

Abstract
Tripeptidyl peptidase II (TPP2) is a serine peptidase involved in various biological processes, including antigen processing, cell growth, DNA repair, and neuropeptide mediated signaling. The underlying mechanisms of how a peptidase can influence this multitude of processes still remain unknown. We identified rapid proteomic changes in neuroblastoma cells following selective TPP2 inhibition using the known reversible inhibitor butabindide, as well as a new, more potent, and irreversible peptide phosphonate inhibitor. Our data show that TPP2 inhibition indirectly but rapidly decreases the levels of active, di-phosphorylated extracellular signal-regulated kinase 1 (ERK1) and ERK2 in the nucleus, thereby down-regulating signal transduction downstream of growth factors and mitogenic stimuli. We conclude that TPP2 mediates many important cellular functions by controlling ERK1 and ERK2 phosphorylation. For instance, we show that TPP2 inhibition of neurons in the hippocampus leads to an excessive strengthening of synapses, indicating that TPP2 activity is crucial for normal brain function.
Tripeptidyl peptidase II (TPP2) is a ubiquitously expressed, mainly cytoplasmic, 138kDa protein that forms homopolymer complexes of ≥6MDa (1). The N-terminal subtilisin-type serine peptidase domain becomes activated upon complex formation (2). The C terminus is important for complex formation but also contains two signature motifs, one of which is a suspected breast cancer gene 1 carboxy-terminal (BRCT) domain (1, 3). TPP2 complex assembly in vitro is enhanced by competitive peptide inhibitors (4). As the name suggests, TPP2 has exopeptidase activity and cleaves aminoterminal tripeptides from substrates (5). However, only a small number of peptide substrates have been identified (6–8). Additionally, TPP2 possesses a weak endopeptidase activity, of which only a few substrates, up to 75 amino acids in length, have been identified by in vitro digests using purified TPP2 (5, 9–11).
In contrast to the low number of polypeptides that have been established as bona fide TPP2 substrates, a myriad of important physiological processes and pathologies have been recognized to be mediated by this protein complex. That TPP2 is an essential protein is supported by the finding that homozygous knock down in mice and Caenorhabditis elegans is lethal (12). TPP2 is involved in antigen processing (13–18), cell growth, DNA damage repair and carcinogenesis (19–22), fat metabolism, feeding behavior, and obesity (12, 23). While the majority of those processes has been linked to TPP2 by describing individual phenotype and expressional changes of TPP2 as well as connected individual proteins (24–26), only antigen processing and feeding behavior have been directly related to TPP2 peptidase activity by identifying involved peptide substrates (6, 9, 10, 23). TPP2 has been shown to generate a few specific epitopes via its endopeptidase activity, for instance, for specific HLA allele mediated peptide presentation (10, 11, 17, 27). It has been suggested a role of TPP2 in obesity relies on exopeptidase cleavage of the satiety signaling neuropeptide cholecystokinin-8, which subsequently regulates feeding behavior (12, 17, 28). The uniform expression of TPP2 in the brain (29, 30), including regions that are not involved in the regulation of feeding behavior, suggests a hitherto general function in neuronal cells. One systematic study of transcriptomic changes following 10 days of TPP2 knock down in immortalized Burkitt lymphoma cells showed an influence of TPP2 on the transcription of genes encoding proteins involved in signal transduction like ERK2 and JNK (26). Until now, no systematic evaluation of proteins affected by TPP2 activity and/or expression has been reported.
As the basis for this study, we determined TPP2-mediated rapid proteomic changes in human neuroblastoma cells as a model system for neurons that are induced by the TPP2 inhibitors butabindide and B6, a novel in-house developed, highly potent, irreversible inhibitor. As indicated by the resulting data, we observed that TPP2 inhibition in mouse hippocampal neurons led to a strengthening of synapses, pointing toward an involvement of TPP2 in learning and memory. Additionally, the proteomic data revealed that TPP2 inhibition changes the expression of proteins that are linked to the ERK2 function, a connection we found to rely on a rapid reduction of phosphorylation level and thereby the activity of ERK1 and ERK2 in the nucleus of neuroblastoma cells. Our data support the view that TPP2 regulates a major signal transduction pathway, thereby influencing many cellular processes like development, proliferation, carcinogenesis and DNA-damage response (31–34), and synaptic strengthening (35, 36), indicating an important role for TPP2 in neuronal fate and function.
EXPERIMENTAL PROCEDURES
B6 Synthesis
Schematic presentation of synthesis strategy is shown in Fig. 1A, and a detailed description of the synthesis can be found in the supplementary file Experimental Procedure.
Fig. 1.

B6—A new irreversible TPP2 inhibitor- synthesis, structure, IC50 value, irreversibility, and stability. (A) Schematic synthesis strategy of B6. Detailed description of B6 synthesis and intermediate products can be found in the supplementary experimental procedure. Structure of B6 (compound 1) is marked in red. (B) IC50 value of B6 for inhibiting human TPP2 (hTPP2) in vitro. Different concentrations of B6 (red) and in parallel butabindide (blue) were incubated in vitro with purified hTPP2. The activity was determined via the TPP2 substrate AAF-pNA and measured as time-dependent fluorescence of free pNA (a.u./min). An average line is shown. A schematic experimental setup is presented below the graph. Activity relative to noninhibited TPP2 in percentage is plotted against inhibitor concentration (n = 3, ±S.D.). IC50, as determined by the tangent equation of the regression function for B6 (red) and as reference butabindide (blue) are indicated. (C) Binding of B6 to TPP2 is irreversible compared with butabindide. Different concentrations of B6 (red curves, left side) and in parallel butabindide (blue curve, right side) were applied to purified human TPP2 and either preincubated (30 min, dashed line) and then diluted 1/20 (v/v) or directly diluted (solid curve). The activity was determined via the TPP2 substrate AAF-pNA and measured as time-dependent fluorescence of free pNA (a.u./min). A schematic experimental setup is presented below the graph. Activity relative to noninhibited TPP2 in percentage (n = 3, ±S.D.) is plotted against final inhibitor concentration after dilution. (D) Stability of B6 under mimicked physiological conditions (310 K, in PBS). Stock solutions of B6 or butabindide (10 mm in DMSO) were diluted in PBS, pH 7.2 (cfinal = 20 μm), an aliquot (200 pmol) was directly(0 h), after 4 h, and after 24 h incubation at 310 K applied to LC-MS. Graph shows signal intensity (MS) of B6 ([1+H]+ m/z 550) and butabindide ([3+H]+ m/z 304) normalized to 0 h value (n = 3, ±S.D.). d.I and d.II represent the chemical reactions of B6 and butabindide as observed under mimicked physiological conditions and determined by identification of the products using mass spectrometry.
TPP2 Purification
TPP2 was purified from human red blood cells as described previously (8), using the reported modifications (37, 38). The enzyme was frozen in liquid nitrogen and stored at −80 °C.
IC50 Value and TPP2 Irreversibility
For determination of the IC50 value, human TPP2 (hsTPP2) was incubated with butabindide (Tocris Biosciences, Bristol, UK) or B6 at concentrations between 5 pm and 1 μm in 0.1 m potassium phosphate buffer, pH 7.5, containing 12% glycerol and 2 mm DTT for 15 min at 37 °C before addition of 100 μm H-Ala-Ala-Phe-pNA (AAF-pNA). The change in absorbance at 405 nm was measured continuously for 45 min (Fig. 1B). For the irreversibility assay, hsTPP2 was incubated with the inhibitor at 20-fold higher concentration for 0 min or 30 min at 37 °C, then diluted 20-fold and added to the 96-well plate. The substrate (AAF-pNA, Bachem, Bubendorf, Switzerland) at 100 μm was added after an additional 5 min preincubation at 37 °C. The final concentration during the activity measurement is indicated (Fig. 1C). Absorbance resulting from pNA (p-nitroaniline, A = 405–410 nm) release due to AAF-pNa cleavage by hsTPP2 was detected over time. The resulting hsTPP2 activity is represented by absorbance in time and was normalized to TPP2 activity in absence of inhibitors. The IC50 value was determined using the tangent equation of the regression function.
TPP2 Activity Assay
neuroblastoma cellline SH-SY5Y cells transduced with shRNA or incubated for 4 h with Dimethyl Sulfoxide (DMSO), butabindide or B6 (concentrations as indicated in Fig. 2A) were lysed in 25 μm digitonin (Sigma Aldrich, Buchs, Switzerland) in KMH buffer (110 mm KAc, 2 mm MgAc, and 20 mm Hepes-KOH, pH 7.2). Organotypic hippocampal slices were isolated from P7 mice, and cultured for 6–13 days in vitro (DIV) in culture medium. After incubation for 6 h with 1% (v/v) DMSO, 10 or 100 μm B6 in slice culture medium, brain slices were homogenized in PBS containing 25 μm digitonin (data not shown). Lysates were incubated for 30 min on ice and centrifuged for 15 min at 14,000 rpm at 4 °C. 50 μm bestatin (Enzo lifesciences AG, Farmingdale, NY) were added to 5 μg protein from the cell lysates in KMH buffer and incubated for 30 min at 4 °C. Subsequently, 100 μm AAF-AMC (Bachem) was added to a total volume of 50 μl. Degradation of AAF-AMC was analyzed at 37 °C using the FLUOstar OPTIMA (BMG Labtec, Ortenberg, Germany). The reaction was incubated for 30 min on ice and centrifuged for 15 min at 14,000 rpm at 4 °C. The remaining TPP2 activity was determined using AAF-AMC degradation assay in presence of bestatin. The graph shows mean relative TPP2 activity normalized to DMSO-treated brain lysates (n = 3, ±S.D.).
Fig. 2.
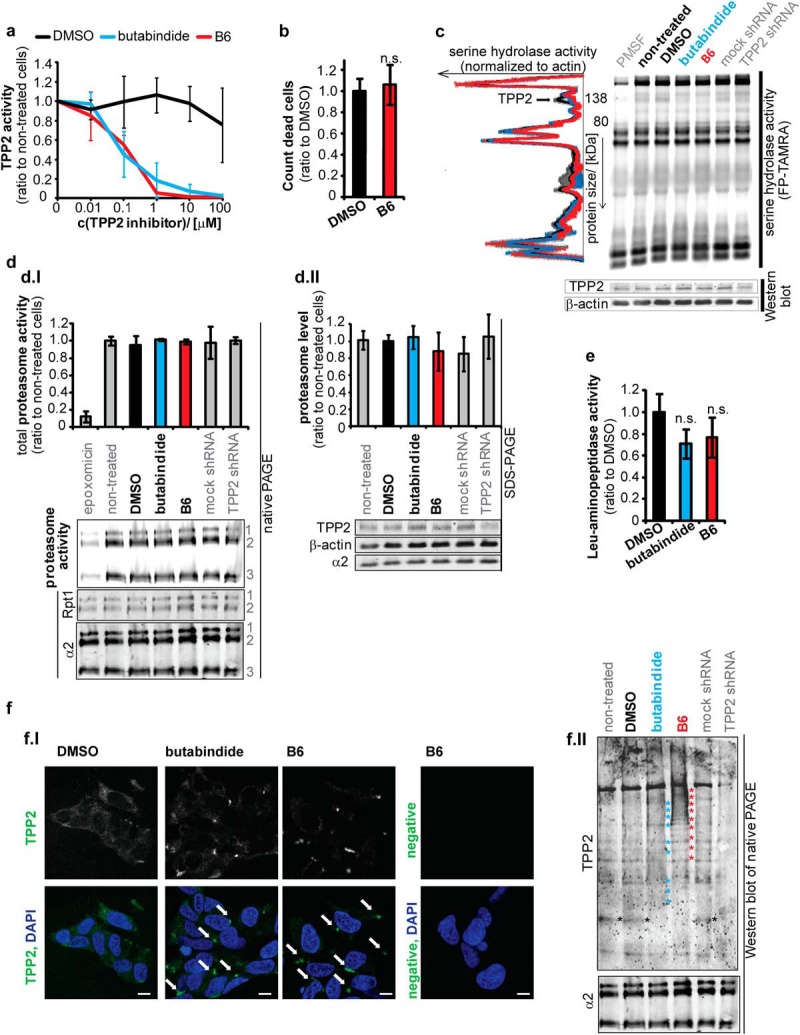
B6 specificity and effects in cellular application. (A) Optimum concentration to inhibit TPP2 is 1 μm B6. SH-SY5Y cells were incubated for 4 h with different concentrations of B6, butabindide or respective amounts of the inhibitor solvent DMSO. TPP2 activity in cell lysates was determined by an AAF-AMC degradation assay (in presence of bestatin). Graph: mean TPP2 activity normalized to nontreated cells (n = 3, ±S.D.). (B)–(F) Effects of 4 h 1 μm TPP2 inhibition and knock down in SH-SY5Y cells. (B) No effect of B6 on cell death. Mean amount of trypan blue positive cells normalized to DMSO control (n = 3, ±S.D.) (C) No significant change of serine hydrolases activity by B6. SDS-PAGE of cell lysates (SH-SY5Y 4 h treated with DMSO or TPP2 inhibitors B6/butabindide) incubated with TAMRA-FP serine hydrolase activity based probe (negative control lysate pretreated with 100 μm PMSF). Representative 12.5% SDS-PAGels (additional 7.5% SDS-PAGels see Supplemental Fig. 2A): serine hydrolase activity (TAMRA signal), β-actin, and TPP2 level (WB) and respective plotted profiles of TAMRA signal normalized to β-actin. Graph: pool of DMSO and nontreated cells (black ±S.D.); butabindide (blue) and B6 (red) treated cells (n = 2, mean ± S.D.). (D) No significant change in proteasome level, activity or complex formation. (D.I) Proteasome activity and complex formation. Bar graph represents mean normalized proteasome activity (sum band 1–3: BODIPY/α2 WB signal) (n = 3, ±S.D.) as quantified from native-PAGE. Below the graph representative native PAGE (4–12% gradient gels) shows proteasome activity (BODIPY signal, negative control epoxomicin treatment previous to activity probe application) and respective Western blot (WB) shows α2 (20S proteasome) and RPT1 (19S proteasome activator) (1) indicates 30S-, (2) PA28–20S-19S- and 20–19S-, (3) PA28–20S- and 20S-proteasome complexes. (D.II) proteasome level. Bar graph represents mean normalized proteasome level (α2/β-actin) (n = 3, ±S.D.) as quantified from SDS-PAGE. Representative Western blot showing α2, TPP2, and β-actin level is presented below the graph (E) No significant effect of B6 leucyl-aminopeptidases. Relative change of l-AMC degradation measured in cell lysates of B6 and butabindide treated cells normalized to DMSO control (n = 3, ±S.D., n.s. = nonsignificant p > .05). (F) B6 influences TPP2 complex assembly/stability and localization. (F.I) Representative confocal pictures of TPP2 immunostain in greyscale and indicated green in overlay with DAPI. Arrows mark cytoplasmic accumulations of TPP2 in presence of both TPP2 inhibitors. White scale bar represents 10 μm. Negative means represents background stain of the secondary antibody and autofluorescence of the cells. Picture contrast for all pictures has been adjusted equally via Adobe Photoshop. (F.II) Representative Western blot of native PAGE stained with TPP2 antibody and as loading control α2. Stars (red and blue) indicate shifts toward lower percentage (bulkier complexes) for butabindide and B6. Black stars indicate small complex or monomeric form of TPP2 that is only present in noninhibited cell lysates. TPP2 shRNA (60% knock down) serves as control for the staining.
Inhibitor Stability
Stock solutions of 10 mm B6 or butabindide in DMSO were diluted in PBS, pH 7.2 (cfinal = 20 μm). An aliquot (200 pmol) was directly applied to LC-MS (0 h). Further aliquots were taken after 4 and 24 h incubation at 37 °C and analyzed (for results, see Fig. 1D). Before samples were applied to LC-MS (see Florea et al. (39) for instrument details not listed), acetonitrile (ACN, end v/v 5%) and formic acid (pHend = 3) were added. The column was packed with BioSphere C18 5-μm 120-Å particles from (Nanoseparations, Nieuwkoop, the Netherlands). Samples (injection volume 10 μl) were separated in the presence of 0.1% formic acid using a 10 min gradient of 20–35% acetonitrile and 80–65% water on a Surveyor-LCQ system (Thermo, Waltham, MA) with a flow rate of 1 ml/min. The samples were analyzed on a LTQ-Orbitrap (Thermo) using the following parameters: source 3.5 kV and 7 μA; capillary 32 V and 275 °C; tube lens 105 V; scan event details: full MS scan with a mass resolution of 30,000 (Fourier Transform Mass Spectrometer (FTMS)+ pnorm) with a m/z range 150–1,200; for MS2 and MS3: m/z range 100–2,000, resolution 15,000, normalized collision energy 35%, activation q = 0.25 and t = 30 ms, IsoW 1.0. Resulting raw data were analyzed using X.calibur software (version 2.0.7 from Thermo Fisher Scientific).
ESI-MS B6 (compound 1) Rt(8.2 min) m/z 550 [3+H]+; MS2 550: 465 (100) [3-C4H7NO+H]+; MS3 465: 231 (100) [(3-C4H7NO)-C12H11O3p + H]+, 320 (23) [(3-C4H7NO)-C9H7NO+H]+; B6 hydrolysis product (compound 3) Rt(6.7 min) m/z: 474 [4+H]+; MS2 474: 380 (100) [4-C6H6O+H]+; 389 (95) [4- C4H7NO+H]+; MS3 380: 298 (100) [(4-C6H6O)-HPO2-H2O+H]+; 213 (33) [(4-C6H6O)-C5H12NPO2-H2O+H]+; 362 (15) [(4-C6H6O)-H2O+H]+; MS3 389: 231 (100) [(4-C4H7NO)-C6H6O3p + H]+; 244 (50) [(4-C4H7NO)-C9H7NO+H]+.
ESI-MS Butabindide (compound 2) Rt(1.2 min) m/z 304 [5+H]+; butabindide cyclization product (compound 4) Rt(4.7 min) m/z 231 [6+H]+.
Generation and Validation of the TPP2 Knockdown Cell Line
The lentiviral shRNA TPP2-specific sequence used was CCGGCCTGAT CCTTTCAGGT CTGAACTCGA GTTCAGACCT GAAAGGATCA GGTTTTTG (Sigma Aldrich, Buchs, Switzerland). The SHC002 scrambled shRNA construct (Sigma-Aldrich) was used as a negative control. All shRNA constructs were in the pLKO.1 vector backbone. shRNA-expressing lentiviral particles were prepared using HEK293T cells and the virus was transduced into SH-SY5Y cells as described previously (40). The knock down of TPP2 was confirmed by qPCR (see section RNA Isolation and Quantitative PCR) on the protein level by Western blotting, and TPP2 activity was measured using the AAF-AMC TPP2 activity assay (see section TPP2 Activity Assay). The TPP2 shRNA transduced cell line that was used here showed a decrease in TPP2 mRNA and protein level as well as TPP2 activity of 60–65% compared with a mock shRNA stable cell line as well as wild-type SH-SY5Y cells (see Supplemental Figs. 2D and 2E). Transductions with other shRNA sequences did not grow and consequently died, as in agreement with literature (12).
Transfection
1 μg plasmid GFP-ERK2 (kindly provided by P. Stork, Oregon Health & Science University, Portland, OR) (41) was transfected into SH-SY5Y cells using Lipofectamine2000 (Invitrogen Thermo Fisher Scientific Inc., Paisley, UK) using the protocol recommended by the company.
Cell Culture
SH-SY5Y cells were incubated at 37 °C and 5% CO2. General culture medium was DMEM high glucose supplemented with 10% FCS, 2 mm l-glutamine, and penicillin/streptomycin (100 U/ml). For stable isotope labelling with amino acids in cell culture Stable isotope labeling by amino acids in cell culture (SILAC) experiments, cells were cultured in DMEM high glucose without K and R (GE Healthcare, Buckinghamshire, UK) supplemented with 10% dialyzed FCS, 2 mm l-glutamine, penicillin/streptomycin (100 U/ml), 0.8 mm l-lysine either K+4Da (H2N(CH2)2(CD2)2CH(NH2)CO2H · 2HCl) or K+8Da (H215N(13CH2)413CH(15NH2)13CO2H · 2HCl), and 0.4 mm l-arginine R+6Da (13C6H14N4O2 · HCl) or R+10Da (13C6H1415N4O2 · HCl) for 48 h. Amino acid stable isotopes were purchased from EUR-ISOTOP. Inhibition of TPP2 was performed by supplementing culture medium with 1 μm butabindide or 1 μm B6 (1/10,000 (v/v) from 10 mm stock solution dissolved in DMSO) and, respectively, 1/10,000 (v/v) DMSO and incubating cells for 4 h (in the case of SILAC, the last 4 h of 48 h labeling), except for concentration optimization experiments. For experiments under stimulated phosphorylation of ERK1 and ERK2, 100 ng/ml of Phorbol-12-myristate 13-acetate (PMA) was added 15 min before harvesting the cells.
Proteome Analysis
Cells were trypsinized. After washing with PBS, cell pellets were lysed (50 mm Tris, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% TritonX-100, complete inhibitor mix (Roche), 100 mm DTT) and directly incubated at 95 °C for 3 min. Afterward, lysates were sonicated and centrifuged (15 min, 10,000 rpm). Protein concentration was determined. Medium heavy labeled lysate (control condition) was pooled with heavy labeled lysate (TPP2 inhibition or knock down) to achieve a protein ratio of 1:1. The ethanol-precipitated protein pellet was dissolved in 6N urea/2N thiourea. After incubation with 55 mm iodoacetamide for 30 min at RT in the dark, proteins were predigested with lysyl-C endopeptidase (WakoChemicals, Richmond, VA) for 3 h at RT then diluted (5xvol 50 mm ammonium bicarbonate (ABC))1 and digested with trypsin (Promega, Fitchburg, WI) at RT overnight. After acidic deactivation of trypsin, peptides were desalted via C18-Stage-Tip purification. (42) Peptides were separated via SCX-chromatography using settings adapted from Brunner et al. (43). The resulting 24 fractions were desalted via C18-Stage-Tip purification (42), and peptide samples were analyzed on a Surveyor nano-LC system (Thermo) connected to an LTQ-Orbitrap mass spectrometer (Thermo). The column was packed with BioSphere C18 5-μm 120-Å particles from Nanoseparations. Instrument settings and measuring parameters were used as described by Florea et al. (39). Overall, three biological replicates for each (a) TPP2 knock down to mock shRNA and (b) TPP2 inhibition by B6/DMSO and four biological replicates for (c) TPP2 inhibition by butabindide/DMSO were prepared and the resulting raw data were analyzed by MaxQuant software (version 1.2.2.5) (44, 45) using the Andromeda search algorithm against the ipi.HUMAN.v3.68 database (both versions from June 2012) in one analysis. Parameters used for peptide based protein identification and quantification in MaxQuant: tryptic cleavage, max. two miscleavages; variable modification of methionines by oxidation and of alpha-aminogroup by acetylation; fixed modification on cysteines by carbamidomethylation; special amino acids K and R; multiplicity of 3; no deisotoping; mass tolerance for precursor ions 20 ppm and for fragmented peptides of 0.5 Da; minimum peptide length of six amino acids; and a max. false discovery rate of 0.01 (peptide and protein level). The quantification was based on nonmodified peptides and peptides with variable modifications (razor and unique), low-scoring versions of peptides were not used; min. ratio count of a replicate was 2. Data from MaxQuant analysis were processed using Perseus software version 1.5.0.15 (44). Quantifications (normalized ratio H/M) were transformed in log2FC. Protein matches from reversed peptide database, contaminants, and only identified hits were deleted. Proteins quantified in less than three replicates of knock down or both inhibitors were deleted. P1 values of knock down (n = 3), butabindide (n = 4) and B6 (n = 4), as well as for all possible combinations of two of these sample sets, were determined. Additionally, Significance B value (44) of the median relative quantifications for knock down, butabindide and B6 was determined (log2FC of each protein toward Intensity of M+H of respective labeled peptides). Only proteins were included in the protein list for TPP2-induced proteomic changes that were more than 1.25-fold changed in the same direction in a minimum of two out of the three screens (two inhibitors and TPP2 knock down), with significance B and P1 value of p ≤ .05 (for relevant quantifications). Heat maps (with produced Perseus software version 1.5.0.15) of those proteins are presented in Fig. 3B; quantification results and statistical values can be found in Supplemental Table 1 (online). A schematic presentation of the experimental procedure can be found in Fig. 3A. A list of proteins significantly changed in only one out of the three screens (that have been stated (numbers) in the Venn diagram Fig. 3A) can be found online in Supplemental Table II.
Fig. 3.

SILAC approach and resulting proteins affected by TPP2 inhibition and/or knock down. (A) Schematic presentation of experimental setup to determine relative proteomic changes. Venn diagrams represents proteins reproducible quantified (P1 and significance B(Log2FC versus Intensity); p < .05) that are minimum 1.25-fold (inhibitors) or 1.5-fold (knock down) increased (upper Venn diagram) or decreased (lower Venn diagram). For details of quantification see online Supplemental Table I for the overlapping proteins and online Supplemental Table II for the proteins significantly changed by only one condition. (B) Heat maps of median Log2FC of B6, butabindide and TPP2 knock down representing targets (first gene name of MaxQuant hit list) showing the same change in minimum two out of three conditions (quantification details and protein names see Supplemental Table I).
Approximate Protein Half-Life Determination
An aliquot of each separate SILAC-labeled (48h labeling) cell lysate was taken for determination of an approximate protein turnover/half-life, prepared and analyzed the same way (MaxQuant multiplicity 2 instead). The half-life/turnover of the proteins TPP2 was calculated from SILAC quantifications of aliquots using the Eqs. (Ia) and (Ib) are in Supplementary Fig. 1). For a schematic presentation of experimental procedure and calculation of the approximate t1/2 (approximate due to expected minor effects of natural isotope recycling in 48 h of SILAC) for TPP2, see Supplemental Fig. 1.
Biological Process and Functional Interaction Annotation Analysis of Proteomic Data
The overlapping proteins influenced by TPP2 as determined by SILAC (Fig. 3B and online Supplemental Table I) and functional interactors or linkers were applied to the pathway analysis tool Cytoscape (version 3.0.2) and overrepresented GO biological processes that were significantly (false discovery rate and p value <.05) influenced by TPP2 were analyzed using FI Reactome app (46, 47). The results are presented in Table I. Furthermore, with using the same proteins and selection criteria overrepresented pathways were determined and are presented online in Supplemental Table III. Functional interactions of proteins increase or decrease by both inhibitors were also analyzed by Cytoscape (see Supplemental Fig. 3 and Supplemental Table IV (online)).
Table I. GO biological pathways (FDR<0.05, p < .05) of overlapping increased & decreased proteins between different SILAC screens (listed in Fig. 3). Gene symbols represent proteins increased or decreased by TPP2 inhibition as well as functional interactors (linker proteins, see Supplemental Fig. 3).
| GO biological process (BP) | Proteins in BP | Proteins changed | p value | FDR | Including linker proteins |
|---|---|---|---|---|---|
| Positive regulation of transcription from RNA polymerase II promoter | 636 | 15 | 0.0E+00 | 5.0E-03 | MYOD1,CTNNB1,MAZ,SOX11,NME2,EP300,JUN,MAVS,SOX4,HAND2,POU3F3,TAF1,SREBF2,HDAC2,SETD3 |
| Noradrenergic neuron differentiation | 5 | 3 | 0.0E+00 | 2.5E-03 | SOX11,SOX4,HAND2 |
| Negative regulation of cardiac muscle cell proliferation | 7 | 3 | 0.0E+00 | 4.7E-03 | CXADR,RBP4,HDAC2 |
| Glood coagulation | 384 | 10 | 1.0E-04 | 2.3E-02 | GNA11,CXADR,KIF2B,PRKAR2B,PRKACA,MAPK1,JMJD1C,HDAC2,FYN,PLCG2 |
| Heart development | 128 | 6 | 1.0E-04 | 2.2E-02 | GNA11,CXADR,EP300,RBP4,SOX4, HAND2 |
| Sympathetic nervous system development | 16 | 3 | 1.0E-04 | 1.9E-02 | SOX11,SOX4,HAND2 |
| Fibroblast growth factor receptor signaling pathway | 131 | 6 | 1.0E-04 | 2.0E-02 | PRKAR2B,SPRY2,PRKACA,MAPK1,PPP2CA,FYN |
| Glial cell proliferation | 3 | 2 | 2.0E-04 | 1.9E-02 | SOX11,SOX4 |
| Glial cell development | 3 | 2 | 2.0E-04 | 1.9E-02 | SOX11,SOX4 |
| Epidermal growth factor receptor signaling pathway | 148 | 6 | 3.0E-04 | 3.0E-02 | PRKAR2B,SPRY2,PRKACA,PIK3C2A, MAPK1,FYN |
| Cardiac ventricle formation | 4 | 2 | 3.0E-04 | 2.6E-02 | SOX11,SOX4 |
| Neuroepithelial cell differentiation | 4 | 2 | 3.0E-04 | 2.6E-02 | SOX11,SOX4 |
| Regulation of secondary heart field cardioblast proliferation | 4 | 2 | 3.0E-04 | 2.6E-02 | CTNNB1,HAND2 |
| UTP biosynthetic process | 6 | 2 | 6.0E-04 | 4.4E-02 | NME4,NME2 |
| GTP biosynthetic process | 6 | 2 | 6.0E-04 | 4.4E-02 | NME4,NME2 |
| Peptidyl-threonine phosphorylation | 29 | 3 | 7.0E-04 | 4.6E-02 | MAPK1,TAF1,MAP2 |
| Learning | 30 | 3 | 8.0E-04 | 4.9E-02 | PRKAR2B,JUN,FYN |
| DNA damage response, detection of DNA damage | 7 | 2 | 8.0E-04 | 4.6E-02 | MRPS9,SOX4 |
| CTP biosynthetic process | 7 | 2 | 8.0E-04 | 4.6E-02 | NME4,NME2 |
| Positive regulation of proteasomal ubiquitin-dependent protein catabolic process | 32 | 3 | 1.0E-03 | 4.8E-02 | TAF1,VCP,CSNK1E |
| Protein heterotrimerization | 8 | 2 | 1.1E-03 | 4.9E-02 | PPP2CA,COL6A1 |
| Neural tube formation | 8 | 2 | 1.1E-03 | 4.9E-02 | SOX11,SOX4 |
| Positive regulation of transcription, DNA-dependent | 436 | 9 | 1.1E-03 | 4.8E-02 | SOX11,MAPK1,JUN,SOX4,HAND2, POU3F3,HDAC2,TCF12,SETD3 |
Serine Hydrolase Activity Determination
SH-SY5Y cells were lysed in 25 μm digitonin (Sigma Aldrich) in PBS. Protein concentration was determined via Bradford assay. 20 μg protein were incubated for 30 min on ice in the presence of 2 μm ActiveX TAMRA-fluorophosphonate (FP) Serine hydrolase probe (Thermo Fisher Scientific). As a negative control, lysate was preincubated with 20 μm PMSF (Bachem) (30 min on ice) before adding the probe. After incubation, 6xSDS-loading buffer was added and the samples were incubated at 95 °C for 5 min and separated via SDS-PAGE (7.5 and 12.5% SDS-PAGels). Gels were scanned using a Typhoon imager (GE Healthcare, Buckinghamshire, UK) with the 580 BP 30 filter to detect the TAMRA-FP probe and afterward Western blotted for evaluating the loading control (Fig. 2C and Supplemental Fig 2A).
Proteasome Activity Determination
SH-SY5Y cells were lysed in TSDG buffer: 20 mm Tris-HCl, pH 7.6, 10 mm KCl, 2 mm MgCl2, 0.1 mm EDTA, 1 mm DTT, 10% glycerol containing 2 mm ATP via three freeze–thaw cycles. Protein concentration was determined by Bradford assay. 20 μg protein were incubated for 1 h at 37 °C in the presence of 0.5 μm activity-based probe BODIPY-epoxomicin (kindly provided by H. Overkleeft (Institute of Chemistry, Leiden)) (39). As a negative control, lysate was preincubated for 30 min on ice with 1 μm epoxomicin (Sigma Aldrich) before adding the activity probe. After incubation, 6x native-loading buffer was added and samples were separated by nativePAGETM NuPAGE® Novex® 4–12% Bis-Tris gels (Invitrogen). Gels were scanned using Typhoon imager (GE Healthcare) with the 580 BP 30 filter to detect the BODIPY-epoxomicin probe and afterward Western blotted for evaluating the loading control (Fig. 2D).
Leucyl-Aminopeptidase Activity Determination
SH-SY5Y cells were lysed in 25 μm digitonin (Sigma Aldrich) in KMH buffer (110 mm KAc, 2 mm MgAc, 20 mm Hepes-KOH, pH 7.2). Cell lysates were incubated for 30 min on ice and centrifuged for 15 min at 14,000 rpm at 4 °C. 100 μm l-AMC (Sigma Aldrich) were subsequently added to 5 μg protein cell lysate in KMH buffer (total volume: 50 μl). Degradation of l-AMC was analyzed at 37 °C using the FLUOstar OPTIMA (BMG Labtec.). See Fig. 2E.
Trypan Blue Assay
Cells were trypsinized and centrifuged (1,200 rpm, RT, 3 min). The cell pellet was resolved in cell culture medium. An aliquot of cell suspension was mixed (1:10) with Trypan blue solution and applied to a cell counter chamber. After 3 min, cells and dead cells (Trypan blue positive) were counted (∼400 cells per experiment counted). See Fig. 2B.
RNA Isolation and Quantitative PCR
RNA from SH-SY5Y cells was isolated using Trizol (Invitrogen, Waltham, MA) and an overnight precipitation in isopropanol. Total RNA (1 μg) was DNase I treated and used to generate cDNA (Maxima First Strand cDNA synthesis kit; Fermentes, Thermo Fisher Scientific) using oligo-dT and random hexamer primers. In short, 1 μg RNA was mixed with 4 μl 5X Reaction and 2 μl Maxima Enzyme Mix, and water was added to a total volume of 20 μl. This solution was incubated at 25 °C for 10 min, followed by 50 °C for 15 min, and the reaction was terminated by heating at 85 °C for 5 min. The resulting cDNA was diluted 1:20 and served as a template in real-time qPCR assays. Real-time qPCR (SYBR® Green PCR Master Mix; Applied Biosystems, Thermo Fisher Scientific) for target proteins was performed using the forward and reverse primer pairs as shown below. Expression levels were normalized against a selection of four reference genes (GAPDH, ACTB, TBP, and YWHAZ) based on a geNorm analysis (Fig. 4A). The normalization factor was the geomean of the four reference genes.
Fig. 4.

(A) Only SOX11 mRNA is increased by 4 h TPP2 inhibition with butabindide but not with B6. Mean relative change in mRNA level (qPCR, n = 3, ±S.D.) of B6/DMSO; butabindide/DMSO; TPP2/mock shRNA stable cell lines. (B) and (C) SOX11 protein level is increased by 4 h TPP2 inhibition. (B) Bar diagram shows SOX11 protein level (normalized to DMSO control). SILAC quantifications (median, n = 3, ±S.D.), WB quantification (mean of summed bands marked with stars, n = 6, ±S.D., WB see Supplemental Fig. 4). (* indicates .05≥p ≥ .01; ** p ≤ .01). (C) Representative Western blots of SOX11 total lysate using an antibody targeting the region where all three SILAC-based quantified peptides are located (see (D)). Stars indicate quantified bands that were summed up. 47 kDa represents SOX11 full length; the other bands are assumed to be posttranslational modifications (>47 kDa) and cleavage products (<47 kDa). (D) Localization of SOX11 peptides as quantified by SILAC and SOX11 antibody binding specificity upper row shows: SILAC-quantified peptides (1–3 green) located in the HMG DNA-binding domain (amino acid 49–117) of SOX11. The middle row shows the amino acid sequence and position in SOX1. The lower row represents the epitope used to generate the SOX11 antibody (Atlas Antibodies). (E) and (F) Increased SOX11 protein level is not a result of increased protein stability. (E) Representative WB of TPP2-inhibitor-treated cells in absence and presence of the protein synthesis inhibitor cycloheximide (CHX). (F) Mean relative changes (WB quantifications of summed bands marked with stars, n = 5, ±S.D., WBs see Supplemental Fig. 4) of protein level in absence and in presence of the protein synthesis inhibitor CHX.
Cycloheximide Chase
The stability of the Sry-related HMG box (SOX)11 protein was assayed by addition of cycloheximide (50 μm) (Sigma Aldrich) to SH-SY5Y cells incubated with either B6 1 μm, butabindide 1 μm or 1/10,000 (v/v) DMSO for 4 h. Subsequently, cells were applied to nuclear-cytoplasmic fractionation and analyzed by Western blotting (Figs. 4C and 4D). All SOX11-positive bands present on the Western blot (marked with * in Fig. 4) were quantified, summed up, and normalized to the respective loading controls. The band at 47 kDa represents SOX11 full-length unmodified protein; the main band ∼60 kDa is assumed to be posttranslational modified and the bands below 47 kDa to be cleavage products of the intronless transcription factor SOX11. Since 2/3 of the three SILAC-quantified SOX11 peptides (Fig. 4D and Supplemental Table VI) are homologous to SOX4 (47.3 kDa) and 1/3 is homologue to SOX12 alias SOX22 (36 kDa) and since the SOX11 antibody epitope sequence (Fig 4D) shows 94% identity to SOX4 and 82% identity to SOX12 alias SOX22 (36 kDa), the SOX11 antibody may also detect SOX4 and SOX12 as the two other SILAC matches. We cannot exclude that some of the quantified bands in the SOX11 bands (indicated with stars in Fig. 4) overlap or are SOX4 or SOX12; however, it is most comparable to the SILAC quantification. We deliberately marked all Western blots with SOX11.
Nuclear-Cytoplasmic Fractionation
Cells were lysed by incubation for 15 min on ice in hypotonic buffer (10 mm MgSO4, 37.5 mm KCl, 5 mm Hepes, pH 8.0) containing complete inhibitor mix (Roche, Basel, Switserland), 50 μm PMSF, 1 μm epoxomicin, 10 mm NEM, 2 mm EDTA. Then 0.6% TritonX-100 was added and lysates were syringed through a 22 gauge needle before lysates were centrifuged (10 min, 10,000 rpm, 4 °C). The supernatant, representing the cytoplasmic fraction, was transferred into a new tube. The pellet was lysed in nuclear extraction buffer (100 mm Tris, pH 7.4; 2 mm Na3VO3; 100 mm NaCl; 1% TritonX-100; 2 mm EDTA; 10% glycerol; 0.1% SDS; 1 mm NaF; 0.5% deoxycholate; 20 mm Na2P2O7; compete inhibitor mix (Roche)) by incubating it for 40 min on ice and vortexing every 10 min for 15 s. Supernatant respective nuclear fraction after centrifugation (10 min, 10,000 rpm) was transferred into a fresh tube. The protein concentration of fractions was determined via Bradford, and 20 μg of each fraction was applied to SDS-PAGE.
Western Blotting
Equal protein amounts obtained from the SH-SY5Y cell lysates were separated on 7.5%, 12.5%, 15% SDS-PAGE gels or Native gels (Invitrogen). After electrophoresis, proteins were transferred to a 0.2 μm pore size nitrocellulose membrane (GE Healthcare). Blots were blocked in 5% milk (w/w) in PBS and incubated with the primary antibodies and subsequently with secondary antibodies from ODYSSEY® Infrared Imaging System anti-goat, anti-mouse, anti-rabbit generated in donkey with IRDye® 800CW or 680CW (1:10,000; LI-COR Biosciences, Lincoln, NE). The signal was detected using the Odyssey imaging system (Licor) and quantified using ImageJ software (version 1.46r Wayne Rasband, NIH, USA).
Antibodies: from Santa Cruz Biotechnology Dalllas, TX: TPP2 (E-17, sc-15148) goat (WB 1:500), PPP2CA/B (FL-309, sc14020) rabbit (WB 1:1,000), β-actin (I-19, sc-1616) goat (WB 1:2,000), ubiquitin (P4D1, sc-8017) mouse (WB 1:1,000); from Atlas Antibodies, Stockholm, Sweden SOX11 (HPA000536) rabbit (WB 1:750); from Sigma Aldrich, Cambridge, UK: pERK1/2 (M8159) mouse (WB 1:10,000); TPP2 (HPA021069) rabbit (ICC 1:50); from abcam: Histone H3 (1791–100) rabbit (WB 1:3,000), α-tubulin (DM1A, ab7291) mouse (WB 1:10,000, ICC 1:200), α2 proteasomal subunit (MCP21, ab22666) mouse (WB 1:1,000); from Biomol, Plymouth Meeting, PA: RPT1/S7 (PW8825) mouse (WB 1:1,000).
Immunostaining
Cells grown on coverslips were treated with respective conditions then washed with PBS and fixed with 2% paraformaldehyde (PFA; Electron Microscopy Sciences, Hatfield, PA) in PBS (20 min, 4 °C). Proteins were denatured with methanol (5 min, RT), cells permeabilized with 0.1% Triton-X100 in PBS (10 min, RT), and unspecific binding sides were blocked with 1% BSA in PBS (10 min, RT). Fixed cells were incubated with antibody against TPP2 generated in goat (1:50, Santa Cruz, E-17, sc-15148) for 1 h at room temperature, washed with PBS, and then incubated for 30 min in the dark with Cy3-tagged anti-goat generated in donkey (1:500, Jackson, Bar Harbor, ME). After washing with PBS, coverslips were mounted with DAPI containing VectaShield® (Vector Laboratories, Inc., Burlingame, CA), and staining was investigated using a Leica TCS SP8 X Confocal Microscope with the software LAS AF Version 2.6.3 (Leica Microsystems, Wetzlar, Germany).
Quantification of Nuclear Protein via Confocal Microscopy
For confocal image based nuclear protein quantification, confocal images (with identical magnification and settings (gain, offset, laser power) were analyzed using Leica AF software by manually marking the nuclear area based on the DAPI staining. The mean of the mean gray values (channel of TPP2 staining) of each nucleus (n = 3 biological replicates, 14 images per sample with >200 nuclei quantified) was determined for DMSO, butabindide, and B6 treated cells as well as cells treated with secondary antibody only. The least named negative control was seen as background fluorescence and subtracted from quantifications of DMSO, butabindide, and B6 before the values were normalized to DMSO (separately for each biological replicate). Exemplar presentation of quantification method is shown in Supplemental Fig. 6.
Neurite Outgrowth Analysis
To measure neurite outgrowth, SH-SY5Y cells were plated on a 96-well plate grown in DMEM 10% FCS and incubated at 37 °C and 5% CO2. The cells were either treated with B6 1 μm, butabindide 1 μm, or 1/10,000 DMSO for 4 h. As a positive control, cells were plated on a 96-well plate coated with poly-l-lysine and BD MatrigelTM Basement Membrane Matrix (growth factor induced) (1 mg/ml) and grown for 10 days in 1 μm retinoid acid dissolved in DMEM 0.5% FCS to induce differentiation. Retinoic acid medium was refreshed on day 3 and 6 after plating. Cells were fixed with 2% PFA in PBS for 20 min, washed with PBS, and subsequently permeabilized in 0.1% Triton-X in PBS for 10 min and blocked with 1% BSA in PBS for 20 min. Then, the primary antibody mouse α-tubulin (1:200 in 1% BSA in PBS) was added for 1 h at room temperature, followed by the secondary antibody Alexa Fluor® 488 chicken anti-mouse (A21200, 1:500, from Invitrogent, Waltham, MA) for 30 min incubation at room temperature. Neurite outgrowth was measured using the Cellomics Arrayscan VTI HCS Reader. Filters used were XF93-FITC for detecting Alexa 488 anti-mouse and XF93-Hoechst for detecting the Hoechst staining. Predefined mask algorithms were used to measure neurite length. For analysis five (four in case of retinoic acid) biological replicates, and for each of them, two technical replicates were used (Figs. 5A and 5B). For each technical replicate, 40 images were acquired, using a 10x objective.
Fig. 5.

(A) and (B) Inhibition of TPP2 induces neurite outgrowth. (A) Graph shows mean total neurite length/cell ± S.D. (n = 6) of SH-SY5Y cells treated for 4 h with 1 μm butabindide, 1 μm B6 or DMSO, and as positive control SH-SY5Y cells that have been treated for 10 days with retinoic acid, as determined by Cellomics Arrayscan of α-tubulin immunostained cells. (B) Representative pictures (one out of 40 for each biological replicate) of B6 and DMSO treated cells fixed and stained for α-tubulin with respective masks of the Cellomics data analysis. (C) and (D) TPP2 inhibition by B6 induces an increase in mESPC amplitude in CA1 neurons in mice organotypic hippocampal slices. Brain slices were incubated for 6 h with 1/100 DMSO or 100 μm B6 (optimized B6 concentration showing complete inhibition). mEPSCs were recorded in presence of tetrodotoxin and picrotoxin. (C) Mean frequency (left graph) and amplitude (right graph) ± S.E. (DMSO: n = 28; B6 n = 26). (D) Representative mEPSC recording. (nonsignificant (n.s): p > .05; significant: * .05≥p ≥ .01, *** p ≤ .001, ***** p ≤ .00001).
Electrophysiology
Organotypic hippocampal slices were isolated from P7 mice and cultured for 6–13 days in vitro (DIV) in culture medium. Slices were incubated with 1/100 (v/v) DMSO or 100 μm B6 in slice culture medium for 6 h. Whole-cell recordings were obtained with Axopatch-1D amplifiers (Molecular Devices, Sunnyvale, CA) using 3- to 5-MΩ pipettes with an internal solution containing (in mm): 115 cesium methanesulfonate, 20 CsCl, 10 Hepes, 2.5 MgCl2, 4 Na2ATP, 0.4 Na3GTP, 10 sodium phosphocreatine, and 0.6 EGTA, at pH 7.25. External perfusion consisted of artificial cerebrospinal fluid containing (in mm): 119 NaCl, 2.5 KCl, 4 CaCl2, 4 MgCl2, 26 NaHCO3, 1 NaH2PO4, and 11 glucose (pH 7.4) and gassed with 5% CO2/95% O2 at 37 i.o. 27 °C. Miniature excitatory postsynaptic currents miniature Excitatory Postsynaptic Current (MEPSCs) were recorded in the presence of 1 μm tetrodotoxin (Tocris) and 100 μm picrotoxin (Sigma Aldrich). Statistical comparisons (P) were performed on log-transformed data. See Figs. 5C and 5D.
Statistical Analysis
All values were obtained from three independent repeated experiments and expressed as mean ± S.D., if not stated differently. Statistical comparisons (P) were performed using nonpaired two-tailed Student's t test. p < .05 was considered statistically significant.
RESULTS
B6—A New, Potent, Irreversible TPP2 Inhibitor
Butabindide is currently the only commercially available inhibitor that is specific for TPP2, but its application is influenced by its reversibility as well as its instability under mimicked physiological conditions (23, 48). We designed and synthesized an irreversible variant of butabindide using diphenylphosphonate as a reactive group targeting the serine residue (49) of the catalytic site of TPP2 (Fig. 1A). The resulting new inhibitor B6 (IC50 = 0.46 ± 0.01 nm) was found to be almost 60 times more potent than butabindide in inhibiting TPP2 (IC50 = 27 ± 7 nm) as determined by a comparative TPP2 substrate degradation assay with purified human TPP2 (hsTPP2) (Fig. 1B). The irreversibility of B6 was experimentally confirmed; in particular preincubation of hsTPP2 with B6 prior to dilution significantly decreased substrate degradation (Fig. 1C) in contrast to the reversible inhibitor butabindide where no significant effect of preincubation was observed. As the described, rapid cyclization of butabindide under mimicked physiological conditions interferes with TPP2 inhibition (48); the stability of B6 was comparatively investigated using the same experimental approach. No respective cyclization product of B6 was detected, likely due to steric hindrance of the bulky phenyl groups. Instead, hydrolysis of the B6 “warhead” was observed (Fig. 1D), which is expected to interfere with passive transport through the plasma membrane due to its negatively charged group at physiological pH. However, this instability has low impact on cellular short time-frame applications of B6 as shown by the data presented below and is probably explained by two observations: first, the hydrophobicity of B6, suggesting higher membrane permeability and thereby rapid access to its target. Second, the half-life of TPP2 was found to be 49 ± 5 h in SH-SY5Y cells (Supplemental Fig. 1), which combined with the fact that B6 is an irreversible binder implies a long-term inhibitory effect.
B6 Is a Specific, Nontoxic TPP2 Inhibitor
Prior to applying the new TPP2 inhibitor to screen for proteomic changes in SH-SY5Y neuroblastoma cells, the concentration of B6 was optimized and several possible side and adaptive effects were investigated. Incubating cells for 4 h with different concentrations of B6 followed by a TPP2 activity assay revealed that 1 μm B6 is sufficient to inhibit TPP2 activity (Fig. 2A). Moreover, under this optimized condition, we did not observe an effect of B6 on cell death neither when performing a trypan blue assay (Fig. 2B) nor during time-lapse microscopy recordings (data not shown). To investigate nonspecific and cell adaptive effects of B6, lysates of TPP2-inhibited and control noninhibited cells were incubated with activity-based probes for serine hydrolases and the proteasome, as well as a fluorogenic substrate for leucyl-aminopeptidases to determine the respective peptidase activities. Besides TPP2, no significant inhibition of the tested peptidases was detected (Figs. 2C-2E and Supplemental Fig. 2A). Additionally, we did not observe significant changes in proteasome levels or proteasome complex formation (Fig. 2D). Interestingly, native gel electrophoresis indicated that there were major changes in TPP2 complex assembly and/or complex stability in presence of B6 and minor changes in presence of butabindide (Fig. 2F). This observation is consistent with described induction of TPP2 complex assembly of TPP2 monomers in vitro in presence of butabindide (4). Examining TPP2 intracellular localization via Western blotting of cytoplasmic and nuclear fractionation revealed a mainly cytoplasmic localization of TPP2 in presence as well as in absence of inhibitor (Supplemental Fig. 2B). Immunocytochemistry showed the formation of cytoplasmic TPP2 accumulations in presence of B6 and to a minor degree by butabindide (Fig. 2F and Supplemental Fig. 2C). These accumulations of TPP2 are consistent with an inhibitor-induced TPP2 complex assembly or complex stabilization.
Proteomic Changes by TPP2 Inhibition and Knock Down
In order to identify the TPP2 client pool. we applied stable isotope labelling with amino acids in cell culture (SILAC) to three independent proteomic screens (for experimental setup, see Fig. 3A), the first comparing the effect of B6 (4 h, 1 μm) to the inhibitor solvent DMSO, the second comparing butabindide (4 h, 1 μm) to DMSO, and the third representing genetic loss of TPP2 by comparing stable lines expressing TPP2 shRNA (∼65% silencing, see Supplemental Fig. 3) with mock shRNA. Consistent with literature (12, 50), it needs to be mentioned that a higher knock down led to growth arrest and cell death. As a filtering criterion, we only considered proteins that were reproducibly (P1, p < .05) more than 1.25-fold (significance B, Intensity versus log2FC, p < .05) (44) increased or decreased as being influenced by TPP2 if they were changed in the same direction in at least two out of the three screens. Heat maps of the quantification of the proteins that fit the above mentioned criteria are shown in Fig 3B (for extended quantification results, see Supplemental Table I online). In total, 37 proteins were detected as increased; 12 as decreased by either both inhibitors, one inhibitor, and TPP2 knock down; or all three conditions when compared with the respective controls. To determine how TPP2 influences the individual expression levels of these proteins, we selected five of these proteins (PIK3C2A, MAP2, COL6A1, SOX11, UBL5) for transcriptional analysis by qPCR (Fig. 4A). Only SOX11 showed an increase of mRNA level after TPP2 inhibition with butabindide but not with B6. The transcription of the other four genes were not significantly influenced by TPP2 inhibition, while COL6A1 and MAP2 showed an increase upon TPP2 stable knock down. The moderate increase in protein level of SOX11 upon TPP2 inhibition could be confirmed by Western blot analysis using a polyclonal antibody that was raised against an epitope covering all three quantified peptides from the SILAC screen (Figs. 4B-4D). The impact of TPP2 inhibition on SOX11 stability was investigated as an example since the half-life time of SOX11 and SOX family transcription factors is below 2 h (51, 52) and thereby smaller than the TPP2 inhibition time period. In the presence of the protein synthesis inhibitor cycloheximide, no significant stabilization of SOX11 was observed (Figs. 4E and 4F and Supplemental Fig. 4), suggesting that TPP2 inhibition did not increase SOX11 expression by altering its degradation but by increasing its translation and in case of butabindide its transcription.
TPP2 Controls Neurite Outgrowth and Synaptic Plasticity
To further study the cellular response to TPP2 inhibition, we analyzed overrepresented biological processes (Table I) and pathways (Supplemental Table III) in the overlapping changed proteins as determined by SILAC (Fig. 3). Neuron differentiation, proliferation, cell differentiation and development, growth factor signaling, learning, DNA damage response, and phosphorylation (Table I) were the main overrepresented biological processes (26, 50, 53). Importantly, the involvement of TPP2 in neuron differentiation and in learning has not been described before, probably mediated by its influence on the transcription factor SOX11 (54–56). Additionally, pathways connected to neuron differentiation like NGF signaling, neural cell adhesion molecule signaling for neurite out-growth, PI3K-Akt signaling, and pathways connected to learning like long-term potentiation and glutamate receptor pathways were found to be significantly affected (Supplemental Table III). In order to specify these indicated effects, we first determined the length of neurites in SH-SY5Y cells as an indicator for neuron differentiation using a Cellomics Arrayscan. Compared with the negative control condition (DMSO), a significant increase in the length of neurites was observed following 4 h of TPP2 inhibition (Figs. 5A and 5B). We consider this increase in neurite length as biologically relevant as it increased after only 4 h cellular treatment with butabindide ∼20% and with B6 even ∼37% of that of the positive control cells cultured for 10 days in the presence of retinoic acid, a commonly used method to induce neurite outgrowth (57, 58). Second, we examined the influence of TPP2 on neuronal communication. Neurons communicate with each other through synapses, and synaptic plasticity underlies learning and memory formation (59, 60). The effect of TPP2 inhibition on synaptic strength was investigated by recording miniature excitatory postsynaptic currents (mEPSC) from CA1 neurons in mouse organotypic hippocampal slices that were incubated with either B6 or DMSO as a control. No change in mEPSC frequency was observed, indicating that TPP2 does not affect presynaptic vesicle release or the number of functional synapses. A significant increase in mEPSC amplitude was observed after inhibition of TPP2 with B6, indicating that synapses were strengthened through an increase of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor-type glutamate receptors (61) at the postsynaptic membrane (Figs. 5C and 5D).
TPP2 Inhibition Reduces Nuclear pERK1 and pERK2, the Linker to Most SILAC Targets
A functional interaction analysis (46, 47) identified mitogen-activated protein kinase (MAPK1) alias extracellular signal-regulated kinase 2 (ERK2) to be the protein functionally linked to most of the proteins that were expressionally affected already after 4 h by both TPP2 inhibitors (Supplemental Fig. 3, Supplemental Table IV). With the evidence of a rapid influence on ERK2 signal transduction pathway, we investigated further on these early changes. Both TPP2 inhibitors had no effect on the protein level of ERK2 as determined by SILAC (Fig. 6A). The activity of ERK2 and its interaction partner ERK1 is regulated by phosphorylation, which is induced by signal transduction downstream of growth factor receptors, like the neuronal or epidermal growth factor receptors. The dual phosphorylation of ERK2, at T185 and Y187, and ERK1, at T202 and Y204, results in their interaction and translocation into the nucleus where they activate transcription of several genes (62–65). Based on the observation that TPP2 inhibition influences phosphorylation as well as expression of phosphatases and kinases (Table I and Supplemental Table I), the effect of TPP2 inhibition on active, di-phosphorylated ERK1 and ERK2 (pERK1 and pERK2) levels in the nucleus and cytoplasm of SH-SY5Y cells was investigated. While no significant changes in the cytoplasm were observed (Supplemental Figs. 6B and 6C), a significant decrease in active pERK2 and pERK1 in the nucleus was observed after 4 h of TPP2 inhibition (Fig. 6B). Determining the ratio of pERK2/ERK2 reveals specifically a decrease of ERK2 phosphorylation in presence of B6 in the nucleus while the cytoplasmic phosphorylation level remained unchanged (Figs. 6C and 6D). A comparable decrease was also visible when stimulating ERK1 and ERK2 phosphorylation with PMA (15 min), an activator of protein kinase C upstream of the ERK2 signaling cascade (Figs. 6C and 6D, and Fig. 6E confocal images). Together, our data reveal a substantial and rapid influence of TPP2 on ERK2-mediated signal transduction. Consistent with our Western blot data, TPP2 inhibition in GFP-ERK2 overexpressing SH-SY5Y cells did not induce an obvious recruitment of GFP-ERK2 to the induced local immunostained accumulations of TPP2 (Supplemental Fig. 5). Although a direct interaction of both proteins cannot be excluded yet, TPP2 has no described kinase or phosphatase activity (3), indicating the involvement of a nuclear phosphatase or kinase as a mediator. One phosphatase that has been described to dephosphorylate nuclear pERK2 is protein phosphatase 2A (PP2A) (32, 66, 67). Interestingly, one regulatory subunit of PP2A was increased in the proteomic screen, PPP2R5C (alias KIAA0044 see Fig. 3 and Supplemental Table I). Additionally, we found an increase of PP2A catalytic subunits on the Western blot (Figs. 7A and 7B). However, we did not observe an increase of PP2A catalytic subunits in the nucleus upon TPP2 inhibition (Figs. 7C-7E and Supplemental Fig. 6), indicating that PP2A is not affecting nuclear phosphorylation of ERK1 and 2 via an up-regulation of total nuclear PP2A catalytic activity.
Fig. 6.

(A) TPP2 inhibition does not affect ERK2 cellular protein level. Median relative ERK2 protein level normalized to DMSO treated cells as determined by SILAC (n = 4 biological replicates, ±S.D. (butabindide n = 3, B6 n = 4 quantified replicates). (B) TPP2 inhibition decreases levels of nuclear, active phosphorylated ERK1 and ERK2. Bar diagram: Mean nuclear di-phosphorylated ERK2 (T185 & Y187, pERK2) and ERK1 (T202 and Y204, pERK1) protein level normalized to histone H3 loading control relative to DMSO control treated cells as determined by quantification of Western blot signals (n = 4, ±S.D., all Western blots are presented in Supplemental Fig. 5, one representative Western blot is shown in the right panel Fig. 6C). (C)–(E) TPP2 inhibition decreases phosphorylation level (active form) of nuclear ERK1 and ERK2 even when stimulating their phosphorylation with phorbol-12-myristate 13-acetate (PMA). (C) Representative Western blots of nuclear-cytoplasmic (cytopl.)-fractionation from DMSO and TPP2-inhibitor-treated cells without (left panel) and with 15min PMA treatment that were stained for pERK2 and pERK1, ERK2 and ERK1, and the loading controls α-tubulin and histone H3. (D) Mean phosphorylation level of ERK1 and ERK2 in nucleus and cytoplasm. Di-phosphorylated ERK2 (T185 & Y187, pERK2) to total ERK2 and di-phosphorylated ERK1 (T202 and Y204, pERK1) to total ERK1 protein level relative to DMSO-control-treated cells as determined by quantification of Western blot signals (n = 4 biological replicates, ±S.D., Western blots used for quantification are shown in Fig. 6C and Supplemental Figs. 5C and 5D). (E) Representative confocal images of PMA-treated cells inhibited with B6 or treated with DMSO as control that were immunostained for pERK1 and pERK2 (one antibody recognizing both, first column gray, second column (overlay) green) and DAPI (blue in overlay second column). White bar represents 50 μm.
Fig. 7.
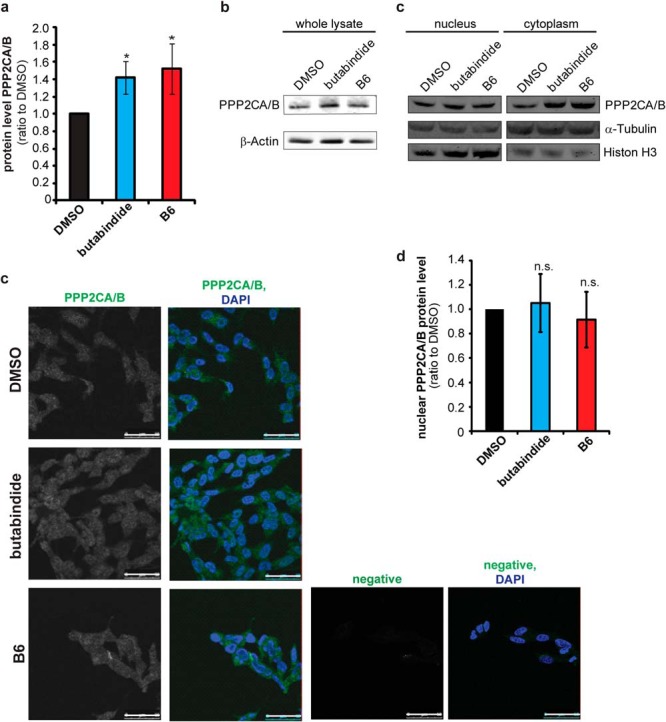
(A) and (B) TPP2 inhibition increases total cellular protein phosphatase 2A (PP2A) level. Bar diagram shows mean quantification results of Western blots of cell lysates stained for PPP2CA/B, the PP2A 36 kDa catalytic subunits, normalized to β-actin relative to DMSO-control-treated cells. (lysis: PBS/digitonin; n = 5 biological replicates ±S.D., one representative blot is shown in 7B, the other four in Supplemental Fig. 6A; significance: * .05≥p ≥ .01) (B) Representative Western blot used for quantification of total cellular PPP2CA/B level. (C)–(E) No change in nuclear phosphatase 2A (PP2A) level. (C) Representative Western blot of nuclear-cytoplasmic (cytopl.)-fractionation stained PPP2CA/B catalytic subunits and the loading controls α-tubulin and histone H3. (Additional Western blots of three further replicates and quantification results are presented in Supplemental Figs. 6B-D) Western blots including quantification is shown in Supplemental Fig. 6) (D) Representative confocal images of cells inhibited with B6 or treated with DMSO as control that were immunostained for PPP2CA/B, the catalytic subunit of PP2A (first column gray, second column (overlay) green) and DAPI (blue in overlay second column). Negative immunostain is of cells (second antibody only) is shown in gray in the third column and in green in the overlay with DAPI (fourth column). White bar represents 50 μm. All pictures have been taken with the same confocal settings (laser power, gain, offset, and for presentation purposes have been processed with Adobe Photoshop with the same settings (contrast, brightness). (E) Graph: mean nuclear PPP2CA/B protein level as determined by confocal microscopy (n = 3 biological replicates ±S.D., n = 200 cells/condition, images/experiment n = 3–8). Mean gray value of the whole nuclear area (as marked by DAPI) was determined after subtracting the background (negative immunostain nuclear mean gray value) and normalizing to DMSO nuclear PPP2CA/B signal. All images used for one experiment were taken with the same confocal settings (laser power, gain, offset). An exemplary quantification is presented in Supplemental Fig. 6D).
DISCUSSION
TPP2 influences numerous important cellular processes (12, 21, 28, 68), but the underlying mechanisms remain unknown. In order to elucidate details of its cellular function and identify proteins that are rapidly influenced by TPP2, we performed a comprehensive proteomic analysis. We determined proteomic changes following the application of B6, a newly developed, specific, irreversible TPP2 inhibitor, in parallel with the commercially available TPP2 inhibitor butabindide (23) and compared the data with the proteomic effects induced by stable TPP2 knock down as a reference. To rule out potential false negatives and positives resulting from unknown nonspecific effects of inhibitors or their metabolites, only proteins that were changed in the same way in at least two out of these three conditions were defined as being influenced by TPP2. In general, TPP2 can affect protein expression either directly or indirectly. A direct consequence of inhibition is expressional changes of TPP2 substrates. However, expressional changes of TPP2 interaction partners can also be suggested since both inhibitors alter TPP2 complex assembly and/or stability as well as localization. This may cause a shielding or exposure of domains, for instance the C-terminal BRCT-domain, that might interfere with a potential protein-stabilizing or destabilizing interaction of TPP2 (3, 19, 24, 69). TPP2 inhibition can also change protein expression indirectly via mediator proteins. This secondary effect can occur at the transcriptional level or by translational changes, as observed for the transcription factor SOX11 (Fig. 4). Alternatively, an indirect change of protein stability can result from altered expression of peptidases and other enzymes, which could not be seen as an effect of TPP2 inhibition.
Analyzing the connections between the proteins that were rapidly changed by TPP2 inhibition revealed ERK2 as a key functional protein hub. Most importantly, we showed that TPP2 inhibition rapidly reduced active, di-phosphorylated ERK1 (pERK1) and ERK2 (pERK2) in the nucleus without affecting their total nuclear and cytoplasmic protein level (Fig. 6) even in the presence of PMA as a stimulator of ERK1 and ERK2 phosphorylation. This indicates a down-regulation of ERK1- and ERK2-mediated signal transduction via their phosphorylation level, suggesting that TPP2 may regulate the ERK2 signal transduction pathway, which is induced by growth factors and mitogenic stimuli and controls transcription of several genes involved in processes like cell cycle, cell growth, proliferation, or angiogenesis (62). An influence on the transcription of genes of the MAPK pathways including ERK2 after 10 days TPP2 knock down in Burkitt lymphoma cells has been observed before (24, 26). However, uncovering a rapid regulating influence of TPP2 inhibition on nuclear phosphorylation level of ERK1 and ERK2 represents a step forward in understanding the observed cellular functions of TPP2. In the presence of TPP2 inhibitors, overexpressed GFP-ERK2 did not show translocation like a recruitment to the observed TPP2 accumulations. However, a direct probably TPP2 complex structure dependent interaction cannot be excluded yet.
Since TPP2 possesses no described kinase or phosphatase activity (3), it is most likely that TPP2 affects phosphorylation of nuclear ERK1 and 2 indirectly by promoting a nuclear phosphatase activity pERK1 and pERK2 (Fig. 8). Protein phosphatase 2A (PP2A) complexes are known to dephosphorylate pERK1 and pERK2 in the nucleus. An increase of the mainly nuclear PP2A regulatory gamma subunit (KIAA0044) by SILAC and the catalytic subunits PPP2CA/B on Western blotting was observed after 4 h of TPP2 inhibition (32, 66, 67, 70) (Figs. 3 and 7A and 7B). Since the nuclear PP2A catalytic subunit levels appeared to be not increased (Figs. 7B-D), the down-regulation of ERK2 activity in the nucleus is not evoked by a general up-regulation of PP2A catalytic activity. However, a shift of the activity of nuclear PP2A complexes with target specific regulatory subunits, like KIAA0044 that has been described to target ERK, cannot be excluded, yet (71, 72).
Fig. 8.

The relationship between TPP2 and ERK1/2 phosphorylation. TPP2 inhibition leads to a decrease of active phosphorylated ERK1 and ERK2 via inducing an activation of (1) nuclear phosphatases that dephosphorylate (-P) pERKs and/or a decrease of (2) nuclear kinase activity that targets ERK1 and ERK2.
The regulation of ERK1 and ERK2 phosphorylation by TPP2 can be connected to a number of biological processes influenced by TPP2. Our proteomic data are consistent with literature and point toward major changes in cell differentiation, proliferation, development, growth factor signaling, learning, DNA damage response, and phosphorylation upon TPP2 inhibition and knock down (20, 26, 68). The ERK2 signal transduction affects at least transcription of genes involved in proliferation and development as well as DNA repair and cell-cycle-related genes. Additionally, our proteomic results (Table I) indicate that TPP2 is upstream of processes involved in learning, a biological process that has not been connected to TPP2 before, probably because we are the first to use a neuronal model system to study TPP2 function. Here, we have linked TPP2 to synaptic transmission (Fig. 5) by showing that TPP2 inhibition in hippocampal neurons led to an increase of currents and consequently AMPA-type receptors at the postsynaptic membrane (61), indicating a strengthening of synapses. This finding implicates a role for TPP2 in neurological processes that has never been described before.
The newly described involvement of TPP2 in neuronal function may relate to its influence on ERK1 and ERK2 phosphorylation since in neurons the ERK2 pathway has been associated with synaptic plasticity and memory formation (35, 36). The observed changes in synaptic plasticity can also be connected to the moderate increase in SOX11 expression (56). It has been shown that an increased expression of SOX11 (Fig. 4B) can activate the transcription of brain-derived neurotrophic factor (BDNF), a protein modulating synaptic plasticity related to memory, learning, and pain signaling (56, 73, 74). BDNF expression was found regulated by the neuronal growth factor NGF, a signaling pathway that was influenced by TPP2 (Supplemental Table III), and furthermore, BDNF expression correlates with neurite outgrowth (75, 76). The latter can be linked to the observed rapid increase of neurite length in presence of TPP2 inhibition. The increase in neurite length seems to be contradictory to the down-regulation of ERK2 signaling downstream of neuronal growth factor receptor. However, it has been shown that phosphatidylinositol 4-phosphate 3-kinase (PI3K) can promote neurite outgrowth independent of ERK2 (77). The proteomic data show an increase of the PI3K subunit PIK3C2A in all three proteomic screens (Fig. 3 and Supplemental Table I), that could explain the observed effect of TPP2 inhibition on neurite outgrowth (Fig. 5).
In conclusion, using three independent approaches, including the application of a novel potent TPP2 inhibitor, we discovered a regulatory influence of TPP2 on the ERK2-mediated signal transduction. We show that TPP2 inhibition down-regulates the ERK2 pathway as it diminishes nuclear active di-phosphorylated ERK1 and 2, most likely via activating its dephosphorylation or inhibiting its phosphorylation in the nucleus (Fig. 8). This finding can be connected to the described influence of TPP2 on multitude biological processes, including our second new finding, the strengthening of synapses by TPP2 inhibition. Since ERK2 mediated signal transduction is increased in one-third of all cancers (62), TPP2 might become an interesting target for upstream control of the ERK pathway in research fields like cancer and neurological or neurodegenerative diseases.
Supplementary Material
Acknowledgments
We would like to acknowledge Nico Meeuwenoord, for his help with SCX-based separation of proteomic samples. We also would like to thank Martin D. Witte, Noam Zelcer and Eric Mul for their scientific contribution and Adam Benham for critically reading the manuscript.
Footnotes
Author contributions: A.W., A.S., W.A.v., J.S., H.W.K., H.O., H.S.O., B.F., and E.A.R. designed the research; A.W., A.S., W.A.v., M.C.R., R.E.v., B.T., H.W.K., B.F., and E.A.R. performed the research; W.A.v., H.S.O., and B.F. contributed new reagents or analytic tools; A.W., A.S., W.A.v., M.C.R., R.E.v., J.S., B.T., H.W.K., B.F., and E.A.R. analyzed data; A.W. wrote the paper; and E.A.J. initiated the project.
* This work was supported by the Prinses Beatrix Fonds (W.OR10-25) and by a grant from the Organization for Scientific Research with a VIDI grant (NWO-Zon-MW, 91796315).
 This article contains supplemental material Supplemental Tables I–IV, and VI and Supplemental Figs. 1–6.
This article contains supplemental material Supplemental Tables I–IV, and VI and Supplemental Figs. 1–6.
1 The abbreviations used are:
- ABC
- ammonium bicarbonate
- Ac
- acetate
- AMC
- 4-methyl-coumaryl-7-amide
- BODIPY
- boron-dipyrromethene
- BRCT-
- breast cancer gene 1 carboxy-terminal domain
- CHX
- cycloheximide
- hsTPP2
- human tripeptidyl peptidase II
- IC50
- half inhibitory concentration
- mEPSC
- miniature excitatory postsynaptic current
- NEM
- N-ethylmaleimide
- PEP
- posterior error probability
- pERK
- di-phosphorylated extracellular signal-regulated kinase
- PFA
- paraformaldehyde
- PMA
- phorbol-12-myristate 13-acetate
- pNA
- p-mitroanilide
- PP2A
- protein phosphatase 2A
- RT
- room temperature
- SCX
- strong cation exchange
- SEM
- standard error of the mean
- shRNA
- short hairpin ribonucleic acid
- SILAC
- stable isotope labeling with amino acids in cell culture
- TAMRA-FP
- carboxytetramethylrhodamine-fluorophosphonate
- Var
- variance.
REFERENCES
- 1. Schönegge A. M., Villa E., Förster F., Hegerl R., Peters J., Baumeister W., Rockel B. (2012) The structure of human tripeptidyl peptidase II as determined by a hybrid approach. Structure 20, 593–603 [DOI] [PubMed] [Google Scholar]
- 2. Seyit G., Rockel B., Baumeister W., Peters J. (2006) Size matters for the tripeptidylpeptidase II complex from Drosophila: The 6-MDa spindle form stabilizes the activated state. J. Biol. Chem. 281, 25723–25733 [DOI] [PubMed] [Google Scholar]
- 3. Eriksson S., Gutiérrez O. A., Bjerling P., Tomkinson B. (2009) Development, evaluation and application of tripeptidyl-peptidase II sequence signatures. Arch. Biochem. Biophys. 484, 39–45 [DOI] [PubMed] [Google Scholar]
- 4. Tomkinson B. (2000) Association and dissociation of the tripeptidyl-peptidase II complex as a way of regulating the enzyme activity. Arch. Biochem. Biophys. 376, 275–280 [DOI] [PubMed] [Google Scholar]
- 5. Peters J., Schönegge A. M., Rockel B., Baumeister W. (2011) Molecular ruler of tripeptidylpeptidase II: Mechanistic principle of exopeptidase selectivity. Biochem. Biophys. Res. Commun. 414, 209–214 [DOI] [PubMed] [Google Scholar]
- 6. Wilson C., Gibson A. M., McDermott J. R. (1993) Purification and characterization of tripeptidylpeptidase-II from post-mortem human brain. Neurochem. Res. 18, 743–749 [DOI] [PubMed] [Google Scholar]
- 7. Bålöw R. M., Ragnarsson U., Zetterqvist O. (1983) Tripeptidyl aminopeptidase in the extralysosomal fraction of rat liver. J. Biol. Chem. 258, 11622–11628 [PubMed] [Google Scholar]
- 8. Bålöw R. M., Tomkinson B., Ragnarsson U., Zetterqvist O. (1986) Purification, substrate specificity, and classification of tripeptidyl peptidase II. J. Biol. Chem. 261, 2409–2417 [PubMed] [Google Scholar]
- 9. Eklund S., Dogan J., Jemth P., Kalbacher H., Tomkinson B. (2012) Characterization of the endopeptidase activity of tripeptidyl-peptidase II. Biochem. Biophys. Res. Commun. 424, 503–507 [DOI] [PubMed] [Google Scholar]
- 10. Seifert U., Marañón C., Shmueli A., Desoutter J. F., Wesoloski L., Janek K., Henklein P., Diescher S., Andrieu M., de la Salle H., Weinschenk T., Schild H., Laderach D., Galy A., Haas G., Kloetzel P. M., Reiss Y., Hosmalin A. (2003) An essential role for tripeptidyl peptidase in the generation of an MHC class I epitope. Nat. Immunol. 4, 375–379 [DOI] [PubMed] [Google Scholar]
- 11. Geier E., Pfeifer G., Wilm M., Lucchiari-Hartz M., Baumeister W., Eichmann K., Niedermann G. (1999) A giant protease with potential to substitute for some functions of the proteasome. Science 283, 978–981 [DOI] [PubMed] [Google Scholar]
- 12. McKay R. M., McKay J. P., Suh J. M., Avery L., Graff J. M. (2007) Tripeptidyl peptidase II promotes fat formation in a conserved fashion. EMBO Rep. 8, 1183–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Firat E., Huai J., Saveanu L., Gaedicke S., Aichele P., Eichmann K., van Endert P., Niedermann G. (2007) Analysis of direct and cross-presentation of antigens in TPPII knockout mice. J. Immunol. 179, 8137–8145 [DOI] [PubMed] [Google Scholar]
- 14. Guil S., Rodriguez-Castro M., Aguilar F., Villasevil E. M., Antón L. C., Del Val M. (2006) Need for tripeptidyl-peptidase II in major histocompatibility complex class I viral antigen processing when proteasomes are detrimental. J. Biol. Chem. 281, 39925–39934 [DOI] [PubMed] [Google Scholar]
- 15. Endert P. (2008) Role of tripeptidyl peptidase II in MHC class I antigen processing - the end of controversies? Eur. J. Immunol. 38, 609–613 [DOI] [PubMed] [Google Scholar]
- 16. Basler M., Groettrup M. (2007) No essential role for tripeptidyl peptidase II for the processing of LCMV-derived T cell epitopes. Eur. J. Immunol. 37, 896–904 [DOI] [PubMed] [Google Scholar]
- 17. York I. A., Bhutani N., Zendzian S., Goldberg A. L., Rock K. L. (2006) Tripeptidyl peptidase II is the major peptidase needed to trim long antigenic precursors, but is not required for most MHC class I antigen presentation. J. Immunol. 177, 1434–1443 [DOI] [PubMed] [Google Scholar]
- 18. Marcilla M., Villasevil E. M., de Castro J. A. (2008) Tripeptidyl peptidase II is dispensable for the generation of both proteasome-dependent and proteasome-independent ligands of HLA-B27 and other class I molecules. Eur. J. Immunol. 38, 631–639 [DOI] [PubMed] [Google Scholar]
- 19. Hong X., Lei L., Künert B., Naredla R., Applequist S. E., Grandien A., Glas R. (2007) Tripeptidyl-peptidase II controls DNA damage responses and in vivo gamma-irradiation resistance of tumors. Cancer Res. 67, 7165–7174 [DOI] [PubMed] [Google Scholar]
- 20. Stavropoulou V., Vasquez V., Cereser B., Freda E., Masucci M. G. (2006) TPPII promotes genetic instability by allowing the escape from apoptosis of cells with activated mitotic checkpoints. Biochem. Biophys. Res. Commun. 346, 415–425 [DOI] [PubMed] [Google Scholar]
- 21. Stavropoulou V., Xie J., Henriksson M., Tomkinson B., Imreh S., Masucci M. G. (2005) Mitotic infidelity and centrosome duplication errors in cells overexpressing tripeptidyl-peptidase II. Cancer Res. 65, 1361–1368 [DOI] [PubMed] [Google Scholar]
- 22. Duensing S., Darr S., Cuevas R., Melquiot N., Brickner A. G., Duensing A., Münger K. (2010) Tripeptidyl peptidase II is required for c-MYC-induced centriole overduplication and a novel therapeutic target in c-MYC-associated neoplasms. Genes Cancer 1, 883–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rose C., Vargas F., Facchinetti P., Bourgeat P., Bambal R. B., Bishop P. B., Chan S. M., Moore A. N., Ganellin C. R., Schwartz J. C. (1996) Characterization and inhibition of a cholecystokinin-inactivating serine peptidase. Nature 380, 403–409 [DOI] [PubMed] [Google Scholar]
- 24. Preta G., de Klark R., Chakraborti S., Glas R. (2010) MAP kinase-signaling controls nuclear translocation of tripeptidyl-peptidase II in response to DNA damage and oxidative stress. Biochem. Biophys. Res. Commun. 399, 324–330 [DOI] [PubMed] [Google Scholar]
- 25. Gavioli R., Frisan T., Vertuani S., Bornkamm G. W., Masucci M. G. (2001) c-myc overexpression activates alternative pathways for intracellular proteolysis in lymphoma cells. Nat. Cell Biol. 3, 283–288 [DOI] [PubMed] [Google Scholar]
- 26. Sompallae R., Stavropoulou V., Houde M., Masucci M. G. (2008) The MAPK signaling cascade is a central hub in the regulation of cell cycle, apoptosis and cytoskeleton remodeling by tripeptidyl-peptidase II. Gene. Regul Syst. Bio. 2, 253–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kawahara M., York I. A., Hearn A., Farfan D., Rock K. L. (2009) Analysis of the role of tripeptidyl peptidase II in MHC class I antigen presentation in vivo. J. Immunol. 183, 6069–6077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Preta G., de Klark R., Gavioli R., Glas R. (2010) The enigma of tripeptidyl-peptidase II: Dual roles in housekeeping and stress. J. Oncol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tomkinson B., Nyberg F. (1995) Distribution of tripeptidyl-peptidase II in the central nervous system of rat. Neurochem. Res. 20, 1443–1447 [DOI] [PubMed] [Google Scholar]
- 30. Radu D., Tomkinson B., Zachrisson O., Weber G., de Belleroche J., Hirsch S., Lindefors N. (2006) Overlapping regional distribution of CCK and TPPII mRNAs in Cynomolgus monkey brain and correlated levels in human cerebral cortex (BA 10). Brain Res. 1104, 175–182 [DOI] [PubMed] [Google Scholar]
- 31. Dumesic P. A., Scholl F. A., Barragan D. I., Khavari P. A. (2009) ERK1/2 MAP kinases are required for epidermal G2/M progression. J. Cell Biol. 185, 409–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hancock C. N., Dangi S., Shapiro P. (2005) Protein phosphatase 2A activity associated with Golgi membranes during the G2/M phase may regulate phosphorylation of ERK2. J. Biol. Chem. 280, 11590–11598 [DOI] [PubMed] [Google Scholar]
- 33. Abbott D. W., Holt J. T. (1999) Mitogen-activated protein kinase kinase 2 activation is essential for progression through the G2/M checkpoint arrest in cells exposed to ionizing radiation. J. Biol. Chem. 274, 2732–2742 [DOI] [PubMed] [Google Scholar]
- 34. Wei F., Xie Y., Tao L., Tang D. (2010) Both ERK1 and ERK2 kinases promote G2/M arrest in etoposide-treated MCF7 cells by facilitating ATM activation. Cell Signal 22, 1783–1789 [DOI] [PubMed] [Google Scholar]
- 35. Sweatt J. D. (2001) The neuronal MAP kinase cascade: A biochemical signal integration system subserving synaptic plasticity and memory. J. Neurochem. 76, 1–10 [DOI] [PubMed] [Google Scholar]
- 36. Giovannini M. G. (2006) The role of the extracellular signal-regulated kinase pathway in memory encoding. Rev. Neurosci. 17, 619–634 [DOI] [PubMed] [Google Scholar]
- 37. Tomkinson B., Wernstedt C., Hellman U., Zetterqvist O. (1987) Active site of tripeptidyl peptidase II from human erythrocytes is of the subtilisin type. Proc. Natl. Acad. Sci. U.S.A. 84, 7508–7512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tomkinson B., Zetterqvist O. (1990) Immunological cross-reactivity between human tripeptidyl peptidase II and fibronectin. Biochem. J. 267, 149–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Florea B. I., Verdoes M., Li N., van der Linden W. A., Geurink P. P., van den Elst H., Hofmann T., de Ru A., van Veelen P. A., Tanaka K., Sasaki K., Murata S., den Dulk H., Brouwer J., Ossendorp F. A., Kisselev A. F., Overkleeft H. S. (2010) Activity-based profiling reveals reactivity of the murine thymoproteasome-specific subunit beta5t. Chem. Biol. 17, 795–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nethe M., Anthony E. C., Fernandez-Borja M., Dee R., Geerts D., Hensbergen P. J., Deelder A. M., Schmidt G., Hordijk P. L. (2010) Focal-adhesion targeting links caveolin-1 to a Rac1-degradation pathway. J. Cell Sci. 123, 1948–1958 [DOI] [PubMed] [Google Scholar]
- 41. Verbeek D. S., Goedhart J., Bruinsma L., Sinke R. J., Reits E. A. (2008) PKC gamma mutations in spinocerebellar ataxia type 14 affect C1 domain accessibility and kinase activity leading to aberrant MAPK signaling. J. Cell Sci. 121, 2339–2349 [DOI] [PubMed] [Google Scholar]
- 42. Rappsilber J., Mann M., Ishihama Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 [DOI] [PubMed] [Google Scholar]
- 43. Brunner E., Ahrens C. H., Mohanty S., Baetschmann H., Loevenich S., Potthast F., Deutsch E. W., Panse C., de Lichtenberg U., Rinner O., Lee H., Pedrioli P. G., Malmstrom J., Koehler K., Schrimpf S., Krijgsveld J., Kregenow F., Heck A. J., Hafen E., Schlapbach R., Aebersold R. (2007) A high-quality catalog of the Drosophila melanogaster proteome. Nat. Biotechnol. 25, 576–583 [DOI] [PubMed] [Google Scholar]
- 44. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnology 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 45. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011) Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 46. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T. (2003) Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Saito R., Smoot M. E., Ono K., Ruscheinski J., Wang P. L., Lotia S., Pico A. R., Bader G. D., Ideker T. (2012) A travel guide to Cytoscape plugins. Nat. Methods 9, 1069–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Breslin H. J., Miskowski T. A., Kukla M. J., Leister W. H., De Winter H. L., Gauthier D. A., Somers M. V., Peeters D. C., Roevens P. W. (2002) Design, synthesis, and tripeptidyl peptidase II inhibitory activity of a novel series of (S)-2,3-dihydro-2-(4-alkyl-1H-imidazol-2-yl)-1H-indoles. J. Med. Chem. 45, 5303–5310 [DOI] [PubMed] [Google Scholar]
- 49. Serim S., Haedke U., Verhelst S. H. (2012) Activity-based probes for the study of proteases: Recent advances and developments. Chem. Med. Chem. 7, 1146–1159 [DOI] [PubMed] [Google Scholar]
- 50. Usukura K., Kasamatsu A., Okamoto A., Kouzu Y., Higo M., Koike H., Sakamoto Y., Ogawara K., Shiiba M., Tanzawa H., Uzawa K. (2013) Tripeptidyl peptidase II in human oral squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 139, 123–130 [DOI] [PubMed] [Google Scholar]
- 51. Beekman J. M., Vervoort S. J., Dekkers F., van Vessem M. E., Vendelbosch S., Brugulat-Panès A., van Loosdregt J., Braat A. K., Coffer P. J. (2012) Syntenin-mediated regulation of Sox4 proteasomal degradation modulates transcriptional output. Oncogene 31, 2668–2679 [DOI] [PubMed] [Google Scholar]
- 52. Clark M. B., Johnston R. L., Inostroza-Ponta M., Fox A. H., Fortini E., Moscato P., Dinger M. E., Mattick J. S. (2012) Genome-wide analysis of long noncoding RNA stability. Genome Res. 22, 885–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tsurumi C., Firat E., Gaedicke S., Huai J., Mandal P. K., Niedermann G. (2009) Viability and DNA damage responses of TPPII-deficient Myc- and Ras-transformed fibroblasts. Biochem. Biophys. Res. Commun. 386, 563–568 [DOI] [PubMed] [Google Scholar]
- 54. Azuma T., Ao S., Saito Y., Yano K., Seki N., Wakao H., Masuho Y., Muramatsu M. (1999) Human SOX11, an upregulated gene during the neural differentiation, has a long 3′ untranslated region. DNA Res. 6, 357–360 [DOI] [PubMed] [Google Scholar]
- 55. Wang Y., Lin L., Lai H., Parada L. F., Lei L. (2013) Transcription factor SOX11 is essential for both embryonic and adult neurogenesis. Dev. Dyn. 242, 638–653 [DOI] [PubMed] [Google Scholar]
- 56. Salerno K. M., Jing X., Diges C. M., Cornuet P. K., Glorioso J. C., Albers K. M. (2012) SOX11 modulates brain-derived neurotrophic factor expression in an exon promoter-specific manner. J. Neurosci. Res. 90, 1011–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Miloso M., Villa D., Crimi M., Galbiati S., Donzelli E., Nicolini G., Tredici G. (2004) Retinoic acid-induced neuritogenesis of human neuroblastoma SH-SY5Y cells is ERK independent and PKC dependent. J. Neurosci. Res. 75, 241–252 [DOI] [PubMed] [Google Scholar]
- 58. Simpson P. B., Bacha J. I., Palfreyman E. L., Woollacott A. J., McKernan R. M., Kerby J. (2001) Retinoic acid evoked-differentiation of neuroblastoma cells predominates over growth factor stimulation: An automated image capture and quantitation approach to neuritogenesis. Anal. Biochem. 298, 163–169 [DOI] [PubMed] [Google Scholar]
- 59. Benfenati F. (2007) Synaptic plasticity and the neurobiology of learning and memory. Acta Biomed. 78, 58–66 [PubMed] [Google Scholar]
- 60. Balietti M., Tamagnini F., Fattoretti P., Burattini C., Casoli T., Platano D., Lattanzio F., Aicardi G. (2012) Impairments of synaptic plasticity in aged animals and in animal models of Alzheimer's disease. Rejuvenation Res. 15, 235–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kessels H. W., Malinow R. (2009) Synaptic AMPA receptor plasticity and behavior. Neuron 61, 340–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Roskoski R., Jr. (2012) ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res. 66, 105–143 [DOI] [PubMed] [Google Scholar]
- 63. Srinivasan R., Zabuawala T., Huang H., Zhang J., Gulati P., Fernandez S., Karlo J. C., Landreth G. E., Leone G., Ostrowski M. C. (2009) ERK1 and ERK2 regulate endothelial cell proliferation and migration during mouse embryonic angiogenesis. PLoS One 4, e8283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shukla S., MacLennan G. T., Fu P., Gupta S. (2012) Apigenin attenuates insulin-like growth factor-I signaling in an autochthonous mouse prostate cancer model. Pharm. Res. 29, 1506–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ahn N. G., Seger R., Bratlien R. L., Krebs E. G. Growth factor-stimulated phosphorylation cascades: Activation of growth factor-stimulated MAP kinase. Ciba Found Symp. 164, 113–126, discussion 126–131 [DOI] [PubMed] [Google Scholar]
- 66. Alessi D. R., Gomez N., Moorhead G., Lewis T., Keyse S. M., Cohen P. (1995) Inactivation of p42 MAP kinase by protein phosphatase 2A and a protein tyrosine phosphatase, but not CL100, in various cell lines. Curr. Biol. 5, 283–295 [DOI] [PubMed] [Google Scholar]
- 67. Millward T. A., Zolnierowicz S., Hemmings B. A. (1999) Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem. Sci. 24, 186–191 [DOI] [PubMed] [Google Scholar]
- 68. Rockel B., Kopec K. O., Lupas A. N., Baumeister W. (2012) Structure and function of tripeptidyl peptidase II, a giant cytosolic protease. Biochim. Biophys. Acta 1824, 237–245 [DOI] [PubMed] [Google Scholar]
- 69. Preta G., de Klark R., Glas R. (2009) A role for nuclear translocation of tripeptidyl-peptidase II in reactive oxygen species-dependent DNA damage responses. Biochem. Biophys. Res. Commun. 389, 575–579 [DOI] [PubMed] [Google Scholar]
- 70. McCright B., Rivers A. M., Audlin S., Virshup D. M. (1996) The B56 family of protein phosphatase 2A (PP2A) regulatory subunits encodes differentiation-induced phosphoproteins that target PP2A to both nucleus and cytoplasm. J. Biol. Chem. 271, 22081–22089 [DOI] [PubMed] [Google Scholar]
- 71. Kawahara E., Maenaka S., Shimada E., Nishimura Y., Sakurai H. (2013) Dynamic regulation of extracellular signal-regulated kinase (ERK) by protein phosphatase 2A regulatory subunit B56gamma1 in nuclei induces cell migration. PLoS One 8, e63729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Letourneux C., Rocher G., Porteu F. (2006) B56-containing PP2A dephosphorylate ERK and their activity is controlled by the early gene IEX-1 and ERK. EMBO J. 25, 727–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Cowansage K. K., LeDoux J. E., Monfils M. H. (2010) Brain-derived neurotrophic factor: A dynamic gatekeeper of neural plasticity. Curr. Mol. Pharmacol. 3, 12–29 [DOI] [PubMed] [Google Scholar]
- 74. Obata K., Noguchi K. (2006) BDNF in sensory neurons and chronic pain. Neurosci. Res. 55, 1–10 [DOI] [PubMed] [Google Scholar]
- 75. Geremia N. M., Pettersson L. M., Hasmatali J. C., Hryciw T., Danielsen N., Schreyer D. J., Verge V. M. (2010) Endogenous BDNF regulates induction of intrinsic neuronal growth programs in injured sensory neurons. Exp. Neurol. 223, 128–142 [DOI] [PubMed] [Google Scholar]
- 76. Matsuoka Y., Yokoyama M., Kobayashi H., Omori M., Itano Y., Morita K., Mori H., Nakanishi T. (2007) Expression profiles of BDNF splice variants in cultured DRG neurons stimulated with NGF. Biochem. Biophys. Res. Commun. 362, 682–688 [DOI] [PubMed] [Google Scholar]
- 77. Sarner S., Kozma R., Ahmed S., Lim L. (2000) Phosphatidylinositol 3-kinase, Cdc42, and Rac1 act downstream of Ras in integrin-dependent neurite outgrowth in N1E-115 neuroblastoma cells. Mol. Cell Biol. 20, 158–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.