

Abstract
Tuberculosis, caused by Mycobacterium tuberculosis, still remains a major global health problem. The main obstacle in eradicating this disease is the ability of this pathogen to remain dormant in macrophages, and then reactivate later under immuno-compromised conditions. The physiology of hypoxic nonreplicating M. tuberculosis is well-studied using many in vitro dormancy models. However, the physiological changes that take place during the shift from dormancy to aerobic growth (reactivation) have rarely been subjected to a detailed investigation. In this study, we developed an in vitro reactivation system by re-aerating the virulent laboratory strain of M. tuberculosis that was made dormant employing Wayne's dormancy model, and compared the proteome profiles of dormant and reactivated bacteria using label-free one-dimensional LC/MS/MS analysis. The proteome of dormant bacteria was analyzed at nonreplicating persistent stage 1 (NRP1) and stage 2 (NRP2), whereas that of reactivated bacteria was analyzed at 6 and 24 h post re-aeration. Proteome of normoxially grown bacteria served as the reference. In total, 1871 proteins comprising 47% of the M. tuberculosis proteome were identified, and many of them were observed to be expressed differentially or uniquely during dormancy and reactivation. The number of proteins detected at different stages of dormancy (764 at NRP1, 691 at NRP2) and reactivation (768 at R6 and 983 at R24) was very low compared with that of the control (1663). The number of unique proteins identified during normoxia, NRP1, NRP2, R6, and R24 were 597, 66, 56, 73, and 94, respectively. We analyzed various biological functions during these conditions. Fluctuation in the relative quantities of proteins involved in energy metabolism during dormancy and reactivation was the most significant observation we made in this study. Proteins that are up-regulated or uniquely expressed during reactivation from dormancy offer to be attractive targets for therapeutic intervention to prevent reactivation of latent tuberculosis.
Tuberculosis (TB)1 remains a major global health problem despite Bacillus Calmette–Guérin (BCG) vaccination and effective drug therapy for more than half a century. Worldwide 8.6 million individuals are infected with the etiologic agent Mycobacterium tuberculosis (MTB) (1). Among the infected individuals, only about 10% develop active TB at some point of their lifetime (2). Majority of MTB infections results in latent TB, where the bacteria remain in a dormant state in granulomas (3). Hypoxia in the fibrotic granulomatus lesions in the lung is one of the factors that triggers dormancy (4–6). Reactivation of dormant bacteria can occur under certain circumstances such as immuno-suppression, diabetes, obesity, and co-infection with human immunodeficiency virus (HIV) (2, 7). The physiology of hypoxic nonreplicating MTB has been studied extensively in vitro, by employing the Wayne's model of dormancy in which MTB is subjected to a self-generated oxygen-depletion in sealed glass tubes (8). Two nonreplicating stages are identified in this model—a microaerophilic stage termed nonreplicating persistence stage 1 (NRP1) that exists between the 8th and 14th days (192–336 h), and an anaerobic stage designated as nonreplicating persistence stage 2 (NRP2) from the 14th day onwards (9). Under these conditions MTB is found to undergo drastic changes in its energy and metabolic status (10, 11). In addition to Wayne's dormancy model, various in vitro hypoxic models are used to study dormancy in MTB (12–15). Environmental stresses such as nutrient deprivation, iron restriction, mild acidity, and reactive nitrogen and oxygen species also induce dormancy (7, 16). However, Wayne's dormancy model has proven to be a very effective and simple method to understand the molecular mechanisms in dormant bacteria, and to discover novel therapeutic agents (8). In addition, Wayne's model is proven to be clinically correlated to human anaerobic latent lesions containing dormant bacilli (17).
Changes in the physiology of MTB during its transition from log phase to dormancy, as well as from dormancy to reactivation, have been studied using genomic, transcriptomic, proteomic, and metabolomic approaches (18–21). Most of the proteomic studies to date have focused on the bacilli grown under normoxia (22, 23), or during transition from normal replicating stage to dormancy (24). Starck et al. used 2-D electrophoresis to compare the proteomes of MTB grown under aerated and anerobic conditions, and found 50 proteins differentially expressed under the latter (12). Wolfe et al. used a probe-based chemo-proteomic approach to selectively profile the ATP-binding proteome in normally growing and hypoxic MTB. They identified 122 ATP-binding proteins of which roughly 60% were reported to be essential for the in vitro survival (14). Extracellular proteins of nutrient-starved MTB were analyzed by Albrethsen et al. They identified 1176 proteins, of which 230 were up-regulated, and 208 were down-regulated (25).
Galagan et al. carried out proteome profiling of dormant and re-aerated MTB using a defined hypoxia model, and identified a total of approximately one thousand proteins (26). The process of reactivation of MTB from dormancy is a critical step in the development of active TB. For understanding the molecular mechanisms involved in the reactivation of MTB, it is important to identify the proteins specifically or differentially expressed during reactivation. In the present study, by re-aerating the medium after establishing the Wayne's dormancy model, we could successfully induce the bacilli to grow actively again. To identify the proteins, we employed a label-free, one-dimensional liquid chromatography coupled with tandem mass spectrometry (LC/MS/MS), to analyze the proteomes of normoxic, dormant, and reactivated MTB H37Rv, the virulent laboratory strain. Genome-based computational analyses were conducted and integrated into the proteomics data.
EXPERIMENTAL PROCEDURES
Culture and Growth Conditions
MTB virulent laboratory strain H37Rv was subcultured on Löwenstein-Jensen slants and incubated at 37 °C for 4–6 weeks. All steps involving handling of MTB were carried out in a biosafety level three (BSL3) facility. Broth cultures were prepared by inoculation of one loopful of bacterial colony into Dubos broth base (Difco, Franklin Lake, NJ) containing 5% (v/v) glycerol, supplemented with Dubos albumin (2%, v/v, Difco). Culture grown to A600 of 0.6 (∼108 bacteria per ml) in 250 ml conical flask on a shaker incubator at 150 rpm and 37 °C was used as the inoculum. To achieve dormancy, we used 16 × 24 mm flat-bottomed glass tubes with a width of 25.5 mm and a liquid holding capacity of 31.0 ml. Dubos Tween-albumin broth containing 2 × 106 bacteria per milliliter was dispensed (20.4 ml) into required number of tubes. Cultures were grown with limited internal agitation of 130 rpm using 8 mm Teflon-coated magnetic bars (Sigma-Aldrich, St. Louis, MO) on multipoint magnetic stirrers (Variomag Poly 15, Thermo Scientific, Walthan, MA). The cap (Pressure compensation set, Duran, Germany) of the glass tube was connected to a 0.2 micron filter using a silicon tubing (3 cm length and 1.6 mm internal diameter). The tubing was closed using a pinchcock clamp (Fig. 1A). The whole setup was placed inside a custom-made 37 °C incubator (Santhom Scientific, Bangalore, India) that could accommodate four multipoint magnetic stirrers simultaneously. The dormant bacteria were re-aerated by removing the pinchcock clamp from the silicone tubing (Fig. 1B). The agitation was increased to 200 rpm to facilitate aeration. The status of self-generated hypoxia and reactivation was monitored visually using methylene blue (1.5 μg/ml). This indicator imparts a greenish blue color to the culture in the presence of oxygen, and turns colorless when oxygen concentration in the medium becomes less than 1% (27). Growth was monitored every 24 h by measuring A600 in a colorimeter (Aimil Photochem, New Delhi, India) and the viability was assessed by plating 100 μl of suitably diluted bacterial culture on 7H10 agar (Difco) in triplicate and incubating at 37 °C for 6–8 weeks.
Fig. 1.
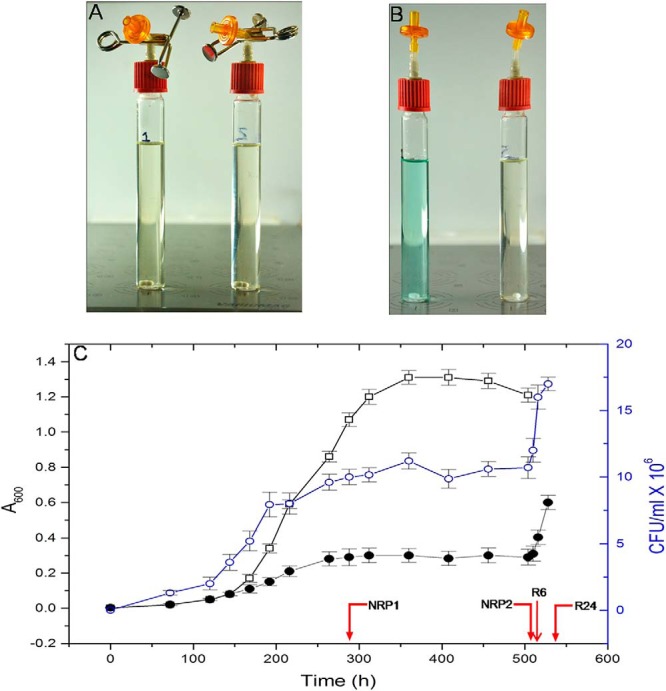
A, Experimental setup for dormancy shows the control tube to monitor the levels of oxygen, and a sample tube. The tube on the left contains dormant MTB and methylene blue indicator; the tube on the right contains only the bacteria. B, Upon aeration methylene blue regains its color. C, Measurement of growth of MTB H37Rv by optical density and CFU (values at each point of time represent an average value from four tubes) –○– CFU values of MTB subjected to dormancy and reactivation. –●– A600 values of MTB subjected to dormancy and reactivation. –□– A600 values of control MTB. Red arrows indicate sampling points.
Cell Lysis and Protein Extraction
Bacteria were harvested at the 288 and 504 h (12 and 21 days) to represent NRP1 and NRP2 stages of dormancy, respectively. Cells harvested at 6 and 24 h after introduction of air represented early and late stages of reactivation (R6 and R24), respectively. The cells from normoxia served as the control. We extracted proteins by employing a modified protocol used by Cho et al. (24). Briefly, 80 ml of MTB cultures were pelleted by centrifuging them at 2500 × g at 4 °C for 15 min, and the pellets were washed three times in 10 ml ice-cold phosphate buffered saline (PBS, pH 7.4) and were resuspended in PBS (1 ml PBS per gram of bacteria). Equal weight of bacteria from different stages were transferred to 2 ml screw-cap microcentrifuge tubes containing glass beads (0.5 mm), protease inhibitor (1 mm PMSF, Sigma-Aldrich) and incubated on ice for 5 mins. The tubes were then placed in a Mini Bead beater (BioSpec Products Inc., Bartlesville, OK) and subjected to three, one-minute pulses at 4200 rpm with 1 min interval. The suspensions were centrifuged for 10 min at 13,000 × g at 4 °C (Eppendorf, Hauppauge, NY) and the supernatants were recovered. Protein concentration was determined by bicinchoninic acid protein assay reagent (Thermo Scientific) (28).
Preparation of Protein for LC/MS/MS
Fifty micrograms of protein from each sample (normoxia, NRP1, NRP2, R6, and R24) was used for in-solution trypsin digestion. The disulfide bonds were reduced by treating the proteins with 10 mm dl-dithiothreitol (Sigma-Aldrich) in 50 mm ammonium bicarbonate (Sigma-Aldrich) buffer at 60 °C for 30 min. The proteins were subsequently alkylated with 200 mm iodoacetamide (Sigma-Aldrich) in the same buffer at 27 °C for 30 min in the dark. Proteins were then digested with trypsin (sequencing-grade modified trypsin, Sigma-Aldrich; 1:25 w/w) in 50 mm ammonium bicarbonate buffer by incubating overnight at 37 °C. Trypsin digestion was terminated by adding formic acid (Sigma-Aldrich; 1% v/v) to the reaction mixture. The digested peptide solutions were centrifuged at 14,000 rpm at 4 °C for 12 min, and the supernatants were stored at −20 °C until the LC/MS/MS analysis.
Liquid Chromatography
The peptide samples were analyzed by nano-LC/MSE (MS at elevated energy) using a nano ACQUITY UPLC® System (Waters, Hertfordshire, UK) coupled to a Quadrupole-Time of Flight (Q/TOF) mass spectrometer (SYNAPT-G2, Waters). Both the systems were operated and controlled by MassLynx4.1 SCN781 software (Waters). In the nano-LC, the peptides were separated by reverse phase column chromatography.
Briefly, 3 μl of each sample, equivalent to 3 μg of protein, was injected in “partial loop” mode and was loaded into the reverse phase column with 0.1% formic acid in water as mobile phase A, and 0.1% formic acid in acetonitrile as mobile phase B, using the binary solvent manager. The sample was then trapped in the trap column (Symmetry® 180 μm × 20 mm C18 5 μm, Waters) to remove any salt by employing a high flow rate (15 μl/minute) with 99.9% mobile phase A and 0.1% mobile phase B for 1 min. The peptide separation was performed on a 75 μm × 100 mm BEH C18 column (Waters), with particle size of 1.7 μm. A gradient elution with 1–40% mobile phase B, for 55.5 min at 300 nl/minute flow rate, was employed. After separation, the column was washed with 80% mobile phase B for 7.5 min and re-equilibrated with 1% mobile phase B for 20 min. The column temperature was maintained at 40 °C. Three biological replicates for each condition were performed and each sample was run in triplicates.
Mass Spectrometry
Peptides eluted from the nano-LC was subjected to mass spectrometric analysis on a SYNAPT® G2 High Definition MS™ System (Waters). The following parameters were used: nano-ESI capillary voltage, 3.3 KV; sample cone, 35 V; extraction cone, 4 V; transfer CE, 4 V; trap gas flow, (2 ml/minute); IMS gas (N2) flow, (90 ml/minute). To perform the mobility separation, the IMS T-Wave™ pulse height was set to 40 V during transmission and the IMS T-Wave™ velocity was set to 800 m/s. The traveling wave height was ramped over 100% of the IMS cycle between 8 and 20 V. The time of flight analyzer (TOF) was calibrated with a solution of 500 fmole/μl of human [Glu1]-Fibrinopeptide B (Sigma-Aldrich), and the lock mass acquisition was performed every 30 s by the same peptide delivered through the reference sprayer of the nano-LockSpray source at a flow rate of 500 nl/minute. This calibration set the analyzer to detect ions in the range of 50–2000 m/z.
The mass spectrometer was operated in the “resolution mode” with a resolving power of 18,000 FWHM, and the data acquisition was performed in “continuum” format. The data was acquired by rapidly alternating between two functions – Function-1 (low energy) and Function-2 (high energy). In Function-1, only low energy mass spectra (MS) were acquired and in Function-2, mass spectra at elevated collision energy (MSE) with ion mobility were acquired. In Function-1, collision energy was set to 4 V in the trap region and 2 V in the transfer region. In Function-2, collision energy was set to 4 V in the trap region and is ramped from 20 V to 45 V in the transfer region. Each spectrum was acquired for 0.9 s with an interscan delay of 0.024 s.
Data Analysis
The LC/MSE data was analyzed using ProteinLynx Global SERVER™ v2.5.3 (PLGS, Waters) for protein identification as well as for the relative protein quantification. Data processing included lock-mass correction post acquisition. Noise reduction thresholds for low energy scan ion, high-energy scan ion, and peptide intensity were fixed at 150, 50, and 500 counts, respectively. The database Mycobacterium tuberculosis H37Rv uid17053 NC_01 8143.faa (November 30/2013) downloaded from NCBI was used for database search. The parameters for protein identification were made in such a way that a peptide was required to have at least one fragment ion match, a protein was required to have at least three fragment ion matches, and at least two peptide matches for identification. Mass tolerance was set to 10 ppm for precursor ions and 20 ppm for fragment ions. Oxidation of methionine was selected as the variable modification and carbamidomethylation of cysteine was selected as the fixed modification. Trypsin was chosen as the enzyme with a specificity of one missed cleavage. The false positive rate (FPR) of the algorithm for identification was set to 4% with a randomized database, appended to the original one. Only those proteins with 50% or more probability to be present in the mixture and detected with a score above 20, as calculated by the software, were selected for proteomic analysis (29). Data sets were normalized using the “auto-normalization” function of PLGS, and label-free quantitative analysis was performed by comparing the normalized peak area/intensity of the peptides identified. Thus, parameters such as score, sequence coverage, and number of peptides were obtained for each protein. Furthermore, only those proteins with a fold change higher than 50% difference (ratio of either <0.50 or >1.5) were considered to be expressed at significantly altered levels.
Bioinformatic and Statistical Analyses
The proteins identified were subjected to Gene Ontology (GO) analysis using DAVID functional ontology analyzer (http://david.abcc.ncifcrf.gov/) (30, 31). Proteins identified in any one of the three technical replicates were included in the analysis. The NCBI IDs of the proteins were used for defining the list of total proteins detected in the dataset. GO terms for the identified proteins were extracted, and over-represented functional categories for differentially abundant proteins were determined. GO groups were selected based on the confidence score (p value < 0.05), and for overlapping GO groups, one representative category was selected within the dataset. For the construction of interaction network BioNet Builder plugin (http://apps.org/apps/bionetbuilder) in the Cytoscape 2.8.3 platform was used (32). The quantitative data from the 1-D LC/MS/MS analysis was incorporated into the cellular network thus created, and the differences in protein levels during dormancy and reactivation in comparison with the control, and with each other were estimated. The proteins were classified based on their function, physical interaction, or location.
The quantitative profiles of proteins that had consistent representation in all the three biological replicates were subjected to statistical comparison employing nonparametric Friedman test (GraphPad Prism V6).
Isolation of RNA, and Validation of the Proteomic Data by qPCR
RNA was isolated following the protocol described by Larsen (33). Fifteen ml of MTB culture (normoxia, NRP1, NRP2, R6, and R24) was centrifuged at 6000 × g for 10 min followed by lysis of cell pellet in ice-cold chloroform/methanol (3:1). The solution was vortexed and 5 ml of Trizol reagent (Sigma-Aldrich) was added, and was centrifuged at 2000 × g for 15 min. To the top layer, an equal volume of ice-cold isopropanol was added, kept at −20 °C overnight, and was centrifuged at 18,000 × g for 30 min. The pellet was resuspended in 70% ice-cold ethanol, and centrifuged at 18,000 × g for 20 min. The pellet thus obtained was dried to remove any trace amount of ethanol left, and was treated with DNase-1 (Sigma-Aldrich) and the RNA was quantified spectrophotometrically (Nanovue, GE Healthcare, Buckinghamshire, UK). Complementary DNA (cDNA) was prepared using Reverse Transcriptase Core kit (Eurogentec, Seraing, Belgium) according to the manufacturer's protocol. Quantitative real-time PCR was performed using gene-specific primers (Bio-Rad, Hercules, CA). The thermal cycling protocol was set as follows – an initial denaturation of 94 °C for 1 min; 35 cycles of denaturation at 94 °C for 30 s, primer annealing at 60 °C for 45 s, and extension at 72 °C for 40 s. Following amplification, a melt curve analysis was performed to confirm the specificity of the amplified product. Relative changes of gene expression were calculated using -ΔΔCt method, with sigA as the housekeeping gene.
RESULTS AND DISCUSSION
A major reason why TB is a global menace, despite effective drugs and BCG vaccine, is the ability of MTB to remain dormant in the human body for a long time, and to be reactivated when the host immune system becomes weak. Study of critical proteins expressed during reactivation may identify novel targets for therapeutic intervention at this stage of development of the disease. Therefore, in this study we aimed to identify differentially and uniquely expressed proteins during transition from dormancy to reactivation. Toward this, we adopted the Wayne's model to induce dormancy in MTB, and subsequently re-aerated the culture to reactivate the dormant bacilli.
Establishment of Dormancy and Reactivation
The self-generated hypoxia was monitored using methylene blue indicator (Figs. 1A and 1B). The initial bluish green color gradually became colorless, to disappear completely on day eight, which indicated attainment of microaerophilic condition. Optical density measurements and plate count assay showed that MTB attained dormancy by this time (192 h/8th day). Growth of the bacteria became static after 264 h (11th day) (Fig. 1C). In accordance with earlier reports we found that nonreplicating persistent stage 1 (NRP1) was attained at 192 h (8th day) and the nonreplicating persistent stage 2 (NRP2) or enduring hypoxia was attained at 336 h (14th day) (9, 34). NRP2 mimics the in vivo situation where MTB attains dormancy inside the macrophages (5). The state of induced-dormancy persisted until the culture was subjected to re-aeration. The reappearance of bluish green color of methylene blue indicated that the aerobic condition had been re-established. The reactivation was confirmed by measuring the A600 and by plate count assay. Dormant MTB upon transfer to fresh medium and subsequent aeration has been shown to undergo synchronized replication (35). Therefore, the physiological changes that we observed in the subsequent studies represent uniform changes in the whole bacterial population subjected to dormancy and reactivation. Upon re-aeration, the bacterial cells underwent replication as indicated by the increase in A600 and CFU values (Fig. 1C). Bacteria were harvested, lysed, and equal quantities of proteins from all the five stages (normoxia, NRP1, NRP2, R6, and R24) were subjected to proteomic analysis. The data is available via ProteomeXchange using the identifier PXD001158.
Proteome Profiles during Normoxia, Dormancy, and Reactivation
MTB grown under normoxia yielded a total of 1663 proteins. This represents 41.4% of the MTB proteome as annotated by Schubert et al. (36). Employing the same protocol used in this study for the extraction of proteins, Cho et al. detected 875 proteins in dormant MTB (24). Another proteomic profiling of subcellular fractions of MTB grown in glycerol alanine salts medium under aerated condition yielded 1044 proteins (23). Albrethsen et al. identified 1176 proteins from extracellular fraction of log phase MTB culture (25).
We detected 764 proteins in NRP1, which represent 19% of the total proteins present in MTB, as the culture progressed to NRP2 (504 h/21st day) the number of proteins decreased to 691 (17.17% of the total proteins). In a similar study, Cho et al. reported 586 proteins from NRP1 and 628 proteins from NRP2 (24). During reactivation, the number of proteins increased to 768 (19.1%) at R6, and 983 (24.46%) at R24. From all these stages, 1871 proteins comprising 47% of the MTB proteome were identified. The distribution of proteins identified during normoxia, dormancy, and reactivation is shown in Fig. 2A. The fold changes in protein levels at different stages of dormancy and reactivation are depicted in Figs. 3A–3D. We found 597 proteins in the control (normoxia) that were absent in both dormancy and reactivation stages. Sixty-six unique proteins were identified at NRP1, and 56 at NRP2. Three proteins (NP_215277, a possible ferredoxin, NP_215682, a probable conserved lipoprotein LpqW and NP_216034, a conserved hypothetical protein) were present only in the two stages of dormancy and absent in all other stages. During reactivation, 73 and 94 proteins were identified as uniquely present at R6 and R24, respectively. Six proteins identified at both time points of reactivation were absent in all other stages. Predicted interactions of unique proteins and list of unique proteins identified in our study are given in supplemental List S1. The complete list of proteins obtained and the difference in their relative quantities are given in the supplemental List S2. Statistical analysis of biological replicates by Friedman test showed a p value of <0.05 for all the four stages when compared with the control.
Fig. 2.

A, Venn diagram shows the number of proteins present in aerobically grown control, 12th day of hypoxia (NRP1), 21st day of dormancy (NRP2), 6 h after reactivation (R6) and 24 h after reactivation (R24). Venn diagram showing the distribution of up-regulated and down-regulated proteins between stages of B, Dormancy (NRP1 and NRP2) and C, Reactivation (R6 and R24). D, Number of proteins in various biological processes obtained by clustering using DAVID.
Fig. 3.

Biological interaction network created by BioNet Builder using proteins identified during A, 12th day of hypoxia (NRP1), B, 21st day of dormancy (NRP2), C, 6 h after reactivation, and D, 24 h after reactivation.
Validation of Quantitative Proteomic Data by qPCR
To validate the proteomic data obtained across the various conditions, we performed a quantitative real time PCR analysis of randomly selected genes (ald, devR, and bfrB). Transcripts of these genes were measured across control, dormancy, and reactivation. When compared with that in the control, ald showed a relative fold expression value of 0.018 at NRP1, 1.9 at NRP2, 12.6 at R6, and 11.2 at R24 (Fig. 4A). The devR transcript showed a consistent up-regulation during all the stages analyzed (12.2-fold at NRP1, 8.3-fold at NRP2, 7.3-fold at R6, and 5.8-fold at R24) (Fig. 4C). The fold expression values of bfrB revealed a marked increase during dormancy but the levels dropped upon entering reactivation even though it was still greater than that of the control (13-fold at NRP1, 10.8-fold at NRP2, 1.3 at R6 and 2 at R24) (Figs. 4E). For these three genes, the fold expression values from qPCR and quantitative proteomic analysis were compared across NRP1, NRP2, R6, and R24 and the results are depicted in Figs. 4A–4F. The qPCR results of ald correlated well with the proteomic profile. Both analyses did not detect ald/Ald at NRP1, it was up-regulated at all other stages. In the case of devR, the correlation was even better, at NRP1 both analyses showed highest fold expression value, which decreased at NRP2, R6, and R24. In the case of bfrB the trend was similar to devR but a dramatic drop in mRNA levels did not result in comparable decline at the protein level during reactivation, which could be attributed to the presence of the residual protein in the cells before they underwent any degradation.
Fig. 4.

Relative fold expression values of Ald, DevR and BfrB through qPCR analysis (A, C, and E) respectively, and quantitative proteomic analysis (B, D, and F) respectively.
Biological and Functional Analyses
Through Gene Ontology analysis using DAVID, the proteins were clustered into various biological processes (Fig. 2D and supplemental List S3). In addition, we used information from UniProt database for manual clustering. Proteins of most of the biological processes except those in electron transport, energy derivation by oxidation of organic compounds and fatty acid metabolism had fewer representatives during dormancy and reactivation than in normoxia.
Besides using DAVID for generating functional clusters, BioNet Builder was used for the construction of cellular networks. The network consisted of 2969 nodes representing the Entrez geneID, and 16,911 edges. The proteins and their quantitative data were merged to this network using “advanced merge” option in Cytoscape 2.8.3. This resulted in the incorporation of 1315 proteins, of the 1871 identified, into the documented interaction network of MTB (Figs. 3A–3D). A decrease in the number of proteins was observed during the initial phase of hypoxia (NRP1), which further decreased during enduring hypoxia (NRP2). A total of 34 proteins were present at down-regulated levels and 242 proteins were up-regulated during NRP1. At NRP2, 58 proteins involved in various biological processes were down-regulated and 192 proteins were found to be up-regulated when compared with the control (supplemental List S4). During initial period of reactivation (R6) the total number of proteins increased to 768, of which 213 were up-regulated, 58 were down-regulated, and 73 were unique. During R24, a total of 983 proteins were identified, of which 224 were up-regulated, 78 were down-regulated, and 93 were detected as unique. The uniquely detected proteins during different stages of dormancy and reactivation are listed in the supplemental List S1.
Energy Metabolism
The central carbon metabolism of MTB was not significantly affected by dormancy when compared with other metabolic processes. Phosphohexose isomerase, triosephosphate isomerase, and dihydrolipoamide dehydrogenase, the enzymes involved in the glycolytic processes, were found to be up-regulated during NRP1. The enzymes present at the entry point of TCA cycle from glycolysis, pyruvate dehydrogenase and citrate synthase were also found to be up-regulated during NRP1. We presume that the up-regulated levels of these proteins might help the bacteria, through substrate level phosphorylation, to meet the ATP requirement. During adaptation to hypoxia, MTB remodels TCA cycle to increase succinate production, which is used to flexibly sustain membrane potential, ATP synthesis, and anaplerosis to survive O2 limitation (19). In our study we detected isocitrate lyase (Icl, Rv1916) only in the control. However, Eoh and Rhee detected Icl-mediated synthesis of succinate during hypoxia which affords MTB with a unique and bio-energetically efficient metabolic means of entry into, and exit from, hypoxia-induced dormancy (19). The probable isocitrate lyases (NP_216431 and NP_216432) detected during hypoxia and reactivation in our study, were present at normal levels. Fumarate hydratase and malate dehydrogenase were also found at normal levels during dormancy and reactivation suggesting a functional glyoxylate pathway. It was interesting to detect the subunits of ATP synthases α (Rv1308), β (Rv1310), γ (Rv1309), δ (Rv1307), and ε (Rv1311) subunits present at normal levels during both phases of dormancy, in contrast to the hypothesis that FO-F1 ATP synthase is likely to be down-regulated upon hypoxia (10). In addition, up-regulation of Rv1623c, a subunit of cytochrome D terminal oxidase complex, during NRP2 is indicative of ATP synthesis through electron transport chain. The homolog of this protein in E. coli is a component of the aerobic respiratory chain that is predominant when cells are grown under low aeration (37, 38). Our study suggests that cytochrome D terminal oxidase of MTB also might perform a similar function when the bacterium gets ready to enter dormancy. Along with the cytochrome D terminal oxidase, we detected 12 other members of the electron transport chain. Nine of them were present at NRP1, seven during NRP2, ten during R6, and nine during R24. Apart from FixB (Rv3028c) and QcrA (Rv2195), all other electron transport chain members identified in our study were present at normal levels during dormancy. Rv3028c, which was found at normal levels during NRP1, was down-regulated at NRP2. Rv2195 was found to be up-regulated during NRP1 and at normal level during all other stages.
Amino Acid Metabolism
We identified 129 proteins functioning in amino acid metabolism out of which majority was found missing during dormancy and the initial phase of reactivation. Sixty-two proteins were detected at NRP1 and 59 during NRP2. Upon re-aeration, the number increased to 61 (at R6) and further increased to 91 at R24 (Fig. 5B). However, only one protein during NRP1 and two during NRP2 were found to be down-regulated. Twenty-nine proteins involved in amino acid metabolism were present at elevated levels during NRP1 and they are involved in the metabolism of methionine, threonine, lysine, leucine, valine, cysteine, serine, glycine, proline, arginine, tryptophan, and aromatic amino acids. The single down-regulated protein HisE identified in the NRP1 is involved in the histidine metabolism. During NRP2, 17 proteins were present at up-regulated levels and are involved in the metabolism of methionine, lysine, glycine, cysteine, homocysteine, arginine, proline, glutamine, aspartic acid, and branched chain amino acids. The two down-regulated proteins (TrpB and MetG) present during NRP2 belong to the biosynthesis of tryptophan and methionine. Rv2458 (MmuM, a probable homocysteine S-methyltransferase) was present only at NRP2 and R6. At R6 and R24 each, three proteins were found down-regulated. The down-regulated proteins observed at R6 belongs to the metabolism of lysine (DapB), glutamine (GlnA1), and proline (ProC), while those detected at R24 belong to the metabolism of glutamine (CobQ2), arginine (ArgH) and cysteine (Csd). Nineteen proteins were present at up-regulated levels during R6, and they belong to the metabolism of glycine, lysine, threonine, arginine, tryptophan, methionine, proline, glutamine, homocysteine, aspartic acid, phenylalanine, and branched chain amino acids. Similarly, at R24, 22 proteins involved in the metabolism of glycine, lysine, cysteine, arginine, proline, homocysteine, aspartic acid, threonine, phenylalanine, histidine, glutamate, and branched chain amino acids were found up-regulated. More studies are required to understand the direction of amino acid metabolism during dormancy and reactivation. However, the up-regulated levels of a 40 kDa l-alanine dehydrogenase (Ald, Rv2780) during NRP2, indicates the maintenance of NAD pool when oxygen, the terminal electron acceptor, becomes limiting, because alanine synthesis is coupled to NADH oxidation (39), and our study shows its up-regulation during reactivation also.
Fig. 5.

Heat maps of proteins present in – A, TCA, glycolysis, glyoxylate, and electron transport chain, B, Amino acid metabolism, C, Lipid biosynthesis, and D, Lipid degradation. Positive values indicate up-regulated proteins, negative values represent down-regulated proteins and zero values represent proteins that are absent in a particular stage, but present in the control.
One of the interesting findings in our study is the presence of sulfate transporters at up-regulated levels during dormancy and reactivation. The sulfate transporters in MTB consist of SubI, CysT, CysW, CysA1, CysA2, and CysA3 (40). Of the six transporters, we detected two (CysA2 and CysA3), and both were found to be up-regulated during dormancy and reactivation. Once sulfate is transported into the cell, it is activated by conversion into adenosine 5′-phosphosulfate (APS) and 3′-phosphoadenosine 5′-phosphosulfate (PAPS) by CysNC, which contains two fused activities, sulfateadenylyl transferase (CysN) and adenylylsulfate kinase (CysC) (41), which are required for the production of methionine and cysteine. These proteins were detected only in control. Thirteen proteins present in the metabolism of sulfur-containing amino acids were detected in our study. Out of them, MetE (Rv1133c), ThrA (Rv1294), SerS (Rv3834c), Asd (Rv3708c), Rv3015c, and Cbs (Rv1077) were detected at up-regulated levels at NRP1. At NRP2, Rv2294, a putative cystathionine beta-lyase and Csd (Rv1464) were present in up-regulated condition. The importance of sulfur-containing amino acids and sulfate transporters during dormancy and reactivation is not clear at present.
Protein Synthesis and Degradation
The protein synthesis machinery was quite subdued during dormancy. By [35S]methionine labeling studies Hu et al., (2006) showed that in dormant MTB culture maintained in unstirred screw-cap tubes for 50 days, protein synthesis decreased by 98% (42). Eighty-six proteins involved in protein synthesis were detected in our study. At NRP1, only 57 proteins were detected of which 40 were present at normal levels, and six were down-regulated whereas, interestingly, 11 proteins were found to be up-regulated. At NRP2, 47 proteins were identified of which 29 were present at normal levels, 17 were found to be down-regulated and only one (AspS) was present at an elevated level (two-fold). At the early phase of re-aeration (R6), the number of proteins detected however, went up slightly to 53 of which 31 were found to be at normal levels, whereas 16 were found to be still down-regulated and six were found to be up-regulated. At R24 the total number of proteins increased significantly to 67, of which 48 were present at normal levels, whereas only seven were down-regulated, and twelve were up-regulated.
Protein degradation in MTB is enhanced during nutrient starvation (25). Our data revealed that during NRP1 and NRP2 the number and levels of proteins decreased, and upon re-aeration both began to show indications of returning to normalcy within 6 h (Fig. 3C). The Clp protease is a key component of the bacterial response to stress and is one of the major cellular proteases responsible for the selective degradation of misfolded proteins under stress (43, 44). It is expressed in MTB under both aerated (45) as well as stressed conditions such as elevated temperatures and phagocytosis by macrophage (46). The Clp protease is a complex consisting of a proteolytic subunit (ClpP) and a regulatory ATPase subunit (ClpC or ClpX) (44). MTB has two homologs of the ClpP proteolytic core (ClpP1 and ClpP2) and three potential Clp ATPases (ClpC1, ClpC2, and ClpX). In our study, we observed normal levels of both ClpP1 and ClpP2 during dormancy and reactivation. Interestingly ClpX (Rv2457c) was present in up-regulated condition during NRP1, NRP2 and during reactivation whereas both ClpP proteolytic core proteins (ClpP1 and ClpP2) were present at normal levels during dormancy and reactivation. The Clp proteases seemed to be playing a significant role in maintaining the protein homeostasis during dormancy and reactivation. Understanding this mechanism might provide us with clues to prevent reactivation of dormant MTB.
Another type of proteolytic pathway, the prokaryotic ubiquitin-like protein (Pup) - proteasome pathway is also present in MTB. In this pathway, Pup binds to the target protein to be degraded in a manner similar to that of eukaryotic ubiquitin (47). Pup is activated by the enzyme deamidase of Pup (Dop, Rv2112c), which deamidates the C-terminal glutamine of Pup to form glutamate. Proteasome accessory factor A (PafA), the Pup ligase, subsequently ligates the newly-formed side chain carboxylate to a lysine residue of the target protein (48). Pupylated proteins are guided into the proteasome through the binding of Pup to the proteasomal ATPase, which unfolds proteins prior to delivery into the proteasome core (49). Dop also functions as a depupylase to remove Pup from substrate proteins prior to proteasomal degradation (50). In our study, Dop was observed to be dramatically up-regulated (15-fold) during NRP2. Upon re-aeration (R24), when the number of proteins began to increase, the level of Dop returned to normal. Probably the change in the total number of proteins at different stages of dormancy and reactivation is determined by the equilibrium between protein synthesis and activation of proteolytic machinery.
Lipid Metabolism
During the early phase of hypoxia (NRP1), while the levels and total number of enzymes involved in fatty acid biosynthesis and their levels were low, those of fatty acid degradation pathways were markedly high (Figs. 5C and 5D). In the lipid degradation pathway a total of 26 proteins was detected. Twenty of them were present at NRP1 of which six were present at up-regulated levels and the remaining proteins at normal levels. At NRP2, nineteen proteins were detected; six of them were up-regulated, twelve at normal levels and one (FadA4) in down-regulated condition. Interestingly, at NRP2, twelve proteins involved in the lipid biosynthesis were found to be down-regulated. As reactivation started (R6) the number of down-regulated proteins in lipid biosynthesis decreased to three (CmaA2, IspG, and FadD15). The total number of proteins in this bioprocess increased to 32 at R6, and further increased to 45 at R24 and, none of them was found down-regulated. During entry into dormancy, it has been shown that mycobacteria undergo cell wall thickening, which indicates changes in lipid metabolism during dormancy (51). During persistent dormancy, MTB seems to maintain a dynamic equilibrium between fatty acid biosynthesis and degradation. During reactivation, biosynthesis of fatty acids was more pronounced, suggesting that a resurgence of biosynthesis of fatty acids is required to make up for their loss during dormancy, and to meet the increased demand for new cell wall synthesis during re-entry into the phase of active growth.
Of the 40 proteins involved in the biosynthesis of mycolic acids in MTB (52), 31 proteins were identified in this study. There are three distinct structural classes of mycolic acids in MTB - methoxy, α-methoxy, and keto-mycolic acids. MTB has both type I fatty acid synthetase (FAS-I) and type II fatty acid synthetase (FAS-II) as in eukaryotes (52). Of the 31 proteins identified in our study, twelve were found to be at normal levels during NRP1, and six were present at up-regulated levels, and none was found to be down-regulated. However, when the bacteria entered the complete hypoxic environment (NRP2), the scenario changed dramatically and we observed sixteen proteins involved in various steps of mycolic acid biosynthesis to be down-regulated, and only five were present at normal levels. Interestingly, we observed three proteins (FabG, KasB, FbpA) to be up-regulated. KasB was found up-regulated in anaerobically grown MTB (12), and its main function is the production of long chain of fatty acids including mycolic acid (53). However, during the initial stage of reactivation (R6) four proteins were found to be up-regulated, sixteen of them returned to normal levels, and only CmaA2 was down-regulated. During R24, 20 proteins were found to be at normal levels and three (KasA, BacA, and FbpA) of them were found to be up-regulated and none was found down-regulated. These observations suggest that during dormancy, mycolic acid could be metabolized for the generation of acetyl-CoA through beta-oxidation for bacterial survival leading to its depletion. This depletion of mycolic acids, we presume, could be one reason why dormant bacteria fail to provoke immune response in the human body as mycolic acids are a major class of TLR activators in initiating innate immune responses.
Cell Division, DNA Replication and Repair
In our analysis, 34 proteins involved in cell division, DNA replication and repair were detected in the control. Only seven of them were detected during early hypoxia (NRP1), and all of them were present at normal levels. However, during complete hypoxia (NRP2), we detected nine proteins of which six (DNA polymerase III β chain DnaN, DNA polymerase I PolA, GyrA, GyrB, large helicase-related protein (Lhr), and single-strand DNA-binding protein, (SSB)) were found to be down-regulated whereas the levels of DNA polymerase III subunit gamma/tau DnaZX and DNA topoisomerase I (TopA) remained unaltered. This is expected because during dormancy most of the prominent cellular functions such as DNA replication and repair are not operational, and therefore synthesis of enzymes involved in these processes would be uneconomical. However, upon re-aeration (R6) the number of proteins in these bioprocesses increased to 13 of which seven were present at normal levels (SSB, Hns, FtsE, ParB, DNA polymerase I PolA, DNA topoisomerase I TopA omega, and DNA polymerase III beta chain DnaN), suggesting the return of DNA metabolic processes and cell division. In addition, we observed the reappearance of RecC, MutT1, and LigA, which are involved in replication as well as in DNA repair. In the later phase of reactivation (R24) the number of proteins further increased to fifteen of which ten (SSB, RecC, MutT1, Hns, DNA polymerase III subunit gamma/tau DnaZX, FtsE, GyrA, ParB, LigA, DNA topoisomerase I TopA omega, and ATP-dependent DNA helicase RecG) were found to be up-regulated. Further, we observed the reappearance of RecG and RecR, which are involved in DNA replication and repair. nrdE and nrdF2 collectively called the nrdEF system, together encode an enzyme that catalyzes the formation of deoxyribonucleotides from ribonucleotides (54). The existence of NrdE in the cell wall fraction of MTB was initially reported by Rosenkrands et al., (2000) (55). Quantitative RT-PCR studies by Murphy et al. (2007) on nrdEF2 using the Wayne's model have shown a ten-fold decrease in the expression of nrdEF2 when MTB culture was shifted from log phase to NRP1, followed by a two to three-fold increase from there as the cells entered NRP2 (56). The absence of the NrdF2 protein at NRP1 in our study correlates well with the drastic decrease in expression of nrdEF2 observed by them. The levels of NrdF2 increased during NRP2 (two-fold) and R6 (three-fold), and returned to normal levels at R24. However NrdE was found highly up-regulated during all stages.
Transcription
MTB is predicted to have ∼214 transcriptional regulators (57). Seventy four of them were detected and identified in our analysis (Fig. 6B). Eight of them (Rv0818, Rv0981, SigK, CspA, Rv2258c, PrrA, WhiA, and DevR) were observed to be up-regulated during NRP1. In NRP2, seven regulators (MRPA, SigK, Rv1019, HrcA, Crp, DevS, and DevR) were found to be up-regulated. Although DevR is a nonessential protein under normoxic free-living conditions, it is an essential factor in dormant bacterium (58). DevR is a constituent of the two-component regulatory system DosR-DosS, which regulates around 50 other genes during dormancy (59). Interestingly this protein was found up-regulated during reactivation also. The relative amount of DevR increased by 15-fold during NRP1 and nine-fold during NRP2. During reactivation this decreased slightly to eight-fold at R6 and six-fold at R24. Rv1674c, and Rv3082c (virS) were present only at NRP1. Similarly, Rv0891c and Rv1395 were present only at NRP2. Seven transcriptional regulators (Rv0818, Rv2258c, NusA, PhoU, HrcA, Rv0474, and DevR) were present at up-regulated levels during R6. Eight proteins, which we found up-regulated at R24, were Rv3058c, EmbR, Rv2175c, Rv1019, Rv2258c, HrcA, DevS, and DevR. EmbR (Rv1267c), a putative transcriptional regulator of arabinan cell wall metabolism (60), and Rv3058c, a probable transcriptional factor that belongs to TetR family (61), were detected only in the control and R24 (five-fold increase at R24). Under unfavorable conditions, bacteria shift their metabolic activities by stringent response (62). In accordance with this hypothesis, Rv2583c, a GTP pyrophosphokinase (RelA) involved in stringent response in MTB was detected only at NRP2. Another protein involved in stringent response Rv2783c, a bifunctional protein guanosine pentaphosphate synthetase/polyribonucleotide nucleotidyl transferase (GpsI) were found to be up-regulated two-fold during dormancy. Rv0386, a transcriptional regulator that belongs to the LuxR family, Rv1219, and Rv0494 (GntR family transcriptional regulator) were detected only at R6. Rv0386 was detected in the cell wall and membrane fractions of log phase proteome of MTB by Mawuenyega et al., (2005) (23). The transcriptional regulators Rv1255c, a probable transcription factor present in RD13 (Regions of Difference) variable region of MTB H37Rv (63), Rv3183, Rv3060, Rv0339c (LuxR family) were present only at R24. The probable interactions of transcriptional regulators identified in our study during different stages of dormancy and reactivation are given in Figs. 7A–7D and the confidence score is shown in supplemental List S5.
Fig. 6.

Heat map of proteins present in – A, DNA replication and repair, B, Transcription, C, Protein synthesis, D, Toxin-antitoxin system, and E, Virulence factors. Positive values indicate up-regulated proteins, negative values represent down-regulated proteins and zero values are proteins that represent absent in a particular stage, but present in the control.
Fig. 7.

String analysis of transcriptional regulators identified from A, NRP1, B, NRP2, C, R6, and D, R24 stages indicating possible interactions.
Changes in the Levels of Proteins Involved in Stress Response
Intracellular pathogens need to overcome severe adverse conditions inside the host for their survival. A number of stress-related proteins were observed to be expressed normally during both dormancy and reactivation. Heat-shock proteins (HSPs) are generally responsible for preventing damage to proteins in response to elevated temperatures and also during other unfavorable conditions. HSPs in MTB include DnaK, DnaJ, GrpE, GroES, GroEL, and other low-molecular weight proteins (64). In addition to its role as a heat-shock protein, GroEL functions as a chaperonin to assist folding of linear amino acid chains into their respective three-dimensional structure (65). Cyclic-AMP-mediated up-regulation of GroEL2 under low-oxygen/CO2-enriched growth conditions was observed by Gazdik and McDonough (2005) (66). A previous study from our laboratory showed that GroEL2 promoter of MTB is highly active in M. smegmatis when subjected to different stress conditions including hypoxia (65). In the present study we observed a normal expression of GroEL2 and all other chaperonins throughout the course of dormancy and reactivation. GroEL1, GroES, and GrpE were also found to be expressed normally during dormancy and reactivation. DNA-associated chaperonins DnaK and DnaJ were also found to be expressed at the same levels as in the control indicating their importance in dormancy and reactivation. Thioredoxin reductase (TrxB2, Rv3913) was found to be up-regulated during dormancy (NRP1 and NRP2). TrxB2 could function as an antioxidant in MTB during anaerobic or microaerophilic conditions and inside human granuloma (67). Zhang et al., (1999) found that in MTB, TrxB2 has the ability to reduce peroxides and dinitrobenzenes. We found its levels to be normal at R6 but was found down-regulated at R24. TrxB2 has been found to be up-regulated in M. smegmatis also during stationary phase (68). The universal stress protein Rv2005c was found to be significantly up-regulated throughout dormancy (14-fold at NRP1 and 46-fold at NRP2) and during reactivation (40-fold at R6 and 17-fold at R24). The heat shock protein HtpX (Rv0563) was uniquely present at R6. Alpha-crystallin 1 (HspX/Acr1/Rv2031c) is a heat shock protein with a large molecular chaperone domain, and is highly induced during dormancy (56). This protein is implicated in the survival of MTB inside macrophages (15). The biology of alpha-crystallin 1 is complex as evidenced by the adverse effect of the gene knockout on growth (69). Overexpression of acr slows down the growth rate of MTB and the autolysis of post-stationary phase cultures (15). Detection of this protein was a key finding in our study. Surprisingly it was found up-regulated 225-fold during NRP1 and 246-fold during NRP2. Interestingly it correlates well with the available microarray data (56). However, its levels decreased to 175-fold during R6 and to 64-fold during R24, which is again very interesting from yet another point of view, because it is an important member under devR regulon, and DevR is up-regulated during reactivation.
Proteins Associated with Reactivation
During reactivation, the total number of proteins and the number of proteins up-regulated increased when compared with those of dormancy (Figs. 2B and 2C). During the initial period of reactivation (R6), 213 proteins were found up-regulated whereas during the later stage (R24), 224 were up-regulated. Of these proteins, 108 were common to both stages of reactivation (Fig. 2C). One hundred and four up-regulated proteins were exclusive to R6, and 113 were exclusive to R24. Altogether 322 proteins were present in up-regulated condition during reactivation. The amino acid metabolism became significantly active upon reactivation. Nineteen proteins in amino acid metabolism were up-regulated at R6, and 22 at R24. During initial stages of reactivation (R6), four proteins in the lipid degradation and nine proteins in the lipid biosynthesis were present at up-regulated levels. At R24, the number of proteins up-regulated increased to seventeen in lipid biosynthesis, and ten in lipid degradation. The levels of proteins involved in lipid biosynthesis were found to be critical for reactivation of MTB from dormancy. The levels of transcriptional regulators DevR, DevS, EmbR, Rv3058c, Rv0474, Rv2175c, PhoU, Rv0818, NusA, HrcA, and Rv2258c were elevated during reactivation. DevR, however, was found up-regulated at all stages of dormancy and reactivation. EmbR and Rv3058c, were found up-regulated only at R24. The transcriptional regulators Rv1219c, Rv0494, and Rv0386 were present only at R6. Similarly four other transcriptional regulators (Rv1255c, Rv0339c, Rv3060c, and Rv3183) were found only at R24. Interestingly we detected the hypothetical protein Rv2817c (Cas1, a CRISPR-associated endonuclease) in the early stage of reactivation (R6). CRISPR-Cas system has gained much attention in recent times because of its involvement in genome editing (70). Therefore detection of Cas1, along with proteins involved in DNA recombination and repair in the early phase of reactivation could indicate that genome editing might play a role during reactivation of dormant MTB. ClpX, the MTB homolog of Clp ATPase involved in the selective degradation of misfolded proteins, was found up-regulated during reactivation. The universal stress protein Rv2005c was found up-regulated 40-fold immediately upon aeration (R6) and decreased to 17-fold at R24. The heat-shock protein alpha crystallin (Acr) was found up-regulated during reactivation. At reactivation, changes in the levels of toxin antitoxin system were observed (Fig. 6D). Toxins Rv0299 and RelE were unique to R6 stage of reactivation whereas antitoxin Rv0298 was found specific to R24. Eight virulence factors were present at up-regulated levels at R6, and nine at R24. At R24 the number of down-regulated virulence factors also decreased to four (Fig. 6E) suggesting the re-establishment of virulence upon reactivation.
Concluding Remarks
The experimental workflow and the key findings from this study are depicted in Fig. 8. We developed a simple in vitro reactivation system based on Wayne's dormancy model to compare the proteomes of dormant and reactivated virulent MTB. We analyzed proteins from the initial microaerophilic stage of dormancy (day 12, NRP1), enduring hypoxia (day 21, NRP2), 6 and 24 h after re-aeration (R6 and R24), and compared them with those of aerobically grown bacilli. We could detect 1871 proteins, the highest number reported from MTB in a quantitative proteomic analysis. During NRP1 the number of proteins, as well as their relative quantities, were observed to be lesser than those of the normoxially grown control. The number and relative quantities of proteins further decreased as MTB reached complete hypoxia (NRP2), and then increased during reactivation. During dormancy, biological processes like cell wall macromolecule biosynthesis, DNA replication and repair were minimal, and these bioprocesses get reactivated upon re-aeration. Alteration in the relative amounts of proteins involved in energy metabolism was observed during dormancy and reactivation. Our study reiterates the effect of lipid degradation pathways and electron transport chain on dormancy, and also shows their roles in reactivation. The up-regulation of sulfate transporters and bacterioferritin proteins was observed during dormancy and reactivation. A very interesting observation was the up-regulation (15-fold) of deamidase of Pup (Dop) protein involved in pupylation at NRP2. Our study implicates that the proteasome-mediated degradation by mycobacterial Pup-Dop system is crucial in attaining a stable state of dormancy, and the up-regulation of Dop is a clear indication of the activation of this pathway. Targeted inhibition of Dop could be an effective therapeutic strategy to eliminate dormant bacteria. Another significant finding is the detection of universal stress protein Rv2005c during reactivation. The Rv2005c is one of the DevR-regulated genes (71) and a predicted vaccine candidate (72). This protein is up-regulated during dormancy and reactivation. We presume that this protein might also play a critical role in MTB during reactivation. Therefore Rv2005c could be a possible drug target for preventing reactivation TB. DevR regulates 50 genes during dormancy (58), however, we found that it is highly up-regulated during reactivation as well. It will be interesting to study the role of this protein in regulating gene expression during the process of reactivation. Detection of Cas1 and the presence of high levels of Acr during reactivation are the other key findings in our study.
Fig. 8.

The experimental workflow of the study.
Supplementary Material
Footnotes
Author contributions: V.G., S.R., R.L.G., L.J., and R.A.K. designed research; V.G., S.R., L.J., A.S., R.R., A.R.P., and R.L.G. performed research; V.G., S.R., R.L.G., L.J., A.S., and A.J. contributed new reagents or analytic tools; V.G., S.R., R.L.G., L.J., R.R., A.R.P., and R.A.K. analyzed data; V.G., S.R., L.J., R.R., A.R.P., S.M., A.J., and R.A.K. wrote the paper.
* This study has been funded by the Department of Biotechnology, Government of India [BT/PR5361/MED/29/507/2012 (RAK)]. SR thanks Department of Science and Technology, Government of India, for INSPIRE fellowship. RLG and LJ acknowledge Council for Scientific and Industrial Research, Government of India, and RR acknowledges University Grants Commission, Government of India, for research fellowship. We are grateful to the PRIDE Team for assistance in MS/MS data distribution.
 This article contains supplemental Lists S1 to S5.
This article contains supplemental Lists S1 to S5.
Additional information: The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD001158 and DOI 10.6019/PXD001158. Supplemental data containing details such as uniquely expressed, up-regulated and down-regulated proteins, and their functional classification are available online through the MCP website.
1 The abbreviations used are:
- TB
- Tuberculosis
- MTB
- Mycobacterium tuberculosis
- BCG
- Bacillus Calmette–Guérin
- NRP1
- Nonreplicating persistent stage 1
- NRP2
- Nonreplicating persistent stage 2
- R6
- 6 hours after Reactivation
- R24
- 24 hours after Reactivation
- LC/MS/MS
- Liquid chromatography – tandem mass spectrometry
- GO
- Gene Ontology
- DAVID
- Database for Annotation, Visualization and Integrated Discovery
- PBS
- Phosphate buffered saline
- MPDS
- MassPREP™ Digestion Standard
- CFU
- Colony forming unit.
REFERENCES
- 1. WHO, T. B. f. s. (2012) World Health Organization (WHO) website http://www.who.int/topics/tuberculosis/en/.
- 2. Gengenbacher M., Kaufmann S. H. E. (2012) Mycobacterium tuberculosis: success through dormancy. FEMS Microbiol. Rev. 36, 514–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gorna A. E., Bowater R. P., Dziadek J. (2010) DNA repair systems and the pathogenesis of Mycobacterium tuberculosis: varying activities at different stages of infection. Clin. Sci. 119, 187–202 [DOI] [PubMed] [Google Scholar]
- 4. Boshoff H., Barry C., 3rd (2005) Tuberculosis – metabolism and respiration in the absence of growth. Nat. Rev. Microbiol. 3, 70–80 [DOI] [PubMed] [Google Scholar]
- 5. Wayne L. G. (1994) Dormancy of Mycobacterium tuberculosis and latency of disease. Eur. J. Clin. Microbiol. Infect. Dis. 13, 908–914 [DOI] [PubMed] [Google Scholar]
- 6. Wayne L. G., Sohaskey C. D. (2001) Nonreplicating persistence of Mycobacterium tuberculosis. Annu. Rev. Microbiol. 55, 139–163 [DOI] [PubMed] [Google Scholar]
- 7. Barry C. E., 3rd, Boshoff H. I., Dartois V., Dick T., Ehrt S., Flynn J., Schnappinger D., Wilkinson R. J., Young D. (2009) The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 7, 845–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dick T. (2001) Dormant tubercle bacilli: the key to more effective TB chemotherapy? J. Antimicrob. Chemother. 47, 117–118 [DOI] [PubMed] [Google Scholar]
- 9. Wayne L. G., Hayes L. G. (1996) An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect. Immun. 64, 2062–2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rao S. P. S., Alonso S., Rand L., Dick T., Pethe K. (2008) The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 105, 11945–11950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shi L., Sohaskey C., Kana B., Dawes S., North R. (2005) Changes in energy metabolism of Mycobacterium tuberculosis in mouse lung and under in vitro conditions affecting aerobic respiration. Proc. Natl. Acad. Sci. U.S.A. 102, 15629–15634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Starck J., Kallenius G., Marklund B. I., Andersson D. I., Akerlund T. (2004) Comparative proteome analysis of Mycobacterium tuberculosis grown under aerobic and anaerobic conditions. Microbiology 150, 3821–3829 [DOI] [PubMed] [Google Scholar]
- 13. Taneja N. K., Dhingra S., Mittal A., Naresh M., Tyagi J. S. (2010) Mycobacterium tuberculosis transcriptional adaptation, growth arrest, and dormancy phenotype development is triggered by vitamin C. PLoS One 5, e10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wolfe L. M., Veeraraghavan U., Idicula-Thomas S., Schurer S., Wennerberg K., Reynolds R., Besra G. S., Dobos K. M. (2013) A chemical proteomics approach to profiling the ATP-binding proteome of Mycobacterium tuberculosis. Mol. Cell. Proteomics 12, 1644–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yuan Y., Crane D. D., Simpson R. M., Zhu Y. Q., Hickey M. J., Sherman D. R., Barry C. E., 3rd (1998) The 16-kDa alpha-crystallin (Acr) protein of Mycobacterium tuberculosis is required for growth in macrophages. Proc. Natl. Acad. Sci. U.S.A. 95, 9578–9583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Deb C., Lee C. M., Dubey V. S., Daniel J., Abomoelak B., Sirakova T. D., Pawar S., Rogers L., Kolattukudy P. E. (2009) A novel in vitro multiple-stress dormancy model for Mycobacterium tuberculosis generates a lipid-loaded, drug-tolerant, dormant pathogen. PLoS One 4, e6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Parrish N. M., Dick J. D., Bishai W. R. (1998) Mechanisms of latency in Mycobacterium tuberculosis. Trends Microbiol. 6, 107–112 [DOI] [PubMed] [Google Scholar]
- 18. Cole S., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S. V., Eiglmeier K., Gas S., Barry C. E., 3rd (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 [DOI] [PubMed] [Google Scholar]
- 19. Eoh H., Rhee K. Y. (2013) Multifunctional essentiality of succinate metabolism in adaptation to hypoxia in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 110, 6554–6559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rosenkrands I., Slayden R. A., Crawford J., Aagaard C., Barry C. E., 3rd, Andersen P. (2002) Hypoxic response of Mycobacterium tuberculosis studied by metabolic labeling and proteome analysis of cellular and extracellular proteins. J. Bacteriol. 184, 3485–3491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Voskuil M. I., Visconti K. C., Schoolnik G. K. (2004) Mycobacterium tuberculosis gene expression during adaptation to stationary phase and low-oxygen dormancy. Tuberculosis 84, 218–227 [DOI] [PubMed] [Google Scholar]
- 22. Kelkar D. S., Kumar D., Kumar P., Balakrishnan L., Muthusamy B., Yadav A. K., Shrivastava P., Marimuthu A., Anand S., Sundaram H., Kingsbury R., Harsha H. C., Nair B., Prasad T. S., Chauhan D. S., Katoch K., Katoch V. M., Chaerkady R., Ramachandran S., Dash D., Pandey A. (2011) Proteogenomic analysis of Mycobacterium tuberculosis by high resolution mass spectrometry. Mol. Cell. Proteomics 10, M111 011627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mawuenyega K. G., Forst C. V., Dobos K. M., Belisle J. T., Chen J., Bradbury E. M., Bradbury A. R., Chen X. (2005) Mycobacterium tuberculosis functional network analysis by global subcellular protein profiling. Mol. Biol. Cell 16, 396–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cho S. H., Goodlett D., Franzblau S. (2006) ICAT-based comparative proteomic analysis of nonreplicating persistent Mycobacterium tuberculosis. Tuberculosis 86, 445–460 [DOI] [PubMed] [Google Scholar]
- 25. Albrethsen J., Agner J., Piersma S. R., Hojrup P., Pham T. V., Weldingh K., Jimenez C. R., Andersen P., Rosenkrands I. (2013) Proteomic profiling of Mycobacterium tuberculosis identifies nutrient-starvation-responsive toxin-antitoxin systems. Mol. Cell. Proteomics 12, 1180–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Galagan J. E., Minch K., Peterson M., Lyubetskaya A., Azizi E., Sweet L., Gomes A., Rustad T., Dolganov G., Glotova I., Abeel T., Mahwinney C., Kennedy A. D., Allard R., Brabant W., Krueger A., Jaini S., Honda B., Yu W. H., Hickey M. J., Zucker J., Garay C., Weiner B., Sisk P., Stolte C., Winkler J. K., Van de Peer Y., Iazzetti P., Camacho D., Dreyfuss J., Liu Y., Dorhoi A., Mollenkopf H. J., Drogaris P., Lamontagne J., Zhou Y., Piquenot J., Park S. T., Raman S., Kaufmann S. H., Mohney R. P., Chelsky D., Moody D. B., Sherman D. R., Schoolnik G. K. (2013) The Mycobacterium tuberculosis regulatory network and hypoxia. Nature 499, 178–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Raffia A., Fahim U. (2010) Photoredox reaction of methylene blue and lactose in alcoholic buffered solution. J. Appl. Chem. Res. 13, 72–84 [Google Scholar]
- 28. Smith P., Krohn R., Hermanson G., Malliya A., Gartner F., Provenzano M., Fujimoto E., Goeke N., Olson B., Klenk D. (1985) Measurement of protein using bicinchoninic acid assay. Anal. Biochem. 150, 76–85 [DOI] [PubMed] [Google Scholar]
- 29. Cockman M. E., Webb J. D., Kramer H. B., Kessler B. M., Ratcliffe P. J. (2009) Proteomics-based identification of novel factor inhibiting hypoxia-inducible factor (FIH) substrates indicates widespread asparaginyl hydroxylation of ankyrin repeat domain-containing proteins. Mol. Cell. Proteomics 8, 535–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huang da W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 31. Huang da W., Sherman B. T., Lempicki R. A. (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Avila-Campillo I., Drew K., Lin J., Reiss D. J., Bonneau R. (2007) BioNetBuilder: automatic integration of biological networks. Bioinformatics 23, 392–393 [DOI] [PubMed] [Google Scholar]
- 33. Larsen M. H. (2000) Some common methods in mycobacterial genetics. Molecular genetics of mycobacteria. ASM Press, Washington, DC: 316 [Google Scholar]
- 34. Rustad T. R., Harrell M. I., Liao R., Sherman D. R. (2008) The enduring hypoxic response of Mycobacterium tuberculosis. PLoS One 3, e1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wayne L. G. (1977) Synchronized replication of Mycobacterium tuberculosis. Infect. Immun. 17, 528–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schubert O. T., Mouritsen J., Ludwig C., Rost H. L., Rosenberger G., Arthur P. K., Claassen M., Campbell D. S., Sun Z., Farrah T., Gengenbacher M., Maiolica A., Kaufmann S. H., Moritz R. L., Aebersold R. (2013) The Mtb proteome library: a resource of assays to quantify the complete proteome of Mycobacterium tuberculosis. Cell Host Microbe 13, 602–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Borisov V. B., Gennis R. B., Hemp J., Verkhovsky M. I. (2011) The cytochrome bd respiratory oxygen reductases. Biochim. Biophys. Acta 1807, 1398–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kana B. D., Weinstein E. A., Avarbock D., Dawes S. S., Rubin H., Mizrahi V. (2001) Characterization of the cydAB-encoded cytochrome bd oxidase from Mycobacterium smegmatis. J. Bacteriol. 183, 7076–7086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hutter B., Dick T. (1998) Increased alanine dehydrogenase activity during dormancy in Mycobacterium smegmatis. FEMS Microbiol. Lett. 167, 7–11 [DOI] [PubMed] [Google Scholar]
- 40. Wooff E., Michell S. L., Gordon S. V., Chambers M. A., Bardarov S., Jacobs W. R., Jr., Hewinson R. G., Wheeler P. R. (2002) Functional genomics reveals the sole sulphate transporter of the Mycobacterium tuberculosis complex and its relevance to the acquisition of sulphur in vivo. Mol. Microbiol. 43, 653–663 [DOI] [PubMed] [Google Scholar]
- 41. Pinto R., Tang Q. X., Britton W. J., Leyh T. S., Triccas J. A. (2004) The Mycobacterium tuberculosis cysD and cysNC genes form a stress-induced operon that encodes a trifunctional sulfate-activating complex. Microbiology 150, 1681–1686 [DOI] [PubMed] [Google Scholar]
- 42. Hu Y. M., Butcher P. D., Sole K., Mitchison D. A., Coates A. R. (1998) Protein synthesis is shutdown in dormant Mycobacterium tuberculosis and is reversed by oxygen or heat shock. FEMS Microbiol. Lett. 158, 139–145 [DOI] [PubMed] [Google Scholar]
- 43. Andersson F. I., Tryggvesson A., Sharon M., Diemand A. V., Classen M., Best C., Schmidt R., Schelin J., Stanne T. M., Bukau B., Robinson C. V., Witt S., Mogk A., Clarke A. K. (2009) Structure and function of a novel type of ATP-dependent Clp protease. J. Biol. Chem. 284, 13519–13532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kress W., Maglica Z., Weber-Ban E. (2009) Clp chaperone-proteases: structure and function. Res. Microbiol. 160, 618–628 [DOI] [PubMed] [Google Scholar]
- 45. Weichart D., Querfurth N., Dreger M., Hengge-Aronis R. (2003) Global role for ClpP-containing proteases in stationary-phase adaptation of Escherichia coli. J. Bacteriol. 185, 115–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gaillot O., Pellegrini E., Bregenholt S., Nair S., Berche P. (2000) The ClpP serine protease is essential for the intracellular parasitism and virulence of Listeria monocytogenes. Mol. Microbiol. 35, 1286–1294 [DOI] [PubMed] [Google Scholar]
- 47. Pearce M. J., Mintseris J., Ferreyra J., Gygi S. P., Darwin K. H. (2008) Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science 322, 1104–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Striebel F., Imkamp F., Sutter M., Steiner M., Mamedov A., Weber-Ban E. (2009) Bacterial ubiquitin-like modifier Pup is deamidated and conjugated to substrates by distinct but homologous enzymes. Nat. Struct. Mol. Biol. 16, 647–651 [DOI] [PubMed] [Google Scholar]
- 49. Burns K. E., Cerda-Maira F. A., Wang T., Li H., Bishai W. R., Darwin K. H. (2010) “Depupylation” of prokaryotic ubiquitin-like protein from mycobacterial proteasome substrates. Mol. Cell. 39, 821–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Imkamp F., Rosenberger T., Striebel F., Keller P. M., Amstutz B., Sander P., Weber-Ban E. (2010) Deletion of dop in Mycobacterium smegmatis abolishes pupylation of protein substrates in vivo. Mol. Microbiol. 75, 744–754 [DOI] [PubMed] [Google Scholar]
- 51. Cunningham A. F., Spreadbury C. L. (1998) Mycobacterial stationary phase induced by low oxygen tension: cell wall thickening and localization of the 16-kilodalton alpha-crystallin homolog. J. Bacteriol. 180, 801–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Takayama K., Wang C., Besra G. S. (2005) Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin. Microbiol. Rev. 18, 81–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schaeffer M. L., Agnihotri G., Volker C., Kallender H., Brennan P. J., Lonsdale J. T. (2001) Purification and biochemical characterization of the Mycobacterium tuberculosis beta-ketoacyl-acyl carrier protein synthases KasA and KasB. J. Biol. Chem. 276, 47029–47037 [DOI] [PubMed] [Google Scholar]
- 54. Elledge S. J., Zhou Z., Allen J. B. (1992) Ribonucleotide reductase: regulation, regulation, regulation. Trends Biochem. Sci. 17, 119–123 [DOI] [PubMed] [Google Scholar]
- 55. Rosenkrands I., Weldingh K., Jacobsen S., Hansen C. V., Florio W., Gianetri I., Andersen P. (2000) Mapping and identification of Mycobacterium tuberculosis proteins by two-dimensional gel electrophoresis, microsequencing, and immunodetection. Electrophoresis 21, 935–948 [DOI] [PubMed] [Google Scholar]
- 56. Murphy D. J., Brown J. R. (2007) Identification of gene targets against dormant phase Mycobacterium tuberculosis infections. BMC Infect. Dis. 7, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Minch K. J., Rustad T. R., Peterson E. J., Winkler J., Reiss D. J., Ma S., Hickey M., Brabant W., Morrison B., Turkarslan S., Mawhinney C., Galagan J. E., Price N. D., Baliga N. S., Sherman D. R. (2015) The DNA-binding network of Mycobacterium tuberculosis. Nat. Commun. 6, 5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Park H. D., Guinn K. M., Harrell M. I., Liao R., Voskuil M. I., Tompa M., Schoolnik G. K., Sherman D. R. (2003) Rv3133c/dosR is a transcription factor that mediates the hypoxic response of Mycobacterium tuberculosis. Mol. Microbiol. 48, 833–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fang H., Yu D., Hong Y., Zhou X., Li C., Sun B. (2013) The LuxR family regulator Rv0195 modulates Mycobacterium tuberculosis dormancy and virulence. Tuberculosis 93, 425–431 [DOI] [PubMed] [Google Scholar]
- 60. Alderwick L. J., Molle V., Kremer L., Cozzone A. J., Dafforn T. R., Besra G. S., Futterer K. (2006) Molecular structure of EmbR, a response element of Ser/Thr kinase signaling in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 103, 2558–2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Warner D. F., Etienne G., Wang X.-M., Matsoso L. G., Dawes S. S., Soetaert K., Stoker N. G., Mizrahi V. (2006) A derivative of< i> Mycobacterium smegmatis</i> mc< sup> 2</sup> 155 that lacks the duplicated chromosomal region. Tuberculosis 86, 438–444 [DOI] [PubMed] [Google Scholar]
- 62. Primm T. P., Andersen S. J., Mizrahi V., Avarbock D., Rubin H., Barry C. E., 3rd (2000) The stringent response of Mycobacterium tuberculosis is required for long-term survival. J. Bacteriol. 182, 4889–4898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Domenech P., Barry Iii C. E., Cole S. T. (2001) Mycobacterium tuberculosis in the postgenomic age. Curr Opin in Microbiol 4, 28–34 [DOI] [PubMed] [Google Scholar]
- 64. Qamra R., Mande S. C., Coates A. R., Henderson B. (2005) The unusual chaperonins of Mycobacterium tuberculosis. Tuberculosis 85, 385–394 [DOI] [PubMed] [Google Scholar]
- 65. Joseph S. V., Madhavilatha G. K., Kumar R. A., Mundayoor S. (2012) Comparative analysis of mycobacterial truncated hemoglobin promoters and the groEL2 promoter in free-living and intracellular mycobacteria. Appl. Environ. Microbiol. 78, 6499–6506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gazdik M. A., McDonough K. A. (2005) Identification of cyclic AMP-regulated genes in Mycobacterium tuberculosis complex bacteria under low-oxygen conditions. J. Bacteriol. 187, 2681–2692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang Z., Hillas P. J., Ortiz de Montellano P. R. (1999) Reduction of peroxides and dinitrobenzenes by Mycobacterium tuberculosis thioredoxin and thioredoxin reductase. Arch. Biochem. Biophys. 363, 19–26 [DOI] [PubMed] [Google Scholar]
- 68. Murugasu-Oei B., Tay A., Dick T. (1999) Up-regulation of stress response genes and ABC transporters in anaerobic stationary-phase Mycobacterium smegmatis. Mol. Gen. Genet. 262, 677–682 [DOI] [PubMed] [Google Scholar]
- 69. Hu Y., Movahedzadeh F., Stoker N. G., Coates A. R. (2006) Deletion of the Mycobacterium tuberculosis alpha-crystallin-like hspX gene causes increased bacterial growth in vivo. Infect Immun 74, 861–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. DiCarlo J. E., Norville J. E., Mali P., Rios X., Aach J., Church G. M. (2013) Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 41, 4336–4343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Voskuil M. I., Schnappinger D., Visconti K. C., Harrell M. I., Dolganov G. M., Sherman D. R., Schoolnik G. K. (2003) Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J. Exp. Med. 198, 705–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zvi A., Ariel N., Fulkerson J., Sadoff J. C., Shafferman A. (2008) Whole genome identification of Mycobacterium tuberculosis vaccine candidates by comprehensive data mining and bioinformatic analysis. BMC Med. Genomics 1, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.