

Summary
We explore cell heterogeneity during spontaneous and transcription-factor-driven commitment for network inference in hematopoiesis. Since individual genes display discrete OFF states or a distribution of ON levels, we compute and combine pairwise gene associations from binary and continuous components of gene expression in single cells. Ddit3 emerges as a regulatory node with positive linkage to erythroid regulators and negative association with myeloid determinants. Ddit3 loss impairs erythroid colony output from multipotent cells, while forcing Ddit3 in granulo-monocytic progenitors (GMPs) enhances self-renewal and impedes differentiation. Network analysis of Ddit3-transduced GMPs reveals uncoupling of myeloid networks and strengthening of erythroid linkages. RNA sequencing suggests that Ddit3 acts through development or stabilization of a precursor upstream of GMPs with inherent Meg-E potential. The enrichment of Gata2 target genes in Ddit3-dependent transcriptional responses suggests that Ddit3 functions in an erythroid transcriptional network nucleated by Gata2.
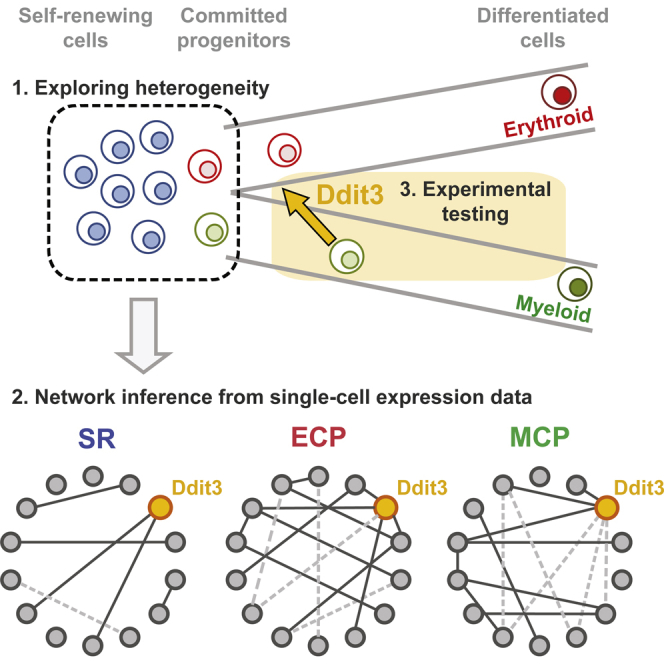
Graphical Abstract
Highlights
-
•
We present a method for inferring gene regulatory networks (GRNs) from single cells
-
•
Lineage cross-antagonism is a key property of GRNs of early lineage commitment
-
•
Ddit3 is a regulatory node in erythroid lineage programming
-
•
A Ddit3-Gata2 regulatory axis antagonizes myeloid and enables erythroid programs
Pina et al. develop a gene regulatory network inference method using single-cell gene expression data and identify Ddit3 as a regulatory node in erythroid lineage programming. The authors explore this inference and show that Ddit3 can antagonize myeloid programming and enable erythroid signatures and forms a regulatory axis with Gata2.
Introduction
Development and differentiation are characterized by genetic circuitry or gene regulatory networks (GRNs) that have inherent forward momentum encoded by a number of regulatory motifs (Davidson, 2010). To self-renew and maintain differentiation potential, stem cells must structure their GRNs so as to arrest or buffer this forward trajectory. Networks at early multipotent stages may bear little relation to those of mature differentiated cells, making comparison between them difficult. Detailed time-series data are useful in this regard (Bruno et al., 2004; May et al., 2013), but there is likely substantial asynchrony between individual cells at any given time point. Cells also may undergo lineage commitment through different initial gene expression trajectories (Pina et al., 2012). Together, these factors can confound attempts to infer network architectures and gain molecular insights into commitment and subsequent lineage specification from averaged gene expression profiles.
Analysis of gene expression in single cells offers a different approach that, in principle, makes use of cellular heterogeneity as a source of variation for establishing gene-gene associations. Recent studies have used all expression data (Guo et al., 2013; Moignard et al., 2013, 2015) or inferred pairwise gene associations using only co-expressing cells (Ståhlberg et al., 2011). It has been suggested that levels of expression are better accounted for in cells that co-express both genes, and may be obscured by presence/absence effects when all cells are considered (Rusnakova et al., 2013). We have tried to address this constraint in our exploration of gene expression networks around the erythroid versus myelo-monocytic lineage choice of multipotent hematopoietic progenitor cells. Additionally, we have focused on capturing networks from closely related cells in the vicinity of the commitment boundary, to gain insight into the evolution of GRNs relevant to lineage specification.
The erythro-myeloid bifurcation is an intensively studied paradigm, and both transcription factors (TFs) and regulatory motifs involved in physiological transcriptional programming of these alternative fates have been described (Wolff and Humeniuk, 2013). Key players include Gata factors, the Ets family protein Pu.1, and C/ebp family members, whose potency has been demonstrated in the experimental reprogramming of blood lineages (Graf and Enver, 2009). We used two distinct cell commitment scenarios to obtain high resolution around the early phase of commitment and lineage specification. First, we identified and prospectively isolated cells spontaneously committing to both lineages under culture conditions that maintain self-renewing (SR) cells and lack pro-differentiative cytokines. Second, we used inducible variants of the lineage-affiliated TFs Gata1 and Pu.1 to drive cells into lineage specification, again in the absence of pro-differentiative cytokines. This approach allows timed sampling after instigation of a discrete lineage trigger, and it may provide a more homogeneous molecular entry into commitment and lineage development.
We describe state-distinct networks in multipotent and early lineage-committed cells and, in particular, highlight the existence of lineage-conflicting programs at the emergence of lineage choice. We further identify an axis involved in lineage specification that includes Gata2 and Ddit3, a C/ebp family member previously implicated in stress response (Zinszner et al., 1998) and described as a potential target of erythropoietin signaling in erythro-leukemic cell lines (Coutts et al., 1999).
Results
We have explored commitment in the non-transformed bone marrow (BM)-derived hematopoietic multipotent cell line FDCPmix. FDCPmix is karyotypically normal, IL-3 dependent, and capable of multilineage differentiation in response to the appropriate environmental cues. Under maintenance culture conditions, SR and lineage-committed cells (erythroid- or myeloid-committed progenitors [ECPs or MCPs]) co-exist in culture (Figure 1A) and can be isolated on the basis of their surface phenotype (Figure 1B). SR cells are Kit+Gr1− cells (Figure 1B) with proliferative capacity in bulk and clonal cultures (Figures S1A and S1B) and are uniquely able to faithfully reconstitute the cellular heterogeneity observed in maintenance cultures (Figure S1C). Lineage-committed cells devoid of SR potential are Gr1+ MCPs, with an early myeloid morphology (Figure S1D) and no erythroid differentiation capacity (Figure S1E), and kit−Gr1− ECPs, with accelerated erythroid differentiation (Figure S1E) but minimal or no contribution to neutrophil cultures (Figure S1F). The transcriptional signatures of SR, MCP, and ECP compartments are readily distinct (Figure S1G) and confirm their lineage affiliation (Table S1).
Figure 1.

Single-Cell Transcriptional Profiling Captures Molecular Spaces of Lineage Commitment
(A) Depiction shows the two modes of commitment surveyed in this study: (top) hierarchically related SR cells, ECPs, and MCPs in equilibrium in a multipotent cell culture system; (bottom) unilineage commitment of SR cells driven by a single TF.
(B) Flow cytometry plot shows co-existing SR, ECP, and MCP cells in an FDCPmix culture under SR conditions.
(C) Heatmap of expression profiles of 26 genes in individual FDCPmix SR, ECP, and MCP cells. The gene panel represents the consensus analyzed in sufficient cell numbers in all compartments between replicate experiments. Data are Z score-normalized ΔCt values; undetectable expression, gray.
(D) Flow cytometry plots show FDCPmix SR cells transduced with control empty vector or with a GATA1-ERT fusion after 21-hr activation with tamoxifen (4-OHT, 1 μM).
(E) Cloning efficiency of control and Gata1-ERT GFP+ cells cultured under SR and neutrophil differentiation (N) conditions during a time course of 4-OHT activation. Sampling times correspond to those of single-cell qRT-PCR analysis. At each time point, 60 individual cells of each genotype were plated into SR or N conditions, and individually seeded wells were inspected at regular intervals for a 7-day period.
(F) PCA plot of the transcriptional profiles of individual FDCPmix cells undergoing distinct modes of lineage commitment. The first two PC explain 31% of the data variance; n = 82 (SR), 60 (ECP), 59 (MCP), 147 (Gata1-ERT), and 103 (Pu.1-ERT). A consensus set of 22 genes was analyzed in all five compartments.
We next explored cellular heterogeneity within these cell compartments using single-cell multiplex qRT-PCR. The results showed substantial cell-to-cell heterogeneity (Figure 1C) and overlap (Figure 1F) within and among all three compartments; nevertheless, the compartments may be robustly identified using single and dual-gene classifiers (Table S1). The transcriptional heterogeneity observed within individual populations may highlight the capture of multiple contemporaneous molecular programs underlying lineage commitment under self-renewal conditions. Since TFs are potent instigators of lineage determination or reprogramming, we next examined cellular and transcriptional heterogeneity in FDCPmix cells driven to erythroid or myeloid lineages through expression of inducible Gata1 or Pu.1 estrogen receptor fusions, respectively (Figure S1H). We analyzed the transcriptional programs of single cells captured at various time points (Figure S1I) after induction. In parallel, we studied the temporal dynamics of lineage commitment in this setting and functionally tested their commitment status by evaluating the following: (1) their retention of self-renewal potential, i.e., their capacity to re-initiate maintenance cultures; and (2) their lineage potential in response to various cytokine cues. This experimental design affords a dynamic appreciation of cellular and molecular mechanisms employed in lineage specification.
Activation of Gata1 in SR cells led to phenotypic changes at as early as 4–6 hr (Figure 1D), accompanied by the loss of neutrophil potential (at 6 hr) and followed by the loss of SR capacity (at 45 hr) (Figure 1E). Enforced Pu.1 activity resulted in the loss of clonogenic SR potential and elicited a myeloid differentiation bias in a more extended time frame (Figures S1J and S1K). Single-cell transcriptional profiling during TF-driven commitment confirmed the lineage identity of the cells obtained, which broadly separated away from the SR state along erythroid (Gata1) and myeloid (Pu.1) axes (Figure 1F). The analysis also revealed significant heterogeneity of molecular programs throughout the process of commitment. Gata1-driven cells co-occupied the same transcriptional space as ECP cells and showed a similar extent of cell-to-cell variation. In contrast, Pu.1-driven cells, while similarly heterogeneous to MCPs, appeared to occupy a distinct territory (data not shown). This presumably reflects the neutrophilic status of MCPs and a monocytic bias of Pu.1-ERT-differentiated cells, consistent with prior reports of Pu.1-driven cell fate (Laslo et al., 2006).
We exploited the heterogeneity of cells at early stages of spontaneous and TF-driven lineage commitment to explore the transcriptional networks controlling lineage specification. Inspection of patterns of expression for individual genes revealed a fraction of cells in which the gene is off, and a fraction of cells expressing the gene to varying levels (on) (Figure 2A). The on/off status can be described as binary while the distribution of on values represents a continuous component of the data. Thus, for any given gene pair, both binary and continuous relationships are possible; this is exemplified for Gata1 and Epor in Figure 2B. We sought to capture both kinds of information to infer putative transcriptional networks. Methodologically, we used odds ratio (OR) to quantify on/off gene-to-gene associations (Figure 2C) and Spearman rank correlation to measure correlations between gene expression levels (Figure 2D). We combined gene associations obtained by both methodologies to infer putative regulatory networks characterizing SR states and the different modes of lineage commitment (Figure 2E).
Figure 2.

Combined Single-Cell Transcriptional Network Inference Methods Implicate Ddit3 in Lineage Specification
(A) Representative gene expression distributions for Epor, Gata1, and Tal1 in ECPs are shown.
(B) Scatterplot of Gata1 and Epor single-cell expression highlights the dual aspect of the data with both binary (on/off) and continuous (expression-level) components.
(C) Contingency table summarizing on/off combination patterns of individual cells for Epor and Gata1. OR quantifies the diagonal versus off-diagonal of this matrix to infer significant positive and negative associations in the binary component of the data. Gata1 and Epor show significant positive association (OR = 3.18; lower95CI > 1).
(D) Scatterplot of Epor and Tal1 expression ranks in co-expressing cells. Epor and Tal1 show significant positive correlation in the continuous component of the data inferred by Spearman rank correlation (r = 0.56; p = 0.002).
(E) Single-cell transcriptional networks in SR, ECP, MCP, Gata1-ERT, and Pu.1-ERT compartments were inferred by combined use of OR and Spearman rank correlation. Solid red lines, positive associations; dashed black lines, negative associations. Node size is proportional to the relative connectivity in each network.
(F) Proportion of negative interactions in the networks in (E) is shown.
(G) Proportion of negative interactions in Gata1-ERT networks at each time point is shown.
At coarse grain, the networks revealed increased connectivity in the lineage-committed compared to the SR state. Also, commitment appeared associated with a higher frequency of negative associations between genes (Figures 2F and S2A), including known lineage-determining factors (Table S1). While this may be, to some extent, a function of the genes analyzed, it also may reflect mechanistically distinct processes governing acquisition of lineage identity versus exit from self-renewal. Negative associations are less prominent in the full-activation time courses of TF-driven commitment, as the networks capture not only the early processes of lineage specification, but also the later consolidation of the differentiation program. This likely increases the proportion of positive associations between lineage-affiliated genes. In contrast, detailed temporal analysis of Gata1-ERT-driven lineage specification revealed that cross-antagonistic associations between lineage determinants peak at 6 hr (Figure 2G), coincident with early loss of neutrophil differentiation potential (Figure 1E) en route to lineage commitment, suggesting that resolution of lineage conflicts is an early step in acquisition of lineage identity. In this respect, Ddit3 emerges as an interesting candidate in lineage cross-antagonism: it is positively associated with Gata2, both in SR and committed cells (Figure S2B), and negatively associated with the neutrophil determinant Cebpa, either directly or through Gata2, in MCPs and at early stages of erythroid lineage commitment (Figure S2B; Table S1). Since Ddit3 has not previously been tabled as a central regulator of erythro-myeloid lineage specification, we functionally tested its impact in loss- and gain-of-function experiments.
Knockdown of Ddit3 (Figures S3A and S3B) in FDCPmix cells resulted in the loss of erythroid and mixed-lineage colonies, with no change to myelo-monocytic potential (Figure 3A). The same loss of erythroid potential in colony-forming assays was observed in stem and progenitor cells (KLS) from mouse BM upon knockdown (Figure 3B) and constitutive knockout (Figure 3C) of Ddit3 expression. The data are compatible with a requirement for Ddit3 in the erythroid lineage, while it is dispensable for the development of the myeloid lineage. The negative association observed between Ddit3 and Cebpa in inferred transcriptional networks from early stages of lineage specification (Figures 2E and S2B; Table S1) suggests that Ddit3 contributes to the erasure of myeloid potential. We tested this in myeloid-committed granulo-monocytic progenitors (GMPs) by enforcing Ddit3 expression (Figure S3C), resulting in a transient re-acquisition of self-renewal potential (Figure 3D) and a dramatic change in the nature of the colonies obtained (Figure 3E), with the predominance of large GM colonies of immature appearance (Figure 3F). Cells in these colonies expressed immature surface markers and were predominantly lineage-negative kit+CD34+CD16/32+, thus presenting an essentially GMP phenotype albeit with variable levels of Sca1 expression; in contrast, cells in control colonies exhibited a differentiated Gr1+Mac1+ phenotype (data not shown). Taken together, the data suggest that ectopic expression of Ddit3 in GMPs blocks lineage progression and transiently re-activates self-renewal capacity.
Figure 3.

Ddit3 Is Required in Early Erythroid Specification and Blocks Myeloid Lineage Progression
(A–C) Lineage potential of multipotent mouse BM cells upon the loss of Ddit3 expression. CFC assays of FDCPmix cells (n = 3) (A) and primary KLS cells (n = 3) (B) upon Ddit3 knockdown and of Ddit3 knockout KLS cells (n = 4) (C) are shown. Error bars, SD.
(D) Re-plating capacity of primary BM GMPs upon enforced expression of Ddit3 read in CFC assays (CSIem, empty vector; n = 4). Colonies were scored 7–10 days after plating of transduced cells (plate 1). The cellular content of the colonies obtained was re-seeded into successive CFC assays (plates 2–4) until the exhaustion of colony production.
(E) Distribution of colony types in CFC plate 1. Most GM colonies obtained upon Ddit3-enforced expression have a blast-like appearance. Error bars, SEM.
(F) Representative images of GM colonies in (E) are shown.
(G) PCA plot of the transcriptional profiles of individual GMPs, either untransduced (WT) or transduced with CSIem- or Ddit3-expressing lentiviral vectors, analyzed for the expression of 44 genes. The first two PC explain 24% of the data variance; n = 114 (CSIem), 84 (Ddit3), and 118 (WT).
(H) Gene loadings of PC1 and PC2 in (G). Genes with the most extreme positions along each axis contribute the most to cell separation along the respective PC.
We used single-cell gene expression profiling of GMPs, either wild-type or transduced with a control vector or a Ddit3-expressing lentivirus, to interrogate the transcriptional program changes imposed by enforced expression of Ddit3 and to inspect its role in remodeling of the transcriptional networks underlying lineage progression and/or identity. Enforcement of Ddit3 changed the expression of two-thirds of genes (Figures S3D and S3E) predicted as its neighbors in our inferred transcriptional networks (Figure S2B), attesting to the robustness of our inference approach. Principal component analysis (PCA) of the populations of individual wild-type and transduced GMP cells separated Ddit3-expressing cells from controls (Figure 3G). This separation is mostly attributable to the increased expression of early erythroid regulators Gata2, Tal1, Zfpm1/Fog1, and Gfi1b and the relative loss of M, GM, and G-CSF receptors (Csf1r, Csf2ra, and Csf3r, respectively) as well as of C/ebp family members (Figure 3H). The relative gain in the expression of erythroid-affiliated genes and loss of myeloid Csf receptors and C/ebp family TFs further developed with prolonged expression of Ddit3 in GMPs under differentiation conditions that support multilineage output (Figures S3F and S3G). These data confirm Ddit3 as a positive regulator of erythroid lineage specification at the expense of myeloid fate, providing an experimental validation of the predictive power of the networks we derived by analyzing the heterogeneity of single cells undergoing lineage specification.
We next asked if the relative gain in importance of erythroid-affiliated regulators in Ddit3-transduced GMPs was associated with a global remodeling of the transcriptional networks underlying GMP lineage identity. Indeed, we observed an overall loss in network connectivity specific to the activity of Ddit3 (Figure 4A) Moreover, there was a relative gain in connectivity of Gata2 at the expense of myeloid hubs, as quantified in Figure S4A. For a broader appreciation of the transcriptional changes induced by Ddit3, we performed RNA sequencing (RNA-seq). GMPs transduced with Ddit3 or control vector (CSIem) were cultured for up to 5 days under conditions supportive of multilineage output. Similarly to cells obtained from colony-forming assays, Ddit3-transduced cells retained a GMP-like phenotype, while control cells acquired differentiated myeloid surface markers (Figure S4B). The global transcriptional profiles of cells with enforced expression of Ddit3 (Figure S4C) were clearly distinct from control-transduced and wild-type GMPs. Gene set enrichment analysis (GSEA) showed that Ddit3 expression is associated with global loss of GMP programs and concomitant upregulation of Meg-E-affiliated signatures (Figure 4B). Interestingly, signatures representative of pre-GM cells, the developmental precursors of GMPs (Figure 4C), also were upregulated (Figure 4B). These data suggest that Ddit3 acts through the development or stabilization of a more primitive precursor with inherent Meg-E potential (Figure 4C). Analysis of the networks derived from wild-type GMPs (Figure S4D) as well as Ddit3-transduced cells exposed to conditions supportive of multilineage output for 2 days (Figure 4A) revealed increased importance of specific erythroid versus myeloid regulatory nodes.
Figure 4.

Ddit3 Remodels the GMP Transcriptional Landscape around Gata2
(A) Transcriptional networks of GMPs transduced with CSIEm and Ddit3 and after 2-day culture under multilineage differentiation conditions. Network inference and representation are as in Figure 2. Highlighted in color are differential regulatory hubs (Gata2, Csf3r, and Mpo); their connectivity is quantified in Figure S4A.
(B) GSEA of early progenitor-affiliated signatures in the transcriptional programs of CSIem- and Ddit3-transduced GMPs is shown.
(C) Diagram shows Ddit3-driven lineage remodeling of GMPs.
(D) Connectivity of the differential regulatory hubs in (A) upon culture of Ddit3-transduced GMPs. Networks are represented in (A) and Figure S4D.
(E) GSEA of Gata2 targets in CSIem- and Ddit3-transduced GMPs. Transduced cells in (B, D, and E) were cultured for 2 days under multilineage differentiation conditions.
Given the association of Gata2 and Ddit3 seen in our network analysis of lineage commitment (Figure 2E) and the increase in activity of Gata2 as a hub in Ddit3-transduced GMPs (Figure 4D), we explored the behavior of Gata2 target genes, previously identified by chromatin immunoprecipitation sequencing (ChIP-seq) in FDCPmix (May et al., 2013), in response to Ddit3 expression. GSEA on the RNA-seq data from Ddit3 and control vector-transduced cells provided evidence for a coincidence of Gata2 and Ddit3-driven gene expression programs (Figure 4E). These data position Ddit3 in an erythroid transcriptional network nucleated by Gata2. To further explore the Ddit3-Gata2 axis in lineage specification, we focused on gene expression programs resident in those single cells that co-expressed the two genes, and derived networks from multipotent and lineage-committed compartments (Table S1). Within the ECP compartment, this analysis revealed the following: (1) an association between the two genes that was not seen when cells were used irrespective of Ddit3/Gata2 status, and (2) the involvement of both these genes in anti-correlations with myeloid-affiliated regulators. A similar picture now also emerges in multipotent cells, highlighting the gains in network information obtainable from interrogating single cells selected on the basis of specific co-expression patterns.
Discussion
Our analysis of cells in the vicinity of commitment highlights the existence of lineage-conflicting programs at the emergence of lineage choice, and it identifies the importance of a Ddit3-Gata2 axis in this process. In revealing this association, it was instrumental that our network inference approach acknowledged and took account of both binary and continuous components of single-cell gene expression. As such, we captured both on/off relationships and the associations seen between distributions of on expression levels in co-expressing cells. Furthermore, having established potentially interesting associations, we zoomed in on single cells where the associations were present to refine their specific network identity. In the case of Ddit3-Gata2, this supported the importance of cross-antagonistic interactions in lineage decisions. While in principle this approach can be applied to any gene-gene interaction in an unsupervised manner, it would require profiling of considerably higher numbers of individual cells to ensure statistical robustness and systematically explore all possible interactions.
To validate our inferred networks, we elected to experimentally test the predicted role of Ddit3 in erythroid lineage specification. Functional experiments revealed an early erythroid effect from Ddit3 loss of function produced by knockdown or genetic knockout. However, we note that the constitutive Ddit3 knockout mice used in this study did not display any discernible erythroid defect (C.F., unpublished data). Thus, lineage-determining networks may be robust to Ddit3 deletion or constitutive deletion may be compensated during development. Forced Ddit3 expression does promote erythroid programming, highlighting a potential role for the Ddit3 node in early development of the erythroid lineage. Previous reports of Ddit3 function in late differentiation of erythro-leukemic cells (Coutts et al., 1999) are compatible, albeit distinct, from our proposed role in erythroid specification. Interestingly, our inspection of CFU-e potential in mouse BM stem and erythroid progenitor compartments upon Ddit3 ablation did not reveal a late erythroid defect (C.F., unpublished data).
The capacity of TFs to re-program cells has been used as a test of their lineage-determining capacity. In the case of Gata1, its introduction into GMPs results in the expression of erythroid potential with the appearance of large blast-like multipotential colonies in vitro (Heyworth et al., 2002). Ddit3 likewise is able to confer erythroid potential on GMPs. Molecular analysis suggests that Ddit3-enforced expression in GMPs leads to expression of erythroid-affiliated genes and an overall transcriptional state similar to that of pre-GM, the precursors of GMPs. One may presume that since pre-GM cells lie upstream of GMPs their transcriptional programs should have diverged less from multipotent cells that retain both GM and E potential. This would explain the E signatures seen in Ddit3-expressing GMPs. By which cellular mechanism does Ddit3 effect these changes? One possibility is that rare pre-GMs exist within prospectively isolated GMP populations and that these are preferentially selected for by Ddit3. This seems unlikely, given the kinetics of the changes in cells and gene expression observed. Alternatively, Ddit3 may regulate a subset of the erythroid program that is simply overlaid on the existing GMP program, resulting in a mixed-lineage program that is reminiscent of pre-GM cells. In such a model, Ddit3 effects a pre-GM state from GMPs, recapitulating its physiological role in lineage programming. Recently, Nerlov and colleagues have suggested that high levels of erythropoietin (EPO) may have instructive effects on lineage specification in vivo (Grover et al., 2014), and interestingly Ddit3 has been suggested as a target of EPO signaling in erythro-leukemic cells (Coutts et al., 1999). The strong network association observed between Ddit3 and Gata2 may indicate that Ddit3 acts on an early erythroid signature primed by Gata2 in cells of mixed-lineage potential (May et al., 2013) and present in the pre-GM state. Elements of this signature may be required for erythroid lineage progression and, thus, explain the erythroid defect observed upon Ddit3 loss of function.
It is interesting to speculate as to the existence of cross-antagonistic interactions between Gata2-centered networks and C/ebp-driven myeloid programs putatively effected through Ddit3. Ddit3 heterodimerizes with C/ebp family members to form complexes that cannot bind DNA, thus blocking activation of C/ebp-driven programs, a mechanism that has been described to block differentiation in mesenchymal lineages (Han et al., 2013; Shirakawa et al., 2006). A combination of Ddit3 structure-function mutant studies and direct investigation of Gata2 and C/ebp DNA binding in Ddit3-expressing GMPs will contribute to clarifying dislodgement of C/ebp complexes from their target genes as a putative mechanism of GMP lineage remodeling.
Experimental Procedures
Mice
B6.129S-Ddit3tm1Dron/J (Ddit3 KO) mice (Jackson ImmunoResearch Laboratories) and C57BL/6 mice were maintained in the John Radcliffe Hospital and CR-UK London Research Institute animal facilities in accordance with Home Office regulations.
Cell Culture and Lentiviral Transductions
FDCPmix culture conditions, lentiviral transductions with Gata1-ERT and Pu.1-ERT constructs, and tamoxifen activation were performed as described previously (May et al., 2013). Lentiviral transductions of FDCPmix cells with Ddit3-small hairpin RNA (shRNA) were performed under maintenance culture conditions; transductions of primary BM cells were performed in serum-free expansion medium (SFEM) (STEMCELL Technologies) supplemented with mouse stem cell factor (SCF) and Flt3L (50 ng/ml). GFP+ cells were sorted after 2 days for downstream assays. In some experiments, GFP+ GMPs were cultured for up to 5 days in Iscove’s modified Dulbecco’s medium (IMDM) + 10% fetal calf serum (FCS) supplemented with mouse SCF (50 ng/ml), mouse IL-3 and IL-6 (10 ng/ml), and human recombinant erythropoietin (EPREX, 10 U/ml). Colony-forming cell (CFC) assays used M3234 supplemented with rat SCF (100 ng/ml); mouse IL-3 (0.01 ng/ml) and EPREX (10 U/ml) (FDCPmix); and M3434 or M3234 supplemented with mouse SCF (100 ng/ml), mouse IL-3, IL-11, GM-CSF, and Tpo (10 ng/ml), and EPREX (10 U/ml). All mouse cytokines were from PeproTech and CFC media were from STEMCELL Technologies.
Single-Cell qRT-PCR
Transcriptional profiling of up to 48 genes in individual cells was performed on a Fluidigm platform and the data retrieved and quality-controlled as described previously (Teles et al., 2014). The ΔCt values were calculated to the mean of the three control genes utilized. Heatmap representation of Z score-normalized ΔCt values was performed in Genesis; PCA plots used the Statistical Toolbox in MATLAB (MathWorks). The Taqman probes used are listed in the Supplemental Experimental Procedures.
Classification and Network Inference
Single-cell gene expression data were linearly transformed as described previously (Teles et al., 2013). Logistic regression linear classifiers were used to infer the best predictor genes in the separation between two cell populations (Teles et al., 2013). Single-cell transcriptional networks were inferred by calculating significant pairwise associations using both continuous (Spearman rank correlations) and binary (OR) components of linearly transformed expression data. Spearman rank correlations were calculated between all pairs of genes co-expressed by a minimum of ten cells in a given population. Correlation coefficients >0.4 with p < 0.01 were considered to be significant. OR and respective 95% confidence intervals (CIs) were calculated based on presence/absence patterns of expression for all pairs of genes in a given population. Significant positive and negative associations were called when Lower95CI > 1 and Upper95CI < 1, respectively. Network representations of significant pairwise associations in both methods were produced using Cytoscape (Smoot et al., 2011)
Additional methods are described in the Supplemental Experimental Procedures.
Author Contributions
C. Pina and C.F. designed and performed experiments and analyzed data. J.T. developed and implemented computational methods and analyzed data. G.M., Y.G., and J.B. performed experiments. D.W. and S.S. analyzed transcriptome data. P.E., M.O., and C. Peterson developed computational methods. T.E. supervised the project. C. Pina and T.E. wrote the paper with input from J.T., C.F., G.M., and C. Peterson.
Acknowledgments
We thank Kevin Clark, Arnold Pizzey, Alex Tipping, and Craig Waugh for expert cell sorting. This work was funded by the Swedish Research Council (C. Peterson and J.T.); the Swedish Foundation for Strategic Research through Create Health (P.E. and M.O.); and the UCL/UCLH Biomedical Research Centre, Leukaemia and Lymphoma Research and Cancer Research UK program grants and Children with Cancer and Great Ormond Street Hospital charity grants (T.E.).
Published: June 4, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, four figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.05.016.
Accession Numbers
The accession numbers for the microarray and RNA-seq data reported in this paper are GEO: GSE68754 and SRA: SRP045672, respectively.
Supplemental Information
References
- Bruno L., Hoffmann R., McBlane F., Brown J., Gupta R., Joshi C., Pearson S., Seidl T., Heyworth C., Enver T. Molecular signatures of self-renewal, differentiation, and lineage choice in multipotential hemopoietic progenitor cells in vitro. Mol. Cell. Biol. 2004;24:741–756. doi: 10.1128/MCB.24.2.741-756.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutts M., Cui K., Davis K.L., Keutzer J.C., Sytkowski A.J. Regulated expression and functional role of the transcription factor CHOP (GADD153) in erythroid growth and differentiation. Blood. 1999;93:3369–3378. [PubMed] [Google Scholar]
- Davidson E.H. Emerging properties of animal gene regulatory networks. Nature. 2010;468:911–920. doi: 10.1038/nature09645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf T., Enver T. Forcing cells to change lineages. Nature. 2009;462:587–594. doi: 10.1038/nature08533. [DOI] [PubMed] [Google Scholar]
- Grover A., Mancini E., Moore S., Mead A.J., Atkinson D., Rasmussen K.D., O’Carroll D., Jacobsen S.E., Nerlov C. Erythropoietin guides multipotent hematopoietic progenitor cells toward an erythroid fate. J. Exp. Med. 2014;211:181–188. doi: 10.1084/jem.20131189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G., Luc S., Marco E., Lin T.W., Peng C., Kerenyi M.A., Beyaz S., Kim W., Xu J., Das P.P. Mapping cellular hierarchy by single-cell analysis of the cell surface repertoire. Cell Stem Cell. 2013;13:492–505. doi: 10.1016/j.stem.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J., Murthy R., Wood B., Song B., Wang S., Sun B., Malhi H., Kaufman R.J. ER stress signalling through eIF2α and CHOP, but not IRE1α, attenuates adipogenesis in mice. Diabetologia. 2013;56:911–924. doi: 10.1007/s00125-012-2809-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyworth C., Pearson S., May G., Enver T. Transcription factor-mediated lineage switching reveals plasticity in primary committed progenitor cells. EMBO J. 2002;21:3770–3781. doi: 10.1093/emboj/cdf368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laslo P., Spooner C.J., Warmflash A., Lancki D.W., Lee H.J., Sciammas R., Gantner B.N., Dinner A.R., Singh H. Multilineage transcriptional priming and determination of alternate hematopoietic cell fates. Cell. 2006;126:755–766. doi: 10.1016/j.cell.2006.06.052. [DOI] [PubMed] [Google Scholar]
- May G., Soneji S., Tipping A.J., Teles J., McGowan S.J., Wu M., Guo Y., Fugazza C., Brown J., Karlsson G. Dynamic analysis of gene expression and genome-wide transcription factor binding during lineage specification of multipotent progenitors. Cell Stem Cell. 2013;13:754–768. doi: 10.1016/j.stem.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moignard V., Macaulay I.C., Swiers G., Buettner F., Schütte J., Calero-Nieto F.J., Kinston S., Joshi A., Hannah R., Theis F.J. Characterization of transcriptional networks in blood stem and progenitor cells using high-throughput single-cell gene expression analysis. Nat. Cell Biol. 2013;15:363–372. doi: 10.1038/ncb2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moignard V., Woodhouse S., Haghverdi L., Lilly A.J., Tanaka Y., Wilkinson A.C., Buettner F., Macaulay I.C., Jawaid W., Diamanti E. Decoding the regulatory network of early blood development from single-cell gene expression measurements. Nat. Biotechnol. 2015;33:269–276. doi: 10.1038/nbt.3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pina C., Fugazza C., Tipping A.J., Brown J., Soneji S., Teles J., Peterson C., Enver T. Inferring rules of lineage commitment in haematopoiesis. Nat. Cell Biol. 2012;14:287–294. doi: 10.1038/ncb2442. [DOI] [PubMed] [Google Scholar]
- Rusnakova V., Honsa P., Dzamba D., Ståhlberg A., Kubista M., Anderova M. Heterogeneity of astrocytes: from development to injury - single cell gene expression. PLoS ONE. 2013;8:e69734. doi: 10.1371/journal.pone.0069734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakawa K., Maeda S., Gotoh T., Hayashi M., Shinomiya K., Ehata S., Nishimura R., Mori M., Onozaki K., Hayashi H. CCAAT/enhancer-binding protein homologous protein (CHOP) regulates osteoblast differentiation. Mol. Cell. Biol. 2006;26:6105–6116. doi: 10.1128/MCB.02429-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoot M.E., Ono K., Ruscheinski J., Wang P.L., Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ståhlberg A., Andersson D., Aurelius J., Faiz M., Pekna M., Kubista M., Pekny M. Defining cell populations with single-cell gene expression profiling: correlations and identification of astrocyte subpopulations. Nucleic Acids Res. 2011;39:e24. doi: 10.1093/nar/gkq1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teles J., Pina C., Edén P., Ohlsson M., Enver T., Peterson C. Transcriptional regulation of lineage commitment—a stochastic model of cell fate decisions. PLoS Comput. Biol. 2013;9:e1003197. doi: 10.1371/journal.pcbi.1003197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teles J., Enver T., Pina C. Single-cell PCR profiling of gene expression in hematopoiesis. Methods Mol. Biol. 2014;1185:21–42. doi: 10.1007/978-1-4939-1133-2_3. [DOI] [PubMed] [Google Scholar]
- Wolff L., Humeniuk R. Concise review: erythroid versus myeloid lineage commitment: regulating the master regulators. Stem Cells. 2013;31:1237–1244. doi: 10.1002/stem.1379. [DOI] [PubMed] [Google Scholar]
- Zinszner H., Kuroda M., Wang X., Batchvarova N., Lightfoot R.T., Remotti H., Stevens J.L., Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.