-transfection. Acquisition of the EMT phenotype in NHEK/ΔNp63α and NHEK/Si-p63 cells was assessed by altered morphology and expression of the EMT markers, e.g. loss of E-Cad, K14, and β-catenin expression and enhanced FN and ZEB1 expression. Although NHEK/ΔNp63α cells required serial subculture and several passages to induce molecular and phenotypic EMT-associated changes, NHEK/Si-p63 cells readily acquired these altered phenotype within 10 days after gene knockdown (Figs. 1C and 3A).
In our previous paper, we showed that NHEK/ΔNp63α cells with the EMT phenotype acquired stemness characteristics and expressed high levels of reprogramming factors, e.g. Lin28 and Nanog (4). Likewise, NHEK/Si-p63 cells showed stem characteristics, e.g. the ability to transdifferentiate into osteogenic and adipocytic lineages (data not shown). We also found that NHEK/Si-p63 cells expressed the cell surface markers CD44, CD73, CD90, and CD105, which are characteristically expressed in the MSC population (Fig. 4) (27, 28). Therefore, our studies demonstrate that EMT in NHEKs is associated with the acquisition of stem characteristics, whether it is triggered by overexpression of ΔNp63α or knockdown of all p63 isoforms. We coined the term “induced mesenchymal stem cells” to describe epithelial cells that have acquired multipotency through EMT to distinguish them from endogenous MSCs or induced pluripotent stem cells, which acquire embryonic stem cell characteristics after stable transduction of the reprogramming factors (29). These induced mesenchymal stem cells may be derived from cultured primary epithelial cells from skin via transient transfection of p63 siRNA and may have tissue engineering potential for cases requiring an autologous cell transplantation approach.
This study reveals GRHL2 as the regulatory partner of p63, which is critical for epithelial cell identity and embryonic morphogenesis (30, 31). GRHL2 is a transcription factor responsible for regulating epithelial proliferation (12), is known to play an important role in inducing many epithelial cancers, and has been found to regulate EMT (14, 21). This study shows that EMT in NHEK/ΔNp63α and NHEK/Si-p63 cells accompanied down-regulation of GRHL2 and E-Cad and up-regulation of ZEB1 and N-Cad expression (Fig. 6A). GRHL2 has also been shown to suppress EMT in breast cancer cells by suppressing ZEB1 signaling, and a reciprocal feedback loop has been found between GRHL2 and ZEB1 in regulating EMT (25, 32). Furthermore, GRHL2 has been found to inhibit ZEB1 expression and EMT by activating miR-200b and miR-200c (32). We found that NHEK/ΔNp63α and NHEK/Si-p63 resulted in down-regulation of miR-200 family gene expression, including miR-200a, miR-200b, miR-200c, miR-141, and miR-429 (Fig. 6B) and that p63 binds directly to the promoter of miR-200 family genes (Fig. 6C). It is important to note that p63 did not bind to the promoter of miR-141 and that miR-141 may be bound and regulated by a downstream target of p63. Therefore, p63 modulation may alter miR-200 family gene activation and induce EMT by facilitating up-regulation of ZEB1 expression. These findings indicate a novel role for p63 in the GRHL2-mediated regulation of EMT. In addition, GRHL2 was found to be required and necessary for the expression of p63. In NHOFs, NHOKs and SCC9 cells, GRHL2 overexpression led to a notable increase in the p63 expression level. Also, GRHL2 knockdown in SCC4 cells abolished the expression of p63, suggesting p63 regulation by GRHL2. This was confirmed by ChIP analyses showing direct GRHL2 binding on the p63 gene promoter and concordant alteration of the histone mark at the gene promoter. We also demonstrated, through a Dual-Luciferase reporter assay, that GRHL2 is required for p63 promoter activity and vice versa. Therefore, we propose a model in which p63 and GRHL2 regulate the expression of each other through direct promoter binding, and this reciprocal regulatory loop between GRHL2 and p63 is critical for maintaining the epithelial phenotype through transcriptional regulation of their target genes (Fig. 9). This model may provide a framework to study the molecular mechanism that regulates epithelial plasticity, a key player in physiologic and disease processes, including cancer metastasis, epithelial wound healing, and tissue damage response to environmental insults.
FIGURE 9.
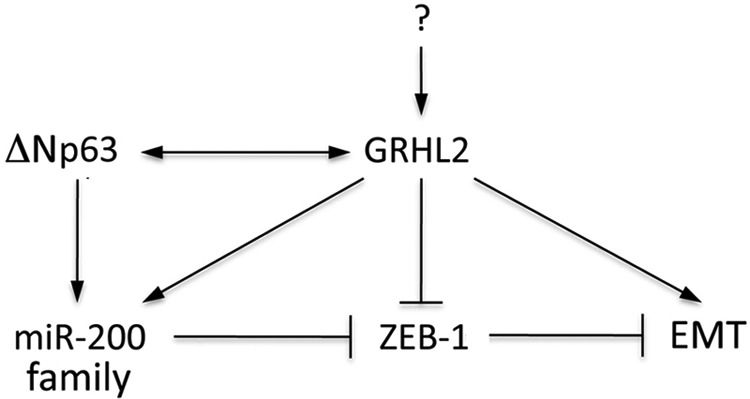
The role of p63 and GRHL2 in epithelial plasticity. Current data establish a reciprocal regulation between ΔNp63 and GRHL2 through direct promoter binding. Both ΔNp63 and GRHL2 are required for the expression of miR-200 family genes, which then suppress EMT through negative regulation of ZEB1. In this model, GRHL2 appears to be intricately involved with many different factors that determine the epithelial phenotype.
Author Contributions
S. M., W. C., and M. K. K. planned the study. R. H. K., K. S., N. H. P., and M. K. K. participated in the design of the experiments. S. M. wrote the paper. S. M. and W. C. performed and analyzed the experiments shown in Figs. 1, 3, 5, 6, and 8. J. E. O. and Z. X. L. performed and analyzed the experiments in Figs. 2 and 4. S. M., W. C., K. L. K., and J. K. Y. performed and analyzed the experiments shown in Fig. 7. All authors analyzed the results, revised the manuscript, and approved the final version of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R01DE18295, K02DE18959, and R56DE024593. This work was also supported by the Jack Weichman Endowed Fund (to M. K. K.). The authors declare no conflict of interest with the content of this article.
- EMT
- epithelial-mesenchymal transition
- HEK
- normal human epithelial keratinocyte
- E-Cad
- E-Cadherin
- FN
- fibronectin
- NHOK
- normal human oral keratinocyte
- NHOF
- normal human oral fibroblast
- qRT-PCR
- quantitative RT-PCR
- PD
- population doubling.
REFERENCES
- 1. Deleted in proof.
- 2. Deleted in proof.
- 3. Deleted in proof.
- 4. Oh J. E., Kim R. H., Shin K. H., Park N. H., Kang M. K. (2011) DeltaNp63-alpha triggers epithelial-mesenchymal transition and confers stem cell properties in normal human keratinocytes. J. Biol. Chem. 286, 38757–38767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pellegrini G., Dellambra E., Golisano O., Martinelli E., Fantozzi I., Bondanza S., Ponzin D., McKeon F., De Luca M. (2001) p63 identifies keratinocyte stem cells. Proc. Natl. Acad. Sci. 98, 3156–3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ghioni P., Bolognese F., Duijf P. H., Van Bokhoven H., Mantovani R., Guerrini L. (2002) Complex transcriptional effects of p63 isoforms: identification of novel activation and repression domains. Mol. Cell. Biol. 22, 8659–8668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hay E. D., Zuk A. (1995) Transformations between epithelium and mesenchyme: normal, pathological, and experimentally induced. Am. J. Kidney Dis. 26, 678–690 [DOI] [PubMed] [Google Scholar]
- 8. Grille S. J., Bellacosa A., Upson J., Klein-Szanto A. J., van Roy F., Lee-Kwon W., Donowitz M., Tsichlis P. N., Larue L. (2003) The protein kinase Akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer Res. 63, 2172–2178 [PubMed] [Google Scholar]
- 9. Moreno-Bueno G., Peinado H., Molina P., Olmeda D., Cubillo E., Santos V., Palacios J., Portillo F., Cano A. (2009) The morphological and molecular features of the epithelial-to-mesenchymal transition. Nat. Protoc. 4, 1591–1613 [DOI] [PubMed] [Google Scholar]
- 10. Docherty N. G., O'Sullivan O. E., Healy D. A., Murphy M., O'neill A. J., Fitzpatrick J. M., Watson R. W. (2006) TGF-β1-induced EMT can occur independently of its proapoptotic effects and is aided by EGF receptor activation. Am. J. Physiol. Renal Physiol. 290, F1202–F1212 [DOI] [PubMed] [Google Scholar]
- 11. Thiery J. P. (2003) Epithelial-mesenchymal transitions in development and pathologies. Curr. Opin. Cell Biol. 15, 740–746 [DOI] [PubMed] [Google Scholar]
- 12. Chen W., Xiao Liu Z., Oh J. E., Shin K. H., Kim R. H., Jiang M., Park N. H., Kang M. K. (2012) Grainyhead-like 2 (GRHL2) inhibits keratinocyte differentiation through epigenetic mechanism. Cell Death Dis. 3, e450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen W., Dong Q., Shin K. H., Kim R. H., Oh J. E., Park N. H., Kang M. K. (2010) Grainyhead-like 2 enhances the human telomerase reverse transcriptase gene expression by inhibiting DNA methylation at the 5′-CpG island in normal human keratinocytes. J. Biol. Chem. 285, 40852–40863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xiang X., Deng Z., Zhuang X., Ju S., Mu J., Jiang H., Zhang L., Yan J., Miller D., Zhang H. G. (2012) Grhl2 determines the epithelial phenotype of breast cancers and promotes tumor progression. PLoS ONE 7, e50781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wellner U., Schubert J., Burk U. C., Schmalhofer O., Zhu F., Sonntag A., Waldvogel B., Vannier C., Darling D., zur Hausen A., Brunton V. G., Morton J., Sansom O., Schüler J., Stemmler M. P., Herzberger C., Hopt U., Keck T., Brabletz S., Brabletz T. (2009) The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 11, 1487–1495 [DOI] [PubMed] [Google Scholar]
- 16. Kang M. K., Bibb C., Baluda M. A., Rey O., Park N. H. (2000) In vitro replication and differentiation of normal human oral keratinocytes. Exp. Cell Res. 258, 288–297 [DOI] [PubMed] [Google Scholar]
- 17. Kang M. K., Guo W., Park N. H. (1998) Replicative senescence of normal human oral keratinocytes is associated with the loss of telomerase activity without shortening of telomeres. Cell Growth Differ. 9, 85–95 [PubMed] [Google Scholar]
- 18. Kang M. K., Park N. H. (2007) Extension of cell life span using exogenous telomerase. Methods Mol. Biol. 371, 151–165 [DOI] [PubMed] [Google Scholar]
- 19. Giannelli G., Falk-Marzillier J., Schiraldi O., Stetler-Stevenson W. G., Quaranta V. (1997) Induction of cell migration by Matrix Metalloprotease-2 cleavage of Laminin-5. Science 277, 225–228 [DOI] [PubMed] [Google Scholar]
- 20. Kang M. K., Kameta A., Shin K. H., Baluda M. A., Park N. H. (2004) Senescence occurs with hTERT repression and limited telomere shortening in human oral keratinocytes cultured with feeder cells. J. Cell. Physiol. 199, 364–370 [DOI] [PubMed] [Google Scholar]
- 21. Kang X., Chen W., Kim R. H., Kang M. K., Park N. H. (2009) Regulation of the hTERT promoter activity by MSH2, the hnRNPs K and D, and GRHL2 in human oral squamous cell carcinoma cells. Oncogene 28, 565–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu J., Lamouille S., Derynck R. (2009) TGF-β-induced epithelial to mesenchymal transition. Cell Res. 19, 156–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gandarillas A., Watt F. M. (1997) c-Myc promotes differentiation of human epidermal stem cells. Genes Dev. 11, 2869–2882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Varma S., Cao Y., Tagne J. B., Lakshminarayanan M., Li J., Friedman T. B., Morell R. J., Warburton D., Kotton D. N., Ramirez M. I. (2012) The transcription factors Grainyhead-like 2 and NK2-homeobox 1 form a regulatory loop that coordinates lung epithelial cell morphogenesis and differentiation. J. Biol. Chem. 287, 37282–37295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cieply B., Riley P., 4th, Pifer P. M., Widmeyer J., Addison J. B., Ivanov A. V., Denvir J., Frisch S. M. (2012) Suppression of the epithelial-mesenchymal transition by Grainyhead-like-2. Cancer Res. 72, 2440–2453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Korpal M., Lee E. S., Hu G., Kang Y. (2008) The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 283, 14910–14914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pittenger M. F., Mackay A. M., Beck S. C., Jaiswal R. K., Douglas R., Mosca J. D., Moorman M. A., Simonetti D. W., Craig S., Marshak D. R. (1999) Multilineage potential of adult human mesenchymal stem cells. Science 284, 143–147 [DOI] [PubMed] [Google Scholar]
- 28. Dominici M., Le Blanc K., Mueller I., Slaper-Cortenbach I., Marini F., Krause D., Deans R., Keating A., Prockop D. J., Horwitz E. (2006) Minimal criteria for defining multipotent mesenchymal stromal cells: the International Society for Cellular Therapy position statement. Cytotherapy 8, 315–317 [DOI] [PubMed] [Google Scholar]
- 29. Takahashi K., Yamanaka S. (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 [DOI] [PubMed] [Google Scholar]
- 30. Barbareschi M., Pecciarini L., Cangi M. G., Macrì E., Rizzo A., Viale G., Doglioni C. (2001) p63, a p53 homologue, is a selective nuclear marker of myoepithelial cells of the human breast. Am. J. Surg. Pathol. 25, 1054–1060 [DOI] [PubMed] [Google Scholar]
- 31. Laurikkala J., Mikkola M. L., James M., Tummers M., Mills A. A., Thesleff I. (2006) p63 regulates multiple signaling pathways required for ectodermal organogenesis and differentiation. Development 133, 1553–1563 [DOI] [PubMed] [Google Scholar]
- 32. Cieply B., Farris J., Denvir J., Ford H. L., Frisch S. M. (2013) Epithelial-mesenchymal transition and tumor suppression are controlled by a reciprocal feedback loop between ZEB1 and Grainyhead-like-2. Cancer Res. 73, 6299–6309 [DOI] [PMC free article] [PubMed] [Google Scholar]