

Abstract
A total synthesis of the aglycone of IB-00208 was accomplished in 22 steps using a newly developed approach towards polycyclic 1,4-dioxygenated xanthones from benzocyclobutenones. The generality of this entry to xanthones was initially established on several model systems before it was successfully applied to the construction of the hexacyclic core of the natural product. A new and potentially general approach towards angularly-fused benzocyclobutenones using ring-closing metathesis (RCM) was also developed.
Keywords: cyclobutenediones, natural products, polycyclic xanthones, Moore rearrangement, total synthesis
Graphical Abstract
To create your abstract, type over the instructions in the template box below. Fonts or abstract dimensions should not be changed or altered.
1. Introduction
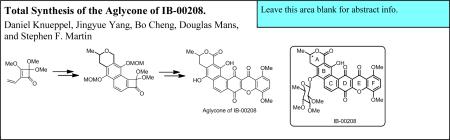
The polycyclic xanthone IB-00208 (1) (Figure 1) was isolated by Romero and coworkers in 2003 from the culture broth of Actinomadura sp.1 This novel compound exhibits potent cytotoxic activity (MIC = 1 nM) against several tumor cell lines, including P388D1, A-549, HT-29 and SK-MEL-28. It also displayed strong antibiotic activity against several Gram-positive bacteria. The hexacyclic core of IB-00208 contains a 1,4-dioxygenated xanthone subunit and is structurally related to other naturally-occuring polycyclic xanthones such as citreamicin α (2),2 kibdelone A (3),3 and cervinomycin A2 (4),4 all of which have been reported to exhibit significant biological activities (Figure 1). The stereochemistry of the sugar moiety in IB-00208 (1) was established by analysis of spin-spin coupling constants and NOESY experiments, but the stereochemistry of the methyl group at C(3) of 1 is unknown.
Figure 1.
Xanthone-Containing Natural Product 1-4
Owing to the complex polycyclic structures and potent biological activities of 1–4, there has been considerable interest in the total synthesis of these and other polycyclic xanthones.5–8 However, to date neither IB-00208 nor citreamicin α have succumbed to total synthesis. A key structural subunit embedded in many of these natural products is a 1,4-dioxygenated xanthone, which itself presents some significant challenges to synthesis.9 In the context of our interest in these and other polycyclic xanthone natural products, we developed a concise and general route to 1,4-dioxygenated xanthones that features a novel extension of the Moore cyclization,10,11 and we recently reported the application of this approach to xanthones to the first total synthesis of the aglycone of IB-00208 (1).12 Herein, we report a detailed account of these efforts.
2. Background for Synthetic Planning
When considering potential approaches to construct the xanthone moiety of IB-00208 (1), we were inspired by the conversion of cyclobutenones to quinones as reported by Moore and coworkers (Scheme 1).10 For example, 5 undergoes a rearrangement upon heating to give quinone 9 by a sequence of reactions that was proposed to be initiated by a 4π-electrocyclic ring opening reaction to provide ketene intermediate 6, which underwent radical cyclization to diradical 7 followed by intermolecular hydrogen abstraction to provide 9.13 The carbon-centered, aryl radical of 7 can also participate in homolytic aromatic substitution reactions, which our group exploited in a concise total synthesis of cribrostatin 6.14
Scheme 1.

In another finding relevant to our own research plan, Fuganti and coworkers reported that 3-en-5-ynoic acids 10 were converted into dibenzofurans 12 when heated under reflux in acetic anhydride in the presence of sodium acetate (Scheme 2).15,16 The newly formed furan and benzene rings were suggested to have been formed in a single annulation step via the ketene intermediate 11.
Scheme 2.

3. The First Generation Approach
3.1. Retrosynthetic Analysis
In our initial consideration of possible approaches towards IB-00208 (1), we envisioned a key step that combines both of these two precedents for the construction of the xanthone core of the natural product (Scheme 3). Specifically, heating of the key intermediate cyclobutenone 15 was expected to produce the acetylenic vinyl ketene 14 in accord with the findings of Moore (cf. Scheme 1). Cyclization of the proximal phenol in 14 via an ionic pathway as proposed by Fuganti (cf. Scheme 2) would then deliver xanthone 13. Elaboration of 13 to the natural product would require oxidation of the hydroquinone ring, construction of the A-ring, and regioselective glycosylation.
Scheme 3.

Compound 15 was envisaged to be accessed by the union of acetylide 17 and fused-cyclobutenone 16 by a 1,2-addition. When we initiated this project, angularly-fused, polycyclic cyclobutenedione derivatives such as 16 were unknown, but we reasoned that 16 might be readily accessible from coupling of arylbromide 18 and the known vinyl squarate 19,17 followed by ring closing metathesis (RCM) to form the benzene ring.18 The synthesis of acetylene 17 would then entail the straightforward addition of an acetylide anion to aldehyde 20, followed by removal of the MOM protecting group and formation of a cyclic silyl bis-ether.
3.2. Initial Model Studies
The synthesis of acetylide 17 commenced with the addition of an alkynyl Grignard reagent to the known aldehyde 2019,20 to produce 21 exclusively (Scheme 4). We welcomed the unexpected, yet fortuitous, loss of the MOM protecting group, because it enabled the direct formation of the requisite cyclic silyl ether that protected both the phenol and secondary alcohol of 21.21 This silyl protecting group was selected because of its robustness to various reaction conditions and its orthogonality to other protecting groups that were envisioned in route to IB-00208 (1).
Scheme 4.

With alkyne 17 in hand, we wanted to validate the proposed reaction sequence for forming the xanthone subunit of IB-00208 (1) using a model system. Toward this end, the known benzocyclobutenedione 22 was selected as a substrate because it possessed an aryl group fused to a cyclobutenone, was symmetrical, and was readily accessible (Scheme 5).22 Deprotonation of alkyne 17 using n-BuLi followed by addition of the resultant anion to 22 provided the 1,2-addition product, which was treated with HF·pyridine to remove the cyclic silyl group to furnish 23 in 48% yield over two steps. Unfortunately, attempts to oxidize the secondary alcohol in 23 were unsuccessful, perhaps owing to the instability of the ynone product. When the cyclobutenol 23 was heated, a simple Moore rearrangement occurred to give 24.10,11 The putative acetylenic vinyl ketene intermediate did not undergo the desired cyclization as expected from the work of Fuganti15 under any of the conditions examined.
Scheme 5.

Notwithstanding our disappointment of the failed cyclization, we reasoned that the ketone 25 might undergo cyclization to give the desired tetracyclic xanthone 26. In the event, addition of IBX to the reaction mixture produced after heating 23 afforded a mixture of the ketone 25 and spirocycle 26. When the crude reaction mixture was stirred with silica gel, complete conversion to spirocycle 26 ensued in 68% overall yield from 23. The regiochemical outcome of the cyclization to form the spirocyclic ring system in 26 rather than the desired 1,4-dioxygenated xanthone ring in 27 was consistent with previous observations from our laboratory in which we found that the substitution pattern on the F-ring of 25 governs the regioselectivity of cylization.11 Microwave heating of 25 resulted in rearrangement of the spirocycle to the thermodynamically favored xanthone, and an aqueous base work-up resulted in oxidation of the hydroquinone intermediate and isolation of quinone 27 in quantitative yield. Although our initially proposed reaction sequence for forming xanthones directly from cyclobutenones was unsuccessful, we devised an alternative approach that allowed us to effectively convert cyclobutenedione 22 into xanthone 27 in only five steps.
Having successfully validated our approach to prepare tetracyclic 1,4-dioxygenated xanthones in a model system, we turned our attention towards accessing the angularly-fused cyclobutenone 16 and transforming it into IB-00208 (1) by the plan adumbrated in Scheme 3.
3.3. Synthesis of Angularly-Fused Benzocyclobutenones
Based on our retrosynthetic approach in Scheme 3, the syntheses of IB-00208 (1) and other polycyclic natural products such as 2–4 required the development of a workable plan to prepare angularly-fused benzocyclobutenones. Toward this end, synthesis of a model system began with conversion of commercially available 2,5-dimethoxybenzaldehyde (28) to an acetal, which would serve as an ortho-directing group for selective lithiation with n-BuLi. (Scheme 6). Quenching of the intermediate aryllithium reagent with 1,2-dibromo-1,1,2,2-tetrachloroethane, followed by deprotection of the acetal provided 29 in 62% yield over three steps. Peterson olefination of the aldehyde moiety in 29 afforded styrene 30. Metal-halogen exchange of the arylbromide followed by coupling of the resulting aryllithium with vinyl squarate 19 following a procedure reported by Moore gave diene 31 in 66% yield.17 In a novel extension of ring closing metathesis (RCM) to form an aromatic ring,18 heating of 31 with 5 mol% of Grubbs II catalyst in toluene gave benzocyclobutenone 32 in 90% yield.
Scheme 6.

Having established a viable new route to angularly-fused benzocyclobutenones, we turned our attention to preparing the requisite cyclobutenone 16 (Scheme 7). Commercially available hydroquinone 33 was regioselectively brominated,23 and the hydroquinone was protected as its bis-MOM ether giving 34 in 92% overall yield. Wittig olefination of the aldehyde moiety in 34 afforded bromide 18.24 Although initial attempts to couple of 18 with squarate 19 using n-BuLi to promote lithium-halogen exchange were plagued by extensive protodelithiation, this problem was easily solved by utilizing t-BuLi giving 35 in 57% overall yield from 34. When diene 35 was heated under reflux in the presence of Grubbs II catalyst, the angular cyclobutenone 16 was isolated in 75% yield.
Scheme 7.

3.4. Synthesis of Pentacyclic Core of IB-00208
In preliminary experiments we discovered that the acetylide anion of 17 did not add to the angular cyclobutenone 32 under a variety of conditions. Attributing this lack of reactivity to increased steric effects of 32 relative to 22, we were required to adopt an alternate approach using a smaller acetylide anion nucleophile (Scheme 8). To this end, 16 was treated with bromomagnesium acetylide to provide alcohol 36. The dianion of 36 added smoothly to the aldehyde 37, which was prepared via silylation20 of the corresponding known phenol,19 to furnish an intermediate adduct that was converted into 38 in 78% yield from 36 by removal of the dimethoxy ketal using p-toluenesulfonic acid.
Scheme 8.

Heating 38 eventuated in the expected Moore rearrangement, and the intermediate secondary alcohol of the resulting product was oxidized using IBX to give the ketone 39 in 41% overall yield (Scheme 9). Treatment of ketone 39 with TBAF in an attempt to affect its cyclization to the pentacycle 41 in analogy with work reported by Theodorakis and coworkers produced only complex mixtures (not shown).25 However, removal of the TBS group using HF·pyridine gave exclusively the spirocycle 40 via spontaneous cyclization of the intermediate phenoxide, in a mode that is consistent with results observed in our model study (cf. Scheme 5) and earlier work.11 Gratifyingly, heating 40 in nitrobenzene led to the rearrangement of the spirocycle to furnish 41 in 58% yield. This critical sequence of reactions established the underlying viability of our novel approach to the pentacyclic core of IB-00208.
Scheme 9.

The structure of 41 was confirmed by X-ray crystallography (Figure 2). The two oxygen atoms O(5) and O(6) (crystal structure numbering) at the angular fusion are in very close proximity (2.55 Å) to each other, which forces the carbonyl group C8–O5 to bend out of plane significantly giving rise to a helix-like structure with the angle between the C(11)-O(6) and C(8)-O(5) bonds being 42.9°.
Figure 2.

X-Ray Crystal Structure of 41
3.5. Attempts to Complete Synthesis of IB-00208 (1)
Having successfully accomplished the synthesis of the pentacycle 41, the stage was set to complete the assembly of the core of IB-00208 (1) by annelating the A-ring of the natural product. Numerous efforts toward this end were met with unanticipated challenges, only a few of which will be discussed here. For example, all strategies for introducing substituents on the B-ring that would enable A-ring formation required removal of the MOM protecting groups. However, endeavours (e.g., TFA, H2O) to effect either selective mono- or di-deprotection of the MOM ethers only afforded complex mixtures with at best trace amounts of the mono-deprotected material being observed (Scheme 10).
Scheme 10.
In view of the difficulties encountered with deprotecting 41, we queried whether the spirocycle 40 might be more accommodating. Accordingly, 40 was readily transformed into 43 under acidic conditions (TFA/H2O, 9:1) to provide exclusively the monodeprotected phenol 43 in quantitative yield (Scheme 11). However, all attempts to brominate (e.g., NBS; Br2; CuBr2) or formylate [e.g., (CHO)n, SnCl4; POCl3, DMF]43 were unavailing.
Scheme 11.

At this stage, it became apparent that neither 40 nor 41 would likely serve as an intermediate in a synthesis of IB-00208 (1). Not only were these compounds uncooperative in our exploratory experiments, but any plans for forming the A-ring on either 40 or 41 would require a disproportionately large number of steps. A new, more concise plan was needed.
4. The Second Generation Approach
4.1. Revised Retrosynthetic Analysis
The lessons of our previous efforts suggested that a better approach to IB-00208 (1) would involve having the A-ring present from the outset. After considering several possibilities, we decided upon incorporating the A-ring as a dihydropyran ring. Such a plan would necessarily lead to an advanced intermediate such as 45 in which the requisite lactone A-ring in 1 would be accessible via a late-stage benzylic oxidation at C(1) (Scheme 12). Compound 45 would be formed from 46 via a sequence involving a Moore rearrangement, cyclizations, and an oxidation in close analogy with the preparation of 41 from 38. The synthesis of 46 required an acetylide stitching sequence that joins aldehyde 37 and benzocyclobutenone 47 in a process similar to that employed to assemble 38 from 16 and 37. The angularly-fused benzocyclobutenone 47 would be accessed by coupling the aryl anion generated from bromide 48 with squarate 19 followed by RCM.
Scheme 12.

4.2. Synthesis of Hexacyclic Core of IB-00208 (1)
The first task that needed to be addressed in this second generation approach to IB-00208 (1) was the synthesis of 48. Because the stereochemistry at C(3) of the natural product was unknown, we sought a route that might be easily adjusted to prepare either enantiomer of 48. Accordingly, commercially available 2-bromo-1,4-dimethoxybenzene (49) was treated with n-BuLi to induce a lithium-halogen exchange, and the resulting aryllithium reagent was allowed to react with propylene oxide to furnish the racemic secondary alcohol 50 (Scheme 13).26 Because either antipode of optically pure propylene oxide is commercially available, this route may be readily adapted to access either enantiomer of 50 as needed once the absolute stereochemistry at C(3) of IB-00208 (1) is known. Bromination of 50 by NBS in the presence of a catalytic amount of ammonium nitrate (NH4NO3),27 followed by formation of the MOM-ether gave 51 in 98% yield over two steps.
Scheme 13.

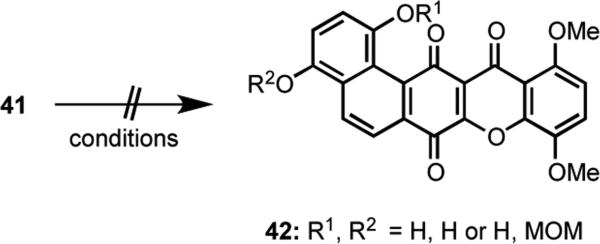
The key conversion of 51 to 52 was first effected by a TMSOTf-induced oxa-Pictet-Spengler cyclization28 to generate the dihydropyran ring followed by oxidation of the aromatic ring using ceric ammonium nitrate [(NH4)2Ce(NO3)6] (CAN) and reduction of the intermediate quinone giving 52 in 43% yield (sequence not shown). This approach, however, required chromatographic purifications after each step because of unavoidable side reactions. For example, when 51 was treated with TMSOTf, some of the MOM ether was lost to generate the secondary alcohol derivative of 51. The oxidation using CAN 27also led to overoxidation giving a product containing a hydroxyl group at C(1).These problems were easily addressed by resequencing the oxidation/reduction and cyclization steps (Scheme 13). Namely, oxidation of 51 using CAN proceeded smoothly without any overoxidation, and TMSOTf-induced cyclization occurred to give 52 in 89% overall yield from 51 that was contaminated with only small amounts (ca 5%) of the deprotected alcohol. A Duff reaction of 52 with hexamethylenetetramine (HMTA)29 gave aldehyde 53 in 65% yield. Following protection of the two phenolic hydroxy groups in 53 as their MOM ethers, a Wittig olefination provided key arylbromide 48 in 73% yield from 53.
The next stage of the synthesis required annelation of a benzocyclobutenone ring on 48 to give 47 (Scheme 14).Toward this end, coupling of the aryllithium reagent generated from 48 with vinyl squarate 19 gave diene 54 in 64% yield. For this reaction, we found that use of Et2O as the solvent rather than THF gave reproducibly better yields. Cyclization of 54 via RCM using Grubbs II catalyst delivered the angularly-fused benzocyclobutenone 47 in 81% yield.
Scheme 14.
The acetylide stitching sequence that would join 47 and aldehyde 37 was now at hand. Although the addition of bromomagnesium acetylide to 47 proceeded smoothly to give 55 in 88% yield (Scheme 15), the subsequent coupling of 55 with 37 proved to be somewhat problematic. Reaction of the dianion of 55, which was generated in situ using EtMgBr as the base, with 37 gave the desired product in up to 81% yield, but the reaction was somewhat capricious because reproducibly eliminating all traces of moisture from 55 was difficult. We discovered, however, that deprotonation of 55 using an excess of the strong, non-nucleophilic base bromomagnesium 2,2,6,6-tetramethylpiperidide (TMPMgBr), followed by reaction with 37 reproducibly gave 56 in 80% yield. Deprotection of the dimethoxy ketal moiety of 56 under acid conditions gave 46 in 79% yield. Heating 46 in DMSO at 100 °C induced a Moore rearrangement to furnish 57, which was then transformed into 58 via oxidation of the intermediate alcohol and fluoride-induced removal of the TBS protecting group, which proceeded with concomitant cyclization. It is perhaps noteworthy that deprotection of the TBS group using HF·pyridine only proceeded in about 10-29% yield. When 58 was heated at increasingly higher temperatures from 150 to 215 °C (see experimental section), rearrangement to 45 proceeded in 67% yield.
Scheme 15.

4.3. Endgame Chemistry to Complete the Synthesis of the Aglycone of IB-00208
In order to complete the synthesis of IB-00208 (1) from hexacycle 45, a benzylic oxidation at C(1) was needed to produce the lactone moiety present in the A-ring of 1. We had found in a much simpler model substrate (not shown) that this oxidation could be successfully performed using a variety of reagents, including RuCl3•H2O and NaIO4,30 RuO2 and NaIO4,31 and CrO3 and 3,5-dimethylpyrazole.32 To our dismay, however, when these and other conditions were applied to 45, only complex reaction mixtures were obtained (Scheme 16). Similar attempts to effect oxidation at C(1) of spirocycle 58 were also unavailing (Scheme 17).
Scheme 16.
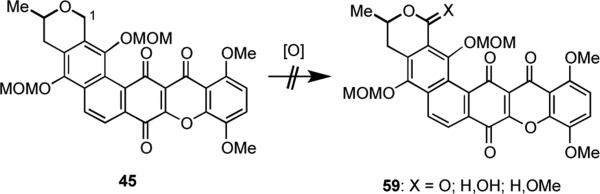
Scheme 17.
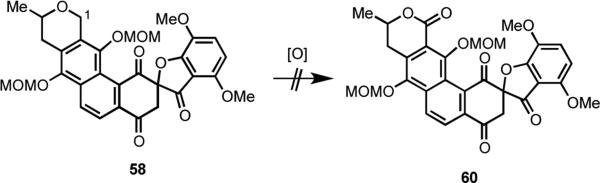
We eventually discovered that oxidation at C(1) could be promoted using DDQ in the presence of MeOH for the earlier-stage intermediate 57 (Scheme 18).33 Three equivalents of DDQ in the presence of BaCO3 provided 61 in 85% yield.34 In an exploratory sequence of experiments, hydrolysis of the mixed acetal at C(1) of 61 using a catalytic amount of p-toluenesulfonic acid gave hemiacetal 62 in 83% yield. Unfortunately, all attempts to oxidize both alcohol groups in 62 using IBX or other oxidants were uniformly unsuccessful and provided only complex mixtures.
Scheme 18.

The failed attempts to convert 62 into 63 dictated the design of a slightly modified approach. We thus discovered that oxidation of the secondary alcohol function in 61 with IBX, proceeded readily to give 64 in 70% yield (Scheme 19). Fluoride-induced removal of the TBS group was accompanied by spontaneous spirocyclization to generate 65 in 87% yield. To our dismay, repeated efforts to rearrange spirocycle 65 to the fused hexacyclic intermediate 66 proved futile.
Scheme 19.

At this stage the options for making adjustments to the sequence were limited. Hopeful that introducing the A-ring lactone prior to performing the thermal rearrangement might prove successful, the acetal moiety at C(1) of 65 was hydrolyzed, and the resulting hemiacetal was oxidized with pyridinium dichromate (PDC) in the presence of 3 Å molecular sieves to give 67 in 81% overall yield from 65 (Scheme 20).35 Gratifyingly, heating spirocycle 67 at 180 °C in a microwave reactor delivered a mixture of hydroquinone 68 and quinone 69, both of which lacked the phenolic MOM protecting group at C(17). Although the oxidation of 68 to 69 was slow when the reaction mixture was simply stirred open to air, continued heating of the reaction mixture in the presence of oxygen resulted in complete conversion to 69, which was isolated in 79% yield from spirocycle 67.
Scheme 20.

Efforts to remove the C(5) phenolic MOM protecting group from 69 under acidic conditions only gave complex mixtures. However, reaction of 69 with freshly distilled TMSBr36 furnished a mixture (~2:1) of compounds for which LC-HRMS and 1H NMR spectral data were consistent with those expected for 70a, the aglycone of IB-00208 (1), and its tautomer 70b. Formation of 70a and 70b in approximately equal amounts is perhaps not unexpected because the redox potentials of the B- and D-rings might be predicted to be similar. Unfortunately, all efforts to separate and purify 70a and 70b were unsuccessful, in part due to their facile interconversion and lability toward decomposition pathways. We were thus neither able to characterize them independently nor to explore glycosylation experiments that would provide IB-00208 (1).
5. Summary
In summary, the first total synthesis of the racemic aglycone of IB-00208 (1) was accomplished in a total of 22 steps from commercially available starting materials. The hexacyclic aglycone 70a was isolated as a mixture of hydroquinone-quinone tautomers, which were both prone toward decomposition and could not be separated or elaborated further. Accordingly, application of our approach to IB-00208 (1) itself will require some adjustments to the protecting group strategy that was implemented in the present synthesis of its aglycone. Several aspects of the synthesis are notable. Facile preparation of polycyclic benzocyclobutenones 16, 32, and 47 is a novel and potentially general extension of the RCM reaction. The scope of this approach toward angularly-fused benzocyclobutenones is currently under investigation in our laboratories and will be reported in due course. Although the cascade reaction that was originally conceived to generate polycyclic xanthones in a one-pot sequence was unsuccessful, a stepwise variant of this process was successfully implemented. Exploitation of the Moore rearrangement to elaborate benzocyclobutenones into highly-functionalized, polycyclic xanthones is significant, and application of this general approach toward the synthesis of other complex xanthone natural products is currently in progress. Results of those investigations will be reported in due course.
6. Experimental
6.1 General
Solvents and reagents were reagent grade and used without purification unless otherwise specified. Tetrahydrofuran (THF) and diethyl ether (Et2O) was passed through two columns of neutral alumina. Methanol (MeOH), acetonitrile (CH3CN), and dimethyl formamide (DMF) were passed through two columns of molecular sieves and stored under argon. Dichloromethane (CH2Cl2) and triethylamine (Et3N) were distilled from calcium hydride and stored under nitrogen. Toluene (PhMe) was passed through a column of Q5 reactant and stored under argon. Reactions involving air- or moisture-sensitive reagents or intermediates were performed under an inert atmosphere of argon or nitrogen in glassware that had been oven dried. Reaction temperatures refer to bath temperatures. Melting points are uncorrected. Infrared (IR) spectra were recorded neat on sodium chloride plates and are reported in wave numbers (cm−1). 1H and 13C NMR spectra were obtained as solutions in CDCl3 unless otherwise noted, and chemical shifts are reported in parts per million (ppm) in reference to CDCl3 (7.24 ppm and 77.0 ppm, respectively). Coupling constants are reported in hertz (Hz). Spectral splitting patterns are designated as follows: s, singlet; d, doublet; t, triplet; q, quartet; app, apparent; br, broad; m, multiplet; comp, overlapping multiplets of magnetically nonequivalent protons.
6.2 Experimental Procedures
3,6-Dimethoxy-2-methoxymethoxybenzaldehyde (20)
A solution of n-BuLi (39.2 mL, 1.6 M, 62.72 mmol) in hexanes was added to a solution of 1,4-dimethoxy-2-methoxymethoxybenzene (10.884 g, 59.73 mmol) in THF (100 mL) at −10 °C. After 30 min, DMF (6.94 mL, 89.60 mmol) was added. The cold bath was removed, and stirring was continued for 4 h at room temperature. Saturated aqueous NH4Cl (40 mL) and Et2O (200 mL) were added, and the layers were separated. The organic phase was washed with H2O (3 × 100 mL) and dried (Na2SO4). The solvent was removed under reduced pressure, and the residue was purified by flash chromatography eluting with hexanes/EtOAc (1:1) to give 7.760 g (57%) of 20 as a yellow oil. Spectral data were consistent with those reported in the literature.19
2-(1-Hydroxyprop-2-ynyl)-3,6-dimethoxyphenol (21)
A solution of 20 (740 mg, 2.78 mmol) in THF (2.8 mL) was added dropwise to a solution ethynylmagnesium bromide (41.7 mL, 0.5 M, 20.85 mmol) in THF at 0 °C. After stirring at 0 °C for 10 min, the cold bath was removed, and the mixture was stirred for 16 h at room temperature. Saturated aqueous NH4Cl (3 mL) was added, and the solution was transferred to a separatory funnel containing H2O (50 mL). The mixture was extracted with Et2O (3 × 30 mL), and the combined organic phases were dried (Na2SO4). The solvent was removed under reduced pressure, and the residue was purified by flash chromatography eluting with hexanes/EtOAc (1:1) to give 482 mg (83%) of 21 as a pale yellow solid: mp 104-105 °C; 1H NMR (500 MHz, CDCl3) δ 6.73 (d, J = 8.9 Hz, 1 H), 6.38 (d, J = 8.9 Hz, 1 H), 6.10 (app s, 1 H), 5.87 (dd, J = 10.9, 2.3 Hz, 1H), 3.98 (app d, J = 11.0 Hz, 1 H), 3.83 (s, 3 H), 3.82 (s, 3 H), 2.46 (d, J = 2.4 Hz, 1 H); 13C NMR (125 MHz, CDCl3) δ 151.5, 143.6, 141.4, 115.5, 110.4, 101.9, 84.2, 71.7, 56.8, 56.5, 56.1; IR (neat) 3486, 3282, 2942, 2838, 1601, 1494, 1247 cm−1; mass spectrum (CI) m/z 209.0816 [C11H13O4 (M+1) requires 209.0814], 209, 191 (base), 181.
2,2-Di-tert-butyl-4-ethynyl-5,8-dimethoxy-4H-benzo[1,3,2]dioxasiline (17)
2,6-Lutidine (0.19 mL, 1.64 mmol) and tert-Bu2Si(OTf)2 (0.24 mL, 0.75 mmol) were added dropwise to a solution of 21 (142 mg, 0.68 mmol) in CH2Cl2 (7 mL) at 0 °C. After 30 min at 0 °C, NH4Cl (6 mL) was added, and the aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic phases were dried (Na2SO4), and the solvent was removed under reduced pressure. The residue was purified by flash chromatography eluting with hexanes/EtOAc (3:1) to give 189 mg (79%) of 17 as a white solid: mp 125-127 °C; 1H NMR (500 MHz, CDCl3) δ 6.76 (d, J = 8.9 Hz, 1 H), 6.37 (d, J = 8.9 Hz, 1 H), 5.99 (d, J = 2.3 Hz, 1 H), 3.80 (s, 3 H), 3.78 (s, 3 H), 2.43 (d, J = 2.3 Hz, 1 H), 1.18 (s, 9 H), 0.92 (s, 9 H); 13C NMR (125 MHz, CDCl3) δ 150.3, 144.5, 144.0, 116.2, 113.5, 102.3, 84.1, 71.8, 60.3, 57.5, 55.9, 27.02, 26.99, 21.7, 20.1; IR (neat) 3284, 2936, 2860, 1596, 1492, 1258 cm−1; mass spectrum (CI) m/z 349.1832 [C19H29O4Si (M+1) requires 349.1835], 349 (base).
8-((2,2-di-tert-Butyl-5,8-dimethoxy-4H-benzo[d][1,3,2]dioxasilin-4-yl)ethynyl)-8-hydroxybicyclo[4.2.0]octa-1(6),2,4-trien-7-one
A solution of n-BuLi (0.045 mL, 2.7 M, 0.12 mmol) in hexanes was added dropwise to a solution of 17 (40 mg, 0.16 mmol) in THF (3 mL) at −78 °C. After 15 min at −78 °C, dione 22 (15 mg, 0.16 mmol) was added, and stirring was continued for 1 h at −78 °C. NH4Cl (3 mL) was added, and the mixture was extracted with EtOAc (3 × 10 mL). The combined organic phases were dried (MgSO4), and the solvent was removed under reduced pressure. The residue was purified by flash chromatography eluting with hexanes/EtOAc (2:1) to give 27 mg (48%) of 8-((2,2-di-tert-butyl-5,8-dimethoxy-4H-benzo[d][1,3,2]dioxasilin-4-yl)ethynyl)-8-hydroxybicyclo[4.2.0]octa-1(6),2,4-trien-7-one as a yellow oil; 1H NMR (500 MHz, CDCl3, mixture of two diastereomers) δ 7.67-7.68 (m, 0.6 H), 7.66-7.63 (m, 0.4 H), 7.62-7.57 (comp, 1 H), 7.56-7.51 (comp, 1 H), 7.50-7.45 (comp, 1 H), 6.78-6.73 (comp, 1 H), 6.38-6.32 (comp, 1 H), 6.04 (s, 0.6 H), 6.02 (s, 0.4 H), 3.79 (s, 1.2 H), 3.77 (s, 1.8 H), 3.74 (s, 1.2 H), 3.73 (s, 1.8 H), 3.20-3.07 (comp, 1 H), 1.10-1.03 (comp, 9 H), 0.93-0.82 (comp, 9 H); 13C NMR (125 MHz, CDCl3, mixture of two diastereomers) δ 186.6, 186.4, 157.88, 157.85, 150.4, 150.3, 148.03, 147.98, 144.5, 144.4, 144.1, 144.0, 136.2, 131.74, 131.73, 122.8, 122.7, 122.5, 122.4, 115.8, 115.6, 113.51, 113.46, 102.45, 102.42, 91.3, 86.5, 79.21, 79.19, 60.5, 60.4, 57.5, 56.0, 27.0, 26.9, 21.69, 21.67, 20.1, 20.0; IR (neat) 3414, 2934, 2860, 1778, 1595, 1492 cm−1; mass spectrum (CI, ESI) m/z 481.2050 [C27H33O6Si (M+1) requires 481.2046], 481 (base).
8-Hydroxy-8-(3-hydroxy-3-(2-hydroxy-3,6-dimethoxyphenyl)prop-1-yn-1-yl)bicyclo[4.2.0]octa-1(6),2,4-trien-7-one (23)
HF·pyridine (6.3 μL, 70% HF, 0.23 mmol) was added using a polypropylene syringe to a solution of 8-((2,2-di-tert-butyl-5,8-dimethoxy-4H-benzo[d][1,3,2]dioxasilin-4-yl)ethynyl)-8-hydroxybicyclo[4.2.0]octa-1(6),2,4-trien-7-one (36 mg, 0.07 mmol) and pyridine (15.1 μL, 0.19 mmol) in THF (1.5 mL) at room temperature. After 10 min at room temperature, the reaction was diluted with Et2O (5 mL). The mixture was washed with sat. aq. NH4Cl (3 × 5 mL) and dried (MgSO4), and the solvent was removed under reduced pressure to 1 mL. The crude mixture was purified by column chromatography eluting with hexanes/EtOAc (1:2) to give 31 mg (100%) of 23 as a clear oil; 1H NMR (400 MHz, CDCl3) δ 7.74 (app d, J = 7.6 Hz, 1 H), 7.62 (app t, J = 7.6 Hz, 1 H), 7.55 (app t, J = 8.4 Hz, 1 H), 7.52-7.47 (comp, 1 H), 6.71 (d, J = 8.8 Hz, 1 H), 6.35 (d, J = 8.8 Hz, 1 H), 6.10 (s, 1 H), 5.90 (br s, 1 H), 3.91 (br s, 1 H), 3.82 (s, 3 H), 3.79-3.73 (comp, 3 H), 3.34 (br s, 1 H); 13C NMR (100 MHz, CDCl3) δ 187.3, 158.4, 151.8, 148.2, 144.0, 141.7, 136.6, 132.0, 123.0, 122.8, 115.4, 110.7, 102.2, 91.3, 86.7, 79.5, 57.1, 56.8, 56.4; IR (neat) 3448, 2922, 1712, 1513, 1263, 1093 cm−1; mass spectrum (CI) m/z 340.0946 [C19H16O6 (M+1) requires 340.0947], 321 (base), 340.
4,7-Dimethoxy-1′H,3H-spiro[benzofuran-2,2′-naphthalene]-1′,3,4′(3′H)-trione (26)
A solution of 23 (12 mg, 0.03 mmol) in THF (1.8 mL) was heated at 120 °C for 30 min in a microwave oven. The reaction was cooled to room temperature, and IBX (15 mg, 0.05 mmol) was added. The reaction was stirred at 60 °C for 4 h and then cooled to room temperature, whereupon hexanes/EtOAc (3:1, 5 mL) was added. The mixture was filtered through Celite and added to CH2Cl2 (2 mL). Silica gel (135 mg) was added, and the reaction was stirred at room temperature for 1.5 h. The solution was filtered through Celite and concentrated under reduced pressure. The residue was purified by flash chromatography eluting with hexanes/EtOAc (2:1) to give 8 mg (68%) of 26 as a yellow oil; 1H NMR (500 MHz, CDCl3) δ 1H NMR (500 MHz, CDCl3) δ 8.19 (app d, J = 7.8 Hz, 1 H), 8.06 (app d, J = 7.8 Hz, 1 H), 7.81 (dt, J = 7.6, 1.2 Hz, 1 H), 7.73 (dt, J = 7.6, 1.2 Hz, 1 H), 7.15 (d, J = 8.8 Hz, 1 H), 6.40 (d, J = 8.8 Hz, 1 H), 3.95 (s, 3 H), 3.82 (s, 3 H), 3.54 (d, J = 16.4 Hz, 1 H), 3.30 (d, J = 16.4 Hz, 1 H); 13C NMR (125 MHz, CDCl3) δ 192.1, 190.4, 186.8, 162.2, 152.2, 139.8, 136.7, 135.2, 134.2, 133.0, 128.2, 126.7, 122.8, 108.9, 103.6, 92.5, 57.1, 56.1, 45.1; IR (neat) 2926, 1698, 1596, 1514, 1271 cm−1; mass spectrum (CI) m/z 339.0865 [C19H15O6 (M+1) requires 339.0869], 339 (base).
1,4-Dimethoxy-6H-benzo[b]xanthene-6,11,12-trione (27)
Spirocycle 26 (1 mg) was added to toluene (3 mL), and the mixture was heated at 250 °C for 2 × 3 h. The solvent was removed and the residue was dissolved in CH2Cl2 (10 mL). The organic layer was shaken with 1 M KOH (10 mL), neutralized with 1 M HCl, and extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure to give 1 mg (100%) of 27 as a red solid; 1H NMR (400 MHz, CDCl3) δ 8.19 (app d, J = 7.2 Hz, 1 H), 8.14 (app d, J = 7.2 Hz, 1 H), 7.79 (dt, J = 7.6, 1.0 Hz, 1 H), 7.73 (dt, J = 7.6, 1.0 Hz, 1 H), 7.16 (d, J = 9.2 Hz, 1 H), 6.78 (d, J = 9.2 Hz, 1 H), 3.95 (s, 3 H), 3.88 (s, 3 H); IR (neat) 2923, 2852, 2360, 1738, 1692, 1489, 1288 cm−1; mass spectrum (CI) m/z 337.0710 [C19H13O6 (M+1) requires 337.0712], 337 (base).
2-(2’,5’-Dimethoxyphenyl)-1,3-dioxane
A solution of 2,5-dimethoxybenzaldehyde (5.00 g, 30.0 mmol), p-TsOH (0.570 g, 3.00 mmol), and 1,3-propane diol (11.0 mL, 150 mmol) were heated under reflux in benzene (60 mL) in a round-bottom flask equipped with a Dean-Stark trap and condenser for 24 h. The mixture was cooled to ambient temperature, Et2O (50 mL) was added, and the mixture was washed with water (3×30 mL) and brine (1×30 mL). The organic layer was dried (MgSO4), filtered, and concentrated to provide an oil. The residue was purified by column chromatography eluting with EtOAc/Hexanes (1:6.7) to provide 6.30 g (94%) of the title compound as a clear colorless oil. Spectral data were in accord with that reported in the literature.37
2-(2’-Bromo-3’,6’-dimethoxyphenyl)-1,3-dioxane
A solution of 2-(2’,5’-dimethoxyphenyl)-1,3-dioxane (6.30 g, 28.0 mmol) dissolved in hexanes (140 mL) was cooled to −25 °C, whereupon benzene (46 mL) was added. A solution of n-BuLi (17.0 mL of a 2.5 M solution in hexanes, 42.0 mmol) was then added dropwise. The mixture was stirred for 18 h at −25 °C. (Note: Care must be taken to keep the internal temperature above −30°C otherwise the mixture freezes. In addition, the use of an overhead stirrer is recommended as the mixture becomes increasingly viscous as the reaction proceeds.) A solution of 1,2-dibromo-1,1,2,2-tetrachloroethane (17.3g, 53.0 mmol) in THF (50 mL) was then added dropwise. The cooling bath was removed, and the mixture stirred for 0.5 h at room temperature. The solvent was removed in vacuo to give a solid, which was purified by column chromatography eluting with EtOAc/hexanes (1:5 to 2:5 gradient) to provide 5.50 g (66%) of the title compound as a pale yellow solid. Spectral data were in accord with that reported in the literature.37
2-Bromo-3,6-dimethoxybenzaldehyde (29)
Concentrated HCl (92.0 mL) was added to a solution of 2-(2’-bromo-3’,6’-dimethoxyphenyl)-1,3-dioxane (5.50 g, 18.0 mmol) in THF (92 mL), and the mixture was stirred at ambient temperature for 20 min. The mixture was extracted with Et2O (3×100 mL) and the combined organic layers were washed with brine (1×100 mL), dried (MgSO4), filtered, and concentrated to provide 4.90 g (100%) of 29 as a yellow solid, which was used without further purification. Spectral data were in accord with that reported in the literature.37
2-Bromo-l,4-dimethoxy-3-vinylbenzene (30)
TMSCH2Cl (0.88 mL, 6.25 mmol) added to a mixture of freshly ground Magnesium turnings (0.61 g, 25 mmol) in THF (50 mL). The mixture was stirred for 15 min then the remaining TMSCH2Cl (2.6 mL, 18.75 mmol) was added and the mixture was stirred rapidly for 12 h. The freshly prepared TMSCH2MgCl was transferred via cannula to a solution of 2-bromo-3,6-dimethoxybenzaldehyde (4.4 g, 18 mmol) in THF (40 mL) over 0.5 h. The solution was stirred for another 0.5 h whereupon 30% H2SO4 (45 mL) was added. The solution was rapidly stirred for 1 h. The mixture was diluted with Et2O (100 mL) and the layers separated. The organic layer was washed with sat. NaHCO3 (1×100 mL), H2O (1×100 mL), and brine (1×100 mL), dried (MgSO4), filtered and concentrated to give 4.2 g (97%) of 30 as an off-white solid.
3-(3’,6’-Dimethoxy-2’-vinylphenyl)-4,4-dimethoxy-2-vinycyclobut-2-enone (31)
A solution of 2.6 M solution of n-BuLi in hexanes (1.0 mL, 2.7 mmol) was added dropwise to a solution of 30 (660 mg, 2.7 mmol) in THF (3 mL) at −78 °C and the solution was stirred for 0.5 h. A solution of 19 (500 mg, 2.7 mmol) in THF (0.5 mL) was then added and the mixture was stirred and allowed to warm to room temperature over 0.5 h. Saturated NaHCO3 (2 mL) was added and the mixture was extracted with Et2O 3×lmL). The combined organic layers were washed with brine (1×3 mL), dried (MgS04), filtered, and concentrated. The crude residue was purified by column chromatography eluting with EtOAc/Hexanes (1:50 to 1:13) gradient to provide 570 mg (66%) of 31 as a yellow solid: 1H NMR (500 MHz, CDCl3) δ 6.88 (d, 1 H, J = 9.0 Hz), 6.78 (d, 1 H, J = 9.0 Hz), 6.64 (dd, 1 H, J = 18.0, 12.0 Hz), 6.09 (dd, 1 H, J = 18.0, 2.0 Hz), 6.02 (dd, 1 H, J = 18.0 Hz, 10.5 Hz), 5.92 (dd, 1 H, J = 18.0, 2.0 Hz), 5.51 (dd, 1 H, J = 10.5, 2.0 Hz), 5.42 (dd, 1 H, J = 12.0, 2.0 Hz), 3.81 (s, 3 H), 3.76 (s, 3 H), 3.45 (s, 6 H); 13C NMR (125 MHz, CDCl3) δ 191.7, 172.1, 152.6, 152.4 149.8, 131.1, 125.8, 125.5, 124.0, 121.1, 117.5, 112.7, 110.4, 56.3, 56.0, 52.8; IR (neat) 2835, 1764, 1578, 1474, 1435, 1256 cm−1; HRMS (CI) calcd 317.1389 for C18H21O5 (M+); found 317.1387.
1,1,5,8-Tetramethoxy-1H-cyclobuta-2-one[3,4a]naphthalene (32)
A mixture of Grubb's II catalyst (19.5 mg, 0.023 mmol) and 31 (37.0 mg, 0.12 mmol) in CH2Cl2 (2.4 mL) was heated to 40 °C for 2 h. The solvent was removed under reduced pressure, and the crude residue was purified by column chromatography eluting with EtOAc/Hexanes (1:10 to 1:5 gradient) to provide 31.0 mg (90%) of 32 as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.41 (d, 1 H, J = 8.5 Hz), 7.50 (d, 1 H, J = 8.5 Hz), 6.99 (d, 1 H, J = 9.0 Hz), 6.93 (d, 1 H, J = 9.0 Hz), 4.00 (s, 3 H), 3.97 (s, 3 H), 3.54 (s, 6 H); 13C NMR (125 MHz, CDCl3) δ 192.9, 160.2, 150.2, 149.6, 146.9, 129.0, 127.6, 121.5, 118.1, 116.7, 108.9, 107.1, 56.1, 56.0, 53.6; IR (neat) 1759, 1617, 1579, 1462, 1265 cm−1; HRMS (CI) calcd 289.1076 for C16H17O5 (M+H)+; found 289.1074.
2-Bromo-3,6-bis(methoxymethoxy)benzaldehyde (34)
MOMCl (9.2 mL, 121.23 mmol) and i-Pr2Net (17.6 mL, 100.94 mmol) were added dropwise to a solution of 2-bromo-3,6-dihydroxybenzaldehyde (8.72 g, 40.38 mmol) in CH2Cl2 (80 mL) at room temperature. After 17 h at room temperature, H2O (100 mL) was added, and the mixture was extracted with Et2O (3 × 100 mL). The combined organic phases were dried (MgSO4), and the solvent was removed under reduced pressure to give 12.32 g (100%) of 34 as a yellow oil; 1H NMR (400 MHz, CDCl3) δ 10.37 (s, 1 H, C7-H), 7.26 (d, J = 9.2 Hz, 1 H, C5-H or C6-H), 7.12 (d, J = 9.2 Hz, 1 H, C5-H or C6-H), 5.18 (s, 4 H, C8-H & C10-H), 3.50 (s, 3 H, C9-H or C11-H), 3.47 (s, 3 H, C9-H or C11-H); 13C NMR (100 MHz, CDCl3) δ 190.5, 153.8, 149.2, 126.0, 121.6, 116.0, 115.2, 95.9, 95.7, 56.5; IR (neat) 2963, 2913, 1700, 1556, 1469, 1394, 1263, 1150 cm−1; mass spectrum (CI) m/z 305.0031 [C11H13O5Br (M+1) requires 305.0025], 305 (base).
2-Bromo-1,4-bis(methoxymethoxy)-3-vinylbenzene (18)
Ph3PCH3Br (28.97 g, 81.36 mmol) and NaH (4.77 g, 119.33 mmol, 60% dispersion in mineral oil) were added to solution of 26 (16.55 g, 54.24 mmol) in THF (540 mL). After 18 h at room temperature, H2O (200 mL) and brine (50 mL) were added, and the mixture was extracted with EtOAc (3 × 100 mL). The combined organic phases were dried (Na2SO4), and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography eluting with hexanes/EtOAc (2:1) to give 16.44 g (100%) of 18 as a pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.03 (d, J = 8.8 Hz, 1 H), 6.98 (d, J = 8.8 Hz, 1 H), 6.78 (dd, J = 17.7, 11.9 Hz, 1 H), 5.86 (dd, J = 17.7, 2.2 Hz, 1 H), 5.59 (dd, J = 11.9, 2.2 Hz, 1 H), 5.16 (s, 2 H), 5.11 (s, 2 H), 3.51 (s, 3 H), 3.46 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 150.7, 149.0, 132.1, 129.2, 121.6, 115.8, 115.4, 115.0, 95.8, 95.4, 56.3, 56.2; IR (neat) 2955, 2901, 2826, 1470, 1251, 1154, 1008 cm−1; mass spectrum (CI) m/z 302.0150 [C12H14O4Br (M+1) requires 302.0154], 302, 273, 271 (base).
3-(3,6-Bis(methoxymethoxy)-2-vinylphenyl)-4,4-dimethoxy-2-vinylcyclobut-2-enone (35)
A solution of t-BuLi (9.1 mL, 14.55 mmol, 1.6 M) in heptane was added quickly to a solution of freshly dried (azeotropic distillation, 2 × 2 mL PhH) 18 (2.052 g, 6.77 mmol) in THF (30 mL) at –78 °C. After 30 sec at –78 °C, a solution of 19 (1.500 g, 8.26 mmol) in THF (5 mL) at –78 °C was added quickly via syringe. After 10 min at –78 °C, the reaction was stirred for 20 min at 0 °C. Upon cooling to – 78 °C, TFAA (2.82 mL, 20.31 mmol) was added dropwise, and the reaction was stirred for 20 min at –78 °C and 5 min at room temperature. Upon addition of saturated aqueous NaHCO3 (15 mL), the reaction was allowed to warm to room temperature. H2O (20 mL) was added, and the mixture was extracted with Et2O (3 × 50 mL). The combined organic phases were dried (Na2SO4), and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography eluting with hexanes/EtOAc (4:1) to give 1.46 g (57%) of 35 as a yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.10 (d, J = 9.2 Hz, 1 H), 7.00 (d, J = 9.2 Hz, 1 H), 6.63 (dd, J = 17.6, 11.7 Hz, 1 H), 6.21-6.00 (comp, 2 H), 5.88 (dd, J = 17.7, 2.1 Hz, 1 H), 5.52 (dd, J = 10.0, 2.9 Hz, 1 H), 5.42 (dd, J = 11.7, 2.1 Hz), 5.14 (s, 2 H), 5.07 (s, 2 H), 3.47 (s, 3 H), 3.44 (app s, 6 H), 3.42 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 191.4, 171.6, 152.5, 150.5, 148.2, 131.0, 126.6, 125.7, 123.9, 121.8, 121.2, 117.3, 117.1, 114.5, 95.21, 95.18, 56.2, 56.0, 52.7; IR (neat) 2946, 2904, 2835, 1766, 149, 1251, 1016 cm−1; mass spectrum (ESI) m/z 3991415 [C20H24O7 (M+23) requires 399.1414], 399 (base).
1,1-Dimethoxy-5,8-bis(methoxymethoxy)cyclobuta[a]naphthalen-2(1H-one (16)
Grubbs second generation catalyst (16 mg, 0.12 mmol) was added to a degassed (Ar, 15 min) solution of diene 35 (145 mg, 0.39 mmol) in toluene (15 mL). The solution was then heated for 5 h at 110 °C, whereupon the reaction was cooled to room temperature. The reaction mixture was concentrated to ca. 1 mL and purified by flash column chromatography eluting with hexanes/EtOAc (4:1) to give 100 mg (75%) of 16 as a yellow oil; 1H NMR (400 MHz, CDCl3) δ 8.41 (d, J = 8.8 Hz, 1 H), 7.49 (d, J = 8.8 Hz, 1 H), 7.28 (d, J = 8.6 Hz, 1 H), 7.18 (d, J = 8.6 Hz, 1 H), 5.33 (s, 2 H), 5.31 (s, 2 H), 3.54 (s, 3 H), 3.50 (app s, 9 H); 13C NMR (100 MHz, CDCl3) δ 193.3, 160.5, 148.4, 147.6, 147.0, 129.4, 127.6, 121.9, 118.4, 116.6, 113.6, 111.6, 95.3, 94.8, 56.22, 56.18, 53.6; IR (neat) 2946, 2906, 2833, 1762, 1458, 1257, 1055 cm−1; mass spectrum (ESI) m/z 371.1102 [C18H20O7 (M+23) requires 371.1101], 719, 371 (base).
2-(tert-Butyldimethylsilanyloxy)-3,6-dimethoxybenzaldehyde (37)
NaH (60% dispersion in mineral oil, 322 mg, 8.05 mmol) was added to a solution of 2-hydroxy-3,6-dimethoxybenzaldehyde19 (1.12 g, 6.19 mmol) in DMF (22 mL) at 0 °C. The mixture was then stirred for 30 min at room temperature and then cooled to 0 °C, whereupon TBSCl (1.40 g, 9.29 mmol) was added. The reaction was stirred for 5 min at 0 °C and 2 h at room temperature, whereupon H2O (50 mL) was added. The mixture was extracted with Et2O (3 × 50 mL), and the combined organic layers were washed with H2O (2 × 50 mL), brine (1 × 50 mL), dried (MgSO4), and concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with hexanes/EtOAc (3:1) to provide 1.55 g (85%) of aldehyde 37 as a yellow oil; 1H NMR (500 MHz, CDCl3) δ 10.49 (app d, J = 0.6 Hz, 1 H), 6.96 (d, J = 9.0 Hz, 1 H), 6.45 (d, J = 8.9 Hz, 1 H), 3.82 (s, 3 H), 3.75 (s, 3 H), 0.96 (s, 9 H), 0.18 (s, 6 H); 13C NMR (125 MHz, CDCl3) δ 190.0, 154.6, 149.1, 144.6, 118.1, 117.4, 103.1, 56.1, 55.7, 25.8, 18.19, −4.1; IR 2933, 2857, 2770, 1693, 1597, 1449, 1283 cm−1; mass spectrum (CI) m/z 297.1522 [C15H25O4Si (M+1) requires 297.1522].
2-Ethynyl-1,1-dimethoxy-5,8-bis(methoxymethoxy)-1,2-dihydrocyclobuta[a]naphthalen-2-ol (36)
A solution of 16 (97 mg, 0.28 mmol) in THF (3.4 mL) was added to a solution of ethynylmagnesium bromide (3.4 mL, 0.5 M, 1.68 mmol) in THF at 0 °C. After 5 min at 0 °C and 2 h at room temperature, saturated aqueous NH4Cl (5 mL) was added. The reaction was diluted with Et2O (50 mL), and the mixture was washed with H2O (3 × 10 mL). The organic phase was dried (MgSO4) and the solvent was removed under reduced pressure to give 80 mg (77%) of 36 as a yellow oil; 1H NMR (400 MHz, CDCl3) δ 8.37 (d, J = 8.4 Hz, 1 H), 7.55 (d, J = 8.4 Hz, 1 H), 7.08 (d, J = 8.4 Hz, 1 H), 7.04 (d, J = 8.4 Hz, 1 H), 5.35-5.21 (comp, 4 H), 3.82 (br s, 1 H), 3.66 (s, 3 H), 3.64 (S, 3 H), 3.55 (s, 3 H), 3.50 (s, 3 H), 2.68 (s, 1 H); 13C NMR (100 MHz, CDCl3) δ 148.5, 146.5, 145.7, 137.4, 127.7, 126.4, 112.9, 118.9, 111.0, 109.4, 107.5, 95.6, 95.2, 81.6, 77.4, 75.6, 56.2, 56.1, 53.0, 52.3; IR (neat) 3439, 3285, 2943, 2839, 1595, 1458, 1258, 1153, 1078, 1009 cm−1; mass spectrum (ESI) m/z 397.1258 [C20H22O7 (M+23) requires 397.1258], 397 (base).
2-(3-(2-(tert-Butyldimethylsilyloxy)-3,6-dimethoxyphenyl)-3-hydroxyprop-1-ynyl)-1,1-dimethoxy-5,8-bis(methoxymethoxy)-1,2-dihydrocyclobuta[a]naphthalen-2-ol
A solution of EtMgBr (0.55 mL, 3 M, 1.64 mmol) in THF was added dropwise to a solution of freshly dried (azeotropic distillation, 2 × 2 mL PhH) 36 (204 mg, 0.545 mmol) in THF (5.4 mL) at 0 °C. After 2 h at room temperature, the reaction was cooled to 0 °C, and a solution of freshly dried (azeotropic distillation, 2 × 2 mL PhH) aldehyde 37 (323 mg, 1.09 mmol) in THF (1 mL) was added dropwise. After 30 min at 0 °C and 10 min at room temperature, saturated aqueous NH4Cl (5 mL) was added. The mixture was extracted with Et2O (3 × 30 mL), and the organic phase was dried (MgSO4). The solvent was removed under reduced pressure, and the residue was purified by flash column chromatography eluting with hexanes/EtOAc (1:1) to give 42 mg (21%) of recovered 36 and 288 mg (79%) of 2-(3-(2-(tert-butyldimethylsilyloxy)-3,6-dimethoxyphenyl)-3-hydroxyprop-1-ynyl)-1,1-dimethoxy-5,8-bis(methoxymethoxy)-1,2-dihydrocyclobuta[a]naphthalen-2-ol as a yellow oil; 1H NMR (400 MHz, CDCl3, mixture of diastereomers) δ 8.33 (d, J = 8.4 Hz, 0.4 H), 8.32 (d, J = 8.4 Hz, 0.6 H), 7.49 (d, J = 8.4 Hz, 0.4 H), 7.48 (d, J = 8.4 Hz, 0.6 H), 7.06 (d, J = 8.6 Hz, 0.4 H), 7.05 (d, J = 8.6 Hz, 0.6 H), 7.02 (d, J = 8.6 Hz, 0.4 H), 7.01 (d, J = 8.6 Hz, 0.6 H), 6.64 (d, J = 8.6 Hz, 0.4 H), 6.63 (d, J = 8.6 Hz, 0.6 H), 6.39 (app d, J = 8.6 Hz, 1 H), 5.86 ( br s, 0.4 H), 5.83 (br s, 0.6 H), 5.47-5.45 (comp, 4 H), 3.73 (s, 1.8 H), 3.69 (s, 1.2 H), 3.68 (s, 1.2 H), 3.67 (s, 1.8 H), 3.59 (app s, 3 H), 3.53 (s, 1.2 H), 3.52 (s, 1.8 H), 3.50 (s, 1.2 H), 3.49 (s, 1.8 H), 3.48 (s, 1.2 H), 3.47 (s, 1.8 H), 0.91 (s, 3.6 H), 0.89 (s, 5.4 H), 0.12 (s, 1.2 H), 0.11 (s, 1.2 H), 0.10 (s, 1.8 H), 0.04 (s, 1.8 H); 13C NMR (100 MHz, CDCl3, mixture of diastereomers) δ 152.4, 148.7, 146.6, 146.3, 144.9, 142.1, 137.4, 127.6, 126.1, 123.1, 121.5, 121.4, 119.2, 111.2, 110.5, 109.3, 107.8, 103.8, 103.6, 95.7, 95.3, 88.3, 88.2, 81.1, 77.6, 57.7, 56.2, 56.1, 55.3, 53.0, 52.1, 26.0, 18.7, −4.2, −4.3; IR (neat) 3515, 2940, 2856, 1490, 1257, 1076 cm−1; mass spectrum (ESI) m/z 693.2700 [C35H46O11Si (M+23) requires 693.2702], 693 (base).
2-(3-(2-(tert-Butyldimethylsilyloxy)-3,6-dimethoxyphenyl)-3-hydroxyprop-1-ynyl)-2-hydroxy-5,8-bis(methoxymethoxy)cyclobuta[a]naphthalen-1(2H-one (38)
p-TsOH (2.6 mg, 0.04 mmol) was added to a solution of 2-(3-(2-(tert-butyldimethylsilyloxy)-3,6-dimethoxyphenyl)-3-hydroxyprop-1-ynyl)-1,1-dimethoxy-5,8-bis(methoxymethoxy)-1,2-dihydrocyclobuta[a]naphthalen-2-ol (91 mg, 0.14 mmol) in acetone (2 mL) at room temperature. After 10 min at room temperature, the reaction was diluted with Et2O (50 mL). The mixture was washed with H2O (2 × 10 mL) and brine (10 mL). The organic phase was dried (MgSO4), and the solvent was removed under reduced pressure to give 84 mg (99%) of 38 as an orange oil; 1H NMR (400 MHz, CDCl3, mixture of diastereomers) δ 8.52 (d, J = 8.5 Hz, 0.4 H), 8.51 (d, J = 8.5 Hz, 0.6 H), 7.71 (app d, J = 8.5 Hz, 1 H), 7.19 (d, J = 8.6 Hz, 0.4 H), 7.28 (d, J = 8.6 Hz, 0.6 H), 7.15 (d, J = 8.6 Hz, 0.4 H), 7.14 (d, J = 8.6 Hz, 0.6 H), 6.67 (d, J = 8.9 Hz, 0.4 H), 6.66 (d, J = 8.9 Hz, 0.6 H), 6.42 (app d, J = 8.9 Hz, 1 H), 5.87 (app s, 1 H), 5.31 (app s, 4 H), 4.07 (app br s, 1 H), 3.79 (s, 1.8 H), 3.77 (s, 1.2 H), 3.69 (s, 1.2 H), 3.68 (s, 1.8 H), 3.54 (s, 1.2 H), 3.53 (s, 1.8 H), 3.50 (app s, 3 H), 0.94 (s, 3.6 H), 0.92 (s, 5.4 H), 0.15 (s, 1.2 H), 0.14 (s, 1.2 H), 0.13 (s, 1.8 H), 0.09 (s, 1.8 H); 13C NMR (100 MHz, CDCl3, mixture of diastereomers) δ 183.3, 159.8, 152.3, 148.4, 147.3, 144.9, 143.0, 142.2, 132.2, 127.7, 120.8, 120.7, 118.6, 113.9, 111.3, 110.8, 103.9, 95.4, 95.3, 91.3, 85.7, 79.6, 57.5, 56.3, 55.3, 26.0, 18.8, −4.09, −4.14; IR (neat) 3375, 2930, 2855, 1774, 1491, 1256, 1078 cm−1; mass spectrum (ESI) m/z 647.2288 [C33H40O10Si (M+23) requires 647.2283], 647 (base).
3-((2-(tert-Butyldimethylsilyloxy)-3,6-dimethoxyphenyl)(hydroxy)methyl)-5,8-bis(methoxymethoxy)phenanthrene-1,4-dione
A solution of benzocyclobutenone 38 (45 mg, 0.07 mmol) in DMSO (3 mL) was heated to 100 °C for 20 min and 120 °C for 10 min. Upon cooling to room temperature, H2O (10 mL) was added, and the mixture was extracted with Et2O (3 × 10 mL). The combined organic phases were washed with H2O (3 × 10 mL) and brine (10 mL) and then dried (Na2SO4). The solvent was removed under reduced pressure, and the residue was purified by flash column chromatography eluting with hexanes/EtOAc (1:2) to give 19 mg (42%) of 3-((2-(tert-butyldimethylsilyloxy)-3,6-dimethoxyphenyl)(hydroxy)methyl)-5,8-bis(methoxymethoxy)phenanthrene-1,4-dione as a burnt orange oil: 1H NMR (400 MHz, CDCl3) δ 8.42 (d, J = 8.8 Hz, 1 H), 8.05 (d, J = 8.8 Hz, 1 H), 7.24 (d, J = 8.6 Hz, 1 H), 7.13 (d, J = 8.6 Hz, 1 H), 6.78 (d, J = 2.3 Hz, 1 H), 6.75 (d, J = 8.8 Hz, 1 H,), 6.46 (d, J = 10.0, 2.3 Hz, 1 H), 6.41 (d, J = 8.8 Hz, 1 H), 5.31 (d, J = 10.0 Hz, 1 H), 5.30 (d, J = 8.4 Hz, 1 H), 5.29 (d, J = 8.4 Hz, 1 H), 4.77 (d, J = 6.8 Hz, 1 H), 4.59 (d, J = 6.8 Hz, 1 H), 3.77 (s, 3 H), 3.62 (s, 3 H), 3.49 (s, 3 H), 3.33 (s, 3 H), 1.00 (s, 9 H), 0.22 (s, 6 H); 13C NMR (100 MHz, CDCl3) δ 187.4, 185.2, 157.1, 152.3, 149.9, 147.8, 145.1, 143.8, 135.1, 132.5, 129.6, 128.1, 126.5, 122.2, 121.1, 120.3, 113.5, 112.1, 110.6, 102.9, 96.0, 95.3, 64.6, 56.2, 55.8, 55.7, 55.1, 26.0, 18.9, −3.8, −4.2; IR (neat) 3425, 2956, 2931, 2856, 1659, 1489, 1454, 1255 cm−1; mass spectrum (ESI) m/z 647.2285 [C33H40O10Si (M+23) requires 647.2283], 647 (base).
3-(2-(tert-Butyldimethylsilyloxy)-3,6-dimethoxybenzoyl)-5,8-bis(methoxymethoxy)phenanthrene-1,4-dione (39)
IBX (81 mg, 0.29 mmol) was added to a solution of 3-((2-(tert-butyldimethylsilyloxy)-3,6-dimethoxyphenyl)(hydroxy)methyl)-5,8-bis(methoxymethoxy)phenanthrene-1,4-dione (60 mg, 0.10 mmol) in DMSO (2.5 mL) at room temperature. After 18 h at room temperature, saturated aqueous Na2S2O3 (5 mL) and saturated aqueous NaHCO3 (5 mL) were added, and the reaction was stirred for 10 min at room temperature. Brine (10 mL) was added, and the mixture was extracted with Et2O (3 × 15 mL). The combined organic phases were washed with saturated aqueous NaHCO3 (10 mL) and H2O (10 mL) and dried (Na2SO4). The solvent was removed under reduced pressure to give 58 mg (97%) of 39 as a red oil; 1H NMR (400 MHz, CDCl3) δ 8.49 (d, J = 8.7 Hz, 1 H), 8.07 (d, J = 8.7 Hz, 1 H), 7.35 (d, J = 8.9 Hz, 1 H), 7.19 (d, J = 8.9 Hz, 1 H), 7.11 (s, 1 H), 6.89 (d, J = 8.9 Hz, 1 H), 6.43 (d, J = 8.9 Hz, 1 H), 5.32 (s, 2 H), 4.74 (s, 2 H), 3.78 (s, 3 H), 3.51 (s, 3 H), 3.51 (s, 3 H), 3.36 (s, 3 H), 0.85 (s, 9 H), 0.15 (s, 6 H); 13C NMR (100 MHz, CDCl3) δ 189.7, 185.8, 185.4, 151.9, 150.1, 148.6, 148.0, 145.4, 144.6, 133.8, 132.6, 132.0, 130.0, 127.1, 122.1, 121.7, 121.1, 114.5, 113.8, 112.5, 102.4, 96.2, 95.3, 56.3, 55.8, 55.7, 25.7, 18.6, −4.4; IR (neat) 2932, 2856, 1662, 1592, 1483, 1254, 1102, cm−1; mass spectrum (ESI) m/z 623.2306 [C33H39O9Si (M+1) requires 623.2307], 645 (base).
4,7-Dimethoxy-5′,8′-bis(methoxymethoxy)-1′H,3Hspiro[benzofuran-2,3′-phenanthrene]-1′,3,4′(2′H)-trione (40)
Pyridine (5 μL, 0.06 mmol) and HF·pyridine (2 drops, 70% wt, 0.07 mmol) were added to a solution of 39 (14 mg, 0.02 mmol) in THF (1 mL) in a plastic tube at room temperature. After 1 day at room temperature, additional HF·pyridine (2 drops) was added, and the reaction was stirred at room temperature. After 1 day, the reaction was diluted with Et2O (30 mL), and the mixture was washed with brine (10 mL), saturated aqueous CuSO4 (2 × 10 mL), and H2O (10 ml). The organic phase was dried (Na2SO4), and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography eluting with hexanes/EtOAc (2:3) to give 10 mg (91%) of 40 as an orange oil; 1H NMR (400 MHz, CDCl3) δ 8.46 (d, J = 8.7 Hz, 1 H), 8.04 (d, J = 8.7 Hz, 1 H), 7.18 (d, J = 8.6 Hz, 1 H), 7.15 (d, J = 8.9 Hz, 1 H), 7.07 (d, J = 8.6 Hz, 1 H), 6.41 (d, J = 8.9 Hz, 1 H), 5.30 (s, 2 H), 4.85 (d, J = 6.8 Hz, 1 H), 4.76 (d, J = 6.8 Hz, 1 H), 3.93 (s, 3 H), 3.81 (s, 3 H), 3.58 (d, J = 17.8 Hz, 1 H), 3.51 (d, J = 17.8 Hz, 1 H), 3.49 (s, 3 H), 3.14 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 191.4, 190.4, 189.2, 161.3, 151.7, 148.5, 147.5, 139.9, 135.6, 135.3, 129.8, 127.2, 122.2, 122.1, 121.3, 112.7, 111.7, 109.7, 103.2, 95.3, 95.1, 93.5, 57.0, 56.2, 56.02, 55.97, 46.1; IR (neat) 2936, 2841, 1710, 1691, 1515, 1273, 1077 cm−1; mass spectrum (ESI) m/z 509.1451 [C27H24O10 (M+1) requires 509.1442], 531, 509 (base).
9,12-Dimethoxy-1,4-bis(methoxymethoxy)-1H-naphtho[1,2-b]xanthene-7,13,14-trione (41)
A solution of spirocycle 40 (10 mg, 0.01 mmol) in PhNO2 was heated at 215 °C for 1.5 h. The reaction was cooled to room temperature, and the majority of the solvent was removed by distillation under high vacuum at 54–56 °C. The residual mixture was diluted with CH2Cl2 (20 mL) and washed with H2O (2 × 10 mL), 1 M KOH (2 × 10 mL). The aqueous KOH layer was extracted with CH2Cl2 (2 × 10 mL). The combined organic phases were dried (Na2SO4), and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography eluting with hexanes/EtOAc (1:3) to give 6 mg (58%) of 41 as a red solid: mp 192-194 °C; 1H NMR (500 MHz, CDCl3) δ 8.47 (d, J = 8.8 Hz, 1 H), 8.12 (d, J = 8.8 Hz, 1 H), 7.37 (d, J = 8.5 Hz, 1 H), 7.25 (d, J = 8.5 Hz, 1 H), 7.21 (d, J = 9.0 Hz, 1 H), 6.82 (d, J = 9.0 Hz, 1 H), 5.33 (s, 2 H), 5.32 (s, 2 H), 4.00 (s, 3 H), 3.95 (s, 3 H), 3.60 (s, 3 H), 3.52 (s, 3 H); 13C NMR (125 MHz, CDCl3) δ 182.2, 178.8, 173.5, 153.3, 152.5, 150.2, 147.8, 147.0, 143.1, 136.4, 130.5, 129.9, 127.1, 122.5, 122.3, 120.6, 117.3, 117.1, 114.0, 113.6, 107.1, 96.7, 95.4, 57.1, 56.8, 56.4, 56.3; IR (neat) 2925, 2854, 1693, 1586, 1489, 1287, 1072 cm−1; mass spectrum (ESI) m/z 507.1290 [C27H22O10 (M+1) requires 507.1286], 529, 507 (base).
5′-Hydroxy-4,7-dimethoxy-8′-(methoxymethoxy)-1′H,3H-spiro[benzofuran-2,3′-phenanthrene]-1′,3,4′(2′H)-trione (43)
A solution of H2O (0.1 mL) in TFA (0.9 mL) was added dropwise to a solution of spirocycle 40 (32 mg, 0.06 mmol) in CH2Cl2 (3 mL) at 0 °C. After 30 min at 0 °C, saturated aqueous NaHCO3 (20 mL) was slowly added (CO2 evolved), and the mixture was extracted with CH2Cl2 (3 × 10 mL). The combined organic phases were was with H2O (2 × 10 mL), dried (Na2SO4), and the solvent was removed under reduced pressure to give 30 mg (100%) of 43 as a dark red oil; 1H NMR (400 MHz, CDCl3) δ 8.83 (d, J = 8.9 Hz, 1 H), 8.82 (s, 1 H), 8.24 (d, J = 8.9 Hz, 1 H), 7.29 (d, J = 8.9 Hz, 1 H), 7.17 (d, J = 8.9 Hz, 1 H), 7.14 (d, J = 8.9 Hz, 1 H), 6.41 (d, J = 8.9 Hz, 1 H), 5.31 (s, 2 H), 3.96 (s, 3 H), 3.80 (s, 3 H), 3.63 d, J = 16.6 Hz, 1 H), 3.51 (s, 3 H), 3.44 (d, J = 16.6 Hz, 1 H); 13C NMR (100 MHz, CDCl3) δ 193.9, 191.0, 190.1, 161.6, 152.1, 148.9, 147.2, 140.0, 139.7, 132.6, 130.5, 129.7, 123.0, 121.6, 121.1, 118.5, 114.2, 108.7, 103.8, 95.5, 93.2, 57.1, 56.3, 56.1, 45.2; IR (neat) 3416, 2953, 1705, 1596, 1515, 1271, 1077 cm−1; mass spectrum (ESI) m/z 487.1000 [C25H20O9 (M+23) requires 487.1000], 951 (base), 465.
1-(4-Bromo-2,5-dimethoxyphenyl)propan-2-ol
A mixture of N-bromosuccinimide (28.12 g, 158.1 mmol) and ammonium nitrate (1.21 g, 15.05 mmol) was added to a stirred solution of 50 (29.54 g, 150.5 mmol)) in CH3CN (75 mL). Stirring was continued for 30 min at room temperature, whereupon H2O (50 mL) was added, and the mixture was extracted with Et2O (3 × 50 mL). The combined organic phases were washed with brine (2 × 50 mL) and dried (MgSO4). The solvent was removed under reduced pressure to give 43.56 g (100%) of crude 1-(4-bromo-2,5-dimethoxyphenyl)propan-2-ol as a light brown solid that was used without further purification. Spectral data were consistent with those reported in the literature for enantiomerically pure (S)-alcohol.28
2,5-Dimethoxy-4-(2-(methoxymethyloxy)propyl)bromobenzene (51)
MOMCl (5.74 mL, 6.08 g, 75.53 mmol) and i-Pr2NEt (17.54 mL, 13.01 g, 100.7 mmol) were added dropwise to a stirred solution of 1-(4-bromo-2,5-dimethoxyphenyl)propan-2-ol (13.85 g, 50.35 mmol) in CH2Cl2 (25 mL) at 0 °C. The solution was then stirred for 20 h at room temperature, and Et2O (200 mL) was added. The layers were separated, and the organic phase was washed with 1 M HCl (50 mL), saturated NaHCO3 (50 mL), and brine (50 mL) and then dried (MgSO4). The solvent was removed under reduced pressure to give 15.69 g (98%) of 51 as a pale yellow oil that was used in the next step without further purification; 1H NMR (400 MHz, CDCl3) δ 7.00 (s, 1 H), 6.75 (s, 1 H), 4.59 (d, J = 6.8 Hz, 1 H), 4.48 (d, J = 6.8 Hz, 1 H), 3.98-3.89 (m, 1 H), 3.82 (s, 3 H), 3.75 (s, 3 H), 3.18 (s, 3 H), 2.76 (ABX, JAB = 13.6 Hz, JAX = 7.2 Hz, JBX = 5.6 Hz, Δν(AB) = 30.2 Hz, 1 H, C9-H), 2.60 (ABX, JAB = 13.6 Hz, JAX = 7.2 Hz, JBX = 5.6 Hz, Δν(AB) = 30.2 Hz, 1 H), 1.15 (d, J = 6.0 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 151.9, 149.5, 127.4, 115.6, 115.5, 108.8, 94.7, 72.2, 56.7, 55.9, 54.8, 38.0, 20.2; IR 2932, 1495, 1464, 1387, 1214, 1035 cm−1; mass spectrum (ESI) m/z 341.0349 [C13H19BrO4 (M+Na)+ requires 341.0359].
2-Bromo-5-(2-(methoxymethoxy)propyl)benzene-1,4-diol
A solution of CAN (54.36 g, 99.20 mmol) in H2O (42 mL) was added to a solution of 51 (10.55 g, 17.14 mmol) in CH3CN (165 mL) at 0 °C, and the mixture was stirred for 25 min at room temperature. H2O (50 mL) and brine (50 mL) were added, and the mixture was extracted with EtOAc (3 × 50 mL). The combined organic phases were washed with brine (10 × 50 mL) and dried (Na2SO4). The solvent was removed under reduced pressure to give 9.53 g (100%) of a substituted benzoquinone as yellow-orange oil, which was dissolved immediately in THF (150 mL) and a stirred solution of Na2S2O4 (28.79 g, 165.37 mmol) in H2O (50 mL) was added. After stirring for 10 min at room temperature, saturated NaHCO3 (50 mL) was added, and the mixture was extracted with EtOAc (3 × 50 mL). The combined organic phases were washed with brine (2 × 50 mL) and dried (MgSO4). The solvent was removed under reduced pressure to give 9.09 g (94%) of 2-bromo-5-(2-(methoxymethoxy)propyl)benzene-1,4-diol as a yellow solid that was used without further purification; mp 88–90 °C; 1H NMR (500 MHz) δ 6.82 (t, J = 1.1 Hz, 1 H), 4.58 (d, J = 7.0 Hz, 1 H), 4.50 (d, J = 7.0 Hz, 1 H), 3.92–3.81 (m, 1 H), 3.23 (s, 3 H), 2.62–2.50 (comp, 2 H), 1.17 (d, J = 6.2 Hz, 3 H); 13C NMR (126 MHz) δ 184.7, 179.4, 146.4, 138.1, 137.1, 133.7, 95.0, 71.5, 55.4, 36.4, 20.6; IR 3316, 2933, 1667, 1591, 1237 cm−1; mass spectrum (ESI) m/z 290.0146 [C11H15BrO4 (M) requires 290.0154].
7-Bromo-3-methylisochroman-5,8-diol (52)
TMSOTf (73 μL, 0.04 mmol) was added dropwise to a stirred solution of 2-bromo-5-(2-(methoxymethoxy)propyl)benzene-1,4-diol (118 mg, 0.41 mmol), which had been dried by azeotropic distillation under reduced pressure from benzene, in CH3CN (2 mL) at 0 °C. The solution was then stirred for 18 h at room temperature, whereupon saturated NaHCO3 (10 mL) was added, and the mixture was extracted with Et2O (3 × 30 mL). The combined organic phases were dried (MgSO4), and the solvent was removed under reduced pressure to give 100 mg (95%) of 52 as a pale yellow solid that was used without further purification; mp 61–63 °C; 1H NMR (500 MHz) δ 6.79 (s, 1 H), 5.10 (s, 1 H), 4.93 (d, J = 16.0 Hz, 1 H), 4.62 (ddd, J = 16.0, 2.4, 1.5 Hz, 1 H), 4.51 (s, 1 H), 3.75 – 3.67 (m, 1 H), 2.64 (ddd, J = 16.6, 3.2, 1.5 Hz, 1 H), 2.35 (dddd, J = 16.6, 10.7, 1.6, 1.1 Hz, 1 H), 1.36 (d, J = 6.2 Hz, 3 H); 13C NMR (126 MHz) δ 146.8, 142.0, 123.7, 122.0, 115.1, 106.0, 69.9, 64.7, 29.9, 21.5; IR 3307, 2974, 1458, 1259 cm−1; mass spectrum (ESI) m/z 257.9883 [C10H11BrO3 (M) requires 257.9892.
7-Bromo-5,8-dihydroxy-3-methylisochroman-6-carbaldehyde (53)
HMTA (1.45 g, 10.37 mmol) was added to a solution of 52 (2.55 g, 9.88 mmol) in TFA (40 mL) in a sealed tube and heated at 100 °C for 16 h. H2O (150 ml) was added, and the mixture was stirred at 60 °C for 4 h. The mixture was then extracted with EtOAc (5 × 100 mL), and the combined organic phases were washed with brine (8 × 100 mL) and dried (MgSO4). The solvent was removed under reduced pressure, and the residue was purified by flash column chromatography eluting with hexanes/EtOAc (2:1) to give 1.85 g (65%) of 53 as a yellow solid: mp 141-143 °C; 1H NMR (400 MHz, CDCl3) δ 11.99 (s, 1 H), 10.17 (s, 1 H), 5.36 (br s, 1 H), 4.95 (d, J = 17.5 Hz, 1 H), 4.64 (d, J = 17.5 Hz, 1 H), 3.78-3.65 (m, 1 H), 2.77 (ddd, J = 17.4, 3.6, 1.6 Hz, 1 H), 2.35 (app dd, J = 17.4, 10.8 Hz, 1 H), 1.37 (d, J = 6.4 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 196.5, 156.1, 141.2, 133.7, 124.6, 113.0, 108.1, 69.9, 64.9, 29.1, 21.4; IR 3362, 2974, 1638, 1423, 1300, 1204 cm−1; mass spectrum (ESI) m/z 284.9764 [C11H10O4Br (M-1) requires 284.9768].
7-Bromo-5,8-bis(methoxymethoxy)-3-methylisochroman-6-carbaldehyde
MOMCl (3.71 g, 3.52 mL, 46.39 mmol) and i-Pr2NEt (4.99 g, 6.73 mL, 38.66 mmol) were added dropwise to a solution of 53 (4.44 g, 15.46 mmol) in CH2Cl2 (50 mL) at room temperature, and the mixture was stirred for 18 h at room temperature. H2O (100 mL) was added, and the mixture was extracted with Et2O (3 × 100 mL). The combined organic phases were dried (MgSO4), and the solvent was removed under reduced pressure to give 5.06 g (87%) of 7-bromo-5,8-bis(methoxymethoxy)-3-methylisochroman-6-carbaldehyde as a yellow solid: mp 47-49 °C; 1H NMR (400 MHz, CDCl3) δ 10.27 (s, 1 H), 5.08 (d, J = 16.8 Hz, 1 H), 5.06 (d, J = 6.2 Hz, 1 H), 5.04 (d, J = 6.2 Hz, 1 H), 5.00 (s, 2 H), 4.76 (d, J = 16.8 Hz, 1 H), 3.73-3.61 (m, 1 H), 3.59 (s, 3 H), 3.55 (s, 3 H), 2.88 (ddd, J = 16.8, 3.0, 1.1 Hz, 1 H), 2.44 (app dd, J = 16.8, 10.6 Hz, 1 H), 1.36 (d, J = 6.4 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 190.7, 153.9, 148.3, 137.2, 130.0, 126.6, 117.1, 101.9, 100.4, 70.1, 65.2, 57.73, 57.70, 30.7, 21.4; IR 2934, 1698, 1360, 1159 cm−1; mass spectrum (ESI) m/z 397.0256 [C15H19O6Br (M+23) requires 397.0257].
7-Bromo-5,8-bis(methoxymethoxy)-3-methyl-6-vinylisochroman (48)
Ph3PCH3Br (7.19 g, 20.19 mmol) and NaH (1.08 g, 26.92 mmol, 60% dispersion in mineral oil) were added to a solution of 7-bromo-5,8-bis(methoxymethoxy)-3-methylisochroman-6-carbaldehyde (5.05 g, 13.46 mmol) in THF (70 mL), and the mixture was stirred for 18 h at room temperature. Brine (100 mL) was slowly added, and the mixture was extracted with EtOAc (3 × 50 mL). The combined organic phases were dried (Na2SO4), and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography eluting with hexanes/EtOAc (2:1) to give 4.24 g (84%) of 48 as a white solid: mp 40-42 °C; 1H NMR (400 MHz, CDCl3) δ 6.68 (dd, J = 17.8, 11.6 Hz, 1 H), 5.75 (dd, J = 17.8, 2.0 Hz, 1 H), 5.56 (dd, J = 11.6, 2.0 Hz, 1 H), 5.05 (d, J = 16.0 Hz, 1 H), 5.03 (d, J = 6.0 Hz, 1 H), 5.01 (d, J = 6.0 Hz, 1 H), 4.87 (d, J = 5.8 Hz, 1 H), 4.85 (d, J = 5.8 Hz, 1 H), 4.74 (d, J = 16.0 Hz, 1 H), 3.74-3.60 (m, 1 H), 2.86 (ddd, J = 16.9, 2.8, 1.2 Hz, 1 H), 2.44 (app dd, J = 17.2, 10.4 Hz, 1 H), 1.35 (d, J = 6.0 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 150.4, 147.6, 132.5, 130.6, 129.9, 128.6, 121.4, 116.2, 100.0, 99.2, 70.2, 65.0, 57.6, 57.5, 31.0, 21.5; IR 2935, 1360, 1159, 965, 927 cm−1; mass spectrum (ESI) m/z 395.0465 [C16H21O5Br (M+23) requires 395.0465].
3-(5,8-Bis(methoxymethoxy)-3-methyl-6-vinylisochroman-7-yl)-4,4-dimethox-y-2-vinylcyclobut-2-enone (54)
tert-BuLi (2.98 mL, 5.06 mmol, 1.7 M in pentane) was added dropwise to a stirred solution of 48 (900 mg, 2.41 mmol), which had been dried by azeotropic distillation of benzene under reduced pressure, in Et2O (14 mL) at −78 °C, and stirring was continued for 5 min at −78 °C. The mixture was then warmed to 0 °C, and a solution of freshly dried (with NaH) 19 (532.7 mg, 2.89 mmol) in Et2O (6 mL) was added dropwise. Stirring was continued for 30 min at 0 °C, whereupon the reaction was cooled to –78 °C, and TFAA (1.26 g, 0.84 mL, 6.02 mmol) was added dropwise. The solution was stirred for 20 min at –78 °C, the cold bath was removed, saturated aqueous NaHCO3 (40 mL) was added, and the reaction was allowed to warm to room temperature. H2O (20 mL) was added, and the mixture was extracted with Et2O (3 × 40 mL). The combined organic phases were washed with brine (50 mL), dried (Na2SO4), and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography eluting with hexanes/EtOAc (4:1→3:1) to give 689.1 mg (64%) of 54 as a yellow oil; 1H NMR (400 MHz, CDCl3) δ 6.55 (dd, J = 17.6, 11.6 Hz, 1 H), 6.11-5.94 (comp, 2 H), 5.67 (dd, J = 17.6, 2.0 Hz, 1 H), 5.50 (dd, J = 10.4, 2.8 Hz, 1 H), 5.37 (dd, J = 11.6, 2.0 Hz, 1 H), 5.00 (d, J = 15.8, Hz, 1 H), 4.85 (d, J = 6.2 Hz, 1 H), 4.83 (d, J = 6.2 Hz, 1 H), 4.76 (d, J = 6.4 Hz, 1 H), 4.74 (d, J = 6.4 Hz, 1 H), 4.66 (d, J = 15.8 Hz, 1 H), 3.70-3.59 (m, 1 H), 3.48 (s, 3 H), 3.41 (s, 3 H), 3.40 (s, 3 H), 3.37 (s, 3 H), 2.89 (dd, J = 17.2, 2.0 Hz, 1 H), 2.47 (dd, J = 17.2, 10.4, Hz, 1 H), 1.31 (d, J = 6.0 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 190.5, 170.7, 152.3, 149.9, 146.8, 131.2, 131.0, 129.6, 128.4, 126.1, 123.9, 122.7, 121.2, 116.6, 100.3, 99.1, 70.1, 64.7, 57.4, 57.2, 52.5, 52.4, 31.2, 21.5; IR 2945, 1767, 1159, 974 cm−1; mass spectrum (ESI) m/z 469.1838 [C24H30O8 (M+Na)+ requires 169.1833].
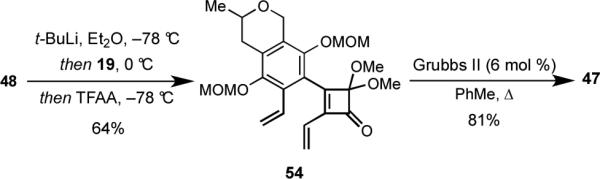
1,1-Dimethoxy-5,10-bis(methoxymethoxy)-7-methyl-6,7-dihydro-1H-cyclobuta[3,4]benzo[1,2-g]isochromen-2(9H)-one (47)
Grubbs second generation catalyst (56.0 mg, 0.066 mmol) was added to a degassed (freeze-pump-thaw, two cycles) solution of diene 54 (491 mg, 1.10 mmol) in toluene (11 mL), and the resulting solution was degassed one more time (freeze-pump-thaw). The solution was then heated under reflux for 5 h 30 min, whereupon the reaction was cooled to room temperature. The reaction mixture was concentrated to ca. 2 mL, and the residue was purified by flash column chromatography eluting with hexanes/EtOAc (4:1) to give 373.5 mg (81%) of 47 as a yellow oil; 1H NMR (400 MHz, CDCl3) δ 8.17 (d, J = 8.6 Hz, 1 H), 7.39 (d, J = 8.6 Hz, 1 H), 5.31 (d, J = 16.0 Hz, 1 H), 5.17 (d, J = 7.2 Hz, 1 H), 5.053 (d, J = 7.2 Hz, 1 H), 5.051 (d, J = 5.6 Hz, 1 H), 5.01 (d, J = 5.6 Hz, 1 H), 4.92 (d, J = 16.0 Hz, 1 H), 3.78-3.66 (m, 1 H), 3.58 (s, 3 H), 3.57 (s, 3 H), 3.49 (s, 3 H), 3.43 (s, 3 H), 3.07 (dd, J = 17.2, 2.8 Hz, 1 H), 2.64 (dd, J = 17.2, 11.2 Hz, 1 H), 1.34 (d, J = 6.0 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 192.2, 159.7, 148.9, 146.8, 146.3, 130.7, 130.4, 130.2, 128.0, 122.7, 118.1, 116.1, 102.0, 100.1, 70.1, 65.2, 57.6, 57.2, 53.56, 53.47, 31.7, 21.5; IR 2945, 1766, 1258, 1157, 960 cm−1; mass spectrum (ESI) m/z 441.1521 [C22H26O8 (M+Na)+ requires 441.1520].
2-Ethynyl-1,1-dimethoxy-5,10-bis(methoxymethoxy)-7-methyl-2,6,7,9-tetrahydro-1H-cyclobuta[3,4]benzo[1,2-g]isochromen-2-ol (55)
A solution of ethynylmagnesium bromide (28 mL, 0.5 M in THF, 14.05 mmol) was added to a stirred solution of 47 (1.68 g, 4.02 mmol) in THF (10 mL). Stirring was continued for 2 h at room temperature, and saturated NH4Cl (5 mL) and H2O (45 mL) were added. The mixture was extracted with Et2O (3 × 100 mL), and the combined organic phases were washed with H2O (2 × 50 mL), and dried (MgSO4). The solvent was removed under reduced pressure to give 1.57 g (88%) of 55 as a pale yellow oil; 1H NMR (400 MHz, CDCl3, mixture of diastereomers) δ 8.14 (d, J = 8.7 Hz, 0.7 H), 8.13 (d, J = 8.7 Hz, 0.3 H), 7.47 (d, J = 8.7 Hz, 0.3 H), 7.46 (d, J = 8.7 Hz, 0.7 H), 5.36 (d, J = 16.0 Hz, 0.7 H), 5.25 (d, J = 16.0 Hz, 0.3 H), 5.19 (d, J = 6.8 Hz, 0.3 H), 5.15 (d, J = 6.8 Hz, 0.7 H), 5.07 (d, J = 5.6 Hz, 0.7 H), 5.06 (d, J = 5.6 Hz, 0.3 H) 5.03 (d, J = 5.6 Hz, 0.3 H), 5.02 (d, J = 5.6 Hz, 0.7 H), 4.00 (d, J = 6.8 Hz, 0.7 H), 4.91 (d, J = 6.8 Hz, 0.3 H), 4.90 (app d, J = 16.0 Hz, 1 H), 3.84 (br s, 0.3 H), 3.83-3.66 (comp, 1 H), 3.71 (br s, 0.7 H), 3.627 (s, 0.9 H), 3.626 (s, 0.9 H), 3.62 (s, 2.1 H), 3.59 (s, 2.1 H), 3.56 (app s, 3 H), 3.55 (s, 0.9 H), 3.548 (s, 2.1 H), 3.13-3.03 (comp, 1 H), 2.71 (s, 0.7 H), 2.68 (s, 0.3 H), 2.65 (app dd, J = 16.8, 10.8 Hz, 1 H), 1.41-1.35 (comp, 3 H); 13C NMR (100 MHz, CDCl3, mixture of diastereomers) δ 148.6, 148.5, 145.3, 144.6, 144.5, 137.0, 136.8, 129.0, 128.91, 128.85, 128.8, 126.8, 126.7, 125.9, 125.7, 123.81, 123.77, 118.5, 107.7, 107.2, 102.00, 101.96, 100.1, 100.0, 81.6, 81.5, 77.6, 77.2, 75.8, 75.5, 70.6, 70.2, 65.6, 65.2, 57.75, 57.69, 57.34, 57.29, 52.8, 52.3, 52.2, 51.6, 31.52, 31.46, 21.8, 21.6; IR 3402, 3294, 2943, 2249, 1594, 1445, 1337, 1158, 1086 cm−1; mass spectrum (ESI) m/z 467.1677 [C24H28O8 (M+Na)+ requires 467.1676].
2-(3-(2-((tert-Butyldimethylsilyl)oxy)-3,6-dimethoxyphenyl)-3-hydroxyprop-1-yn-1-yl)-1,1-dimethoxy-5,10-bis(methoxymethoxy)-7-methyl-2,6,7,9-tetrahydro-1H-cyclobuta[3,4]benzo[1,2-g]isochromen-2-ol (56)
Method A. A solution of EtMgBr (3.53 mL, 3 M in THF 10.60 mmol) was added dropwise to a stirred solution of 55 (1.57 g, 3.53 mmol), which had been dried by azeotropic distillation of benzene under reduced pressure, in THF (15 mL) at 0 °C, and the solution was stirred for 2 h at room temperature. A solution of aldehyde 37 (2.09 g, 7.07 mmol), which had been dried by azeotropic distillation of benzene under reduced pressure, in THF (3 mL) was then added, and stirring was continued for 30 min at room temperature. Saturated NH4Cl (5 mL) and H2O (50 mL) were added, and the mixture was extracted with Et2O (3 × 30 mL). The combined organic layers were dried (Na2SO4), and the solvent was removed under reduced pressure and the residue purified by flash column chromatography eluting with hexanes/EtOAc (2:1→1:1) to give 209 mg (13%) of recovered 55 and 2.11 g (81%) of 56 as a yellow oil; 1H NMR (400 MHz, CDCl3, mixture of diastereomers) δ 8.14-8.06 (comp, 1 H), 7.44-7.38 (comp, 1 H), 6.66-6.61 (comp, 1 H), 6.42-6.35 (comp, 1 H), 5.90-5.82 (comp, 1 H), 5.40-5.21 (comp, 1 H), 5.20-5.13 (comp, 1 H), 4.95-4.82 (comp, 2 H), 4.09-3.95 (comp, 1 H), 3.83-3.71 (comp, 1 H), 3.75-3.68 (comp, 3 H), 3.68-3.65 (comp, 3 H), 3.63-3.61 (comp, 3 H), 3.56-3.50 (comp, 6 H), 3.47-3.40 (comp, 3 H), 3.14-3.03 (comp, 1 H), 2.70-2.59 (comp, 3 H), 1.39-1.35 (comp, 3 H), 0.93-0.87 (comp, 9 H), 0.12-0.02 (comp, 6 H); 13C NMR (100 MHz, CDCl3, mixture of diastereomers) δ 152.3, 148.5, 145.8, 144.9, 144.6, 144.5, 142.0, 136.7, 136.6, 128.8, 128.6, 128.5, 126.4, 126.3, 125.5, 125.4, 123.8, 123.7, 121.4, 118.8, 110.4, 107.8, 107.4, 103.73, 103.67, 102.0, 99.99, 99.95, 88.3, 88.2, 88.0, 87.8, 81.0, 80.9, 77.6, 77.3, 70.6, 70.1, 65.6, 65.1, 57.7, 57.6, 57.3, 57.2, 56.0, 55.2, 52.7, 52.4, 51.7, 51.2, 31.5, 31.4, 25.9, 21.8, 21.6, 18.6, −4.3, −4.4; IR 3528, 3429, 2940, 1595, 1491, 1257, 1078 cm−1; mass spectrum (ESI) m/z 763.3121 [C39H52O12Si (M+Na)+ requires 763.3120].
Method B
A solution of EtMgBr (2.77 mL, 0.9 M in THF, 2.49 mmol) was added to a solution of tetramethylpiperidine (0.45 mL, 2.64 mmol) in THF (2.23 mL) at room temperature. The resulting mixture was stirred under reflux for 6.5 h and cooled to room temperature. A portion of this solution (2.8 mL) was added to the solution of 55 (123 mg, 0.275 mmol), which had been dried by azeotropic distillation of benzene under reduced pressure, and aldehyde 37 (179 mg, 0.605 mmol) in THF (2.8 mL). The solution was stirred for 6.5 h at room temperature, and saturated NH4Cl (5 mL) and H2O (10 mL) were added. The mixture was extracted with Et2O (3 × 15 mL), and the combined organic phases were washed with brine (20 mL) and dried (Na2SO4). The solvent was removed under reduced pressure, and the residue was purified by flash column chromatography eluting with hexanes/EtOAc (2:1→1:1) to give 163 mg (80%) of 56 as a yellow oil.
2-(3-(2-((tert-Butyldimethylsilyl)oxy)-3,6-dimethoxyphenyl)-3-hydroxyprop-1-yn-1-yl)-2-hydroxy-5,10-bis(methoxymethoxy)-7-methyl-2,6,7,9-tetrahydro-1H-cyclobuta[3,4]benzo[1,2-g]isochromen-1-one (46)
p-TsOH (54 mg, 0.29 mmol) was added to a solution of 56 (2.11 g, 2.85 mmol) in acetone (50 mL). After 15 min at room temperature, the reaction was diluted with Et2O (100 mL), and the mixture was washed with brine (3 × 50 mL). The organic phase was dried (MgSO4), and the solvent was removed under reduced pressure to give 1.95 g (98%) of 46 as an orange foam; 1H NMR (400 MHz, CDCl3, complex mixture of diastereomers) δ 8.37-8.24 (comp, 1 H), 7.68-7.60 (comp, 1 H), 6.66-6.59 (comp, 1 H), 6.38 (app d, J = 9.2 Hz, 1 H), 5.84 (app br s, 1 H), 5.32-4.73 (comp, 6 H), 4.48-4.25 (br 2, 1 H), 4.15-3.88 (br s, 1 H), 3.84-3.69 (comp, 1 H), 3.78-3.72 (comp, 1 H), 3.69-3.59 (comp, 6 H), 3.59-3.48 (comp, 3 H), 3.03 (app t, J = 17.8 Hz, 1 H), 2.73-2.56 (comp, 1 H), 1.38 (app d, J = 6.0 Hz, 3 H), 0.93-0.88 (comp, 9 H), 0.18-0.05 (comp, 6 H); 13C NMR (100 MHz, CDCl3, mixture of diastereomers) δ 184.2, 183.8, 161.0, 160.4, 152.2, 148.5, 148.3, 145.7, 145.5, 144.8, 142.1, 141.7, 141.6, 133.2, 133.0, 131.1, 130.7, 129.1, 129.0, 127.8, 127.6, 120.8, 120.7, 120.6, 118.3, 118.2, 110.7, 103.9, 101.6, 101.4, 100.2, 91.4, 91.2, 85.7, 85.5, 79.2, 70.5, 70.1, 65.2, 65.1, 57.9, 57.8, 57.5, 57.4, 56.3, 55.2, 31.5, 31.3, 25.9, 21.5, 21.4, 18.7, −4.16, −4.19; IR (neat) 3532, 3347, 2934, 1767, 1491, 1256, 1158 cm−1; mass spectrum (ESI) m/z 717.2699 [C37H46O11Si (M+23) requires 717.2702].
2-((2-((tert-Butyldimethylsilyl)oxy)-3,6-dimethoxyphenyl)(hydroxy)methyl)-7,12-bis(methoxymethoxy)-9-methyl-8,9-dihydro-1H-naphtho[2,1-g]isochromene-1,4(11H)-dione (57)
A solution of benzocyclobutenone 46 (124 mg, 0.178 mmol) in DMSO (36 mL) was heated at 80 °C for 5 min and then 100 °C for 25 min in dark. After cooling to room temperature, EtOAc (40 ml) was added, and the mixture was washed with brine (3 × 30 mL). The organic phase was dried (Na2SO4) and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography eluting with hexanes/EtOAc (1:1) to give 123.6 mg (67%) of 57 as a red amorphous solid; 1H NMR (500 MHz, CDCl3, mixture of diastereomers) δ 8.30 (d, J = 8.7 Hz, 0.5 H), 8.29 (d, J = 8.7, 0.5 H), 7.99 (d, J = 8.7 Hz, 1 H), 6.76 (d, J = 9.0 Hz, 0.5 H), 6.755 (d, J = 9.0 Hz, 0.5 H), 6.57 (dd, J = 10.0, 1.8 Hz, 0.5 H), 6.55 (dd, J = 10.0, 1.8 Hz, 0.5 H), 6.48-6.43 (comp, 1.5 H), 6.36 (d, J = 1.8 Hz, 0.5 H), 5.36 (d, J = 16.1 Hz, 0.5 H), 5.27 (d, J = 16.3 Hz, 0.5 H), 5.09-5.03 (comp, 2 H), 4.92-4.83 (comp, 1.5 H), 4.72 (d, J = 6.7 Hz, 0.5 H), 4.49 (d, J = 6.8 Hz, 0.5 H), 4.31 (d, J = 6.7 Hz, 0.5 H), 4.11 (d, J = 10.2 Hz, 0.5 H), 3.98 (d, J = 10.3 Hz, 0.5 H), 3.78-3.68 (comp, 7 H), 3.62 (s, 3 H), 3.35 (s, 1.5 H), 3.31 (s, 1.5 H), 3.14-3.06 (m, 1 H), 2.70-2.62 (m, 1 H), 1.39 (d, J = 6.0 Hz, 1.5 H), 1.38 (d, J = 6.1 Hz, 1.5 H), 0.94 (s, 4.5 H), 0.92 (s, 4.5 H), 0.203 (s, 1.5 H), 0.196 (s, 1.5 H), 0.19 (s, 1.5 H), 0.15 (s, 1.5 H). 13C NMR (126 MHz, CDCl3) δ 186.3, 186.2, 185.1, 185.1, 155.0, 154.7, 152.4, 152.3, 147.64, 147.57, 147.4, 147.3, 145.1, 145.0, 143.8, 143.7, 134.06, 134.05, 132.4, 132.3, 131.07, 131.05, 130.3, 130.2, 129.3, 129.1, 129.0, 127.8, 127.7, 121.4, 121.3, 120.8, 118.9, 118.5, 111.0, 110.9, 103.03, 102.95, 100.34, 100.32, 99.4, 99.2, 70.2, 70.1, 65.67, 65.65, 65.1, 65.0, 57.8, 57.7, 57.5, 57.4, 55.72, 55.71, 55.1, 31.53, 31.49, 26.03, 25.97, 21.62, 21.58, 18.9, 18.8, −3.75, −3.77, −4.0. IR 3504, 2934, 2857, 1660, 1627, 1594, 1489, 1467, 1443, 1388, 1359, 1338, 1303, 1256, 1227, 1158, 1127, 1105, 1056, 1029, 1001. Mass spectrum (ESI) m/z 717.2713 [C37H46O11NaSi (M+Na)+ requires 747.2702].
2-(2-(tert-Butyldimethylsilyloxy)-3,6-dimethoxybenzoyl)-7,12-bis(methoxymethoxy)-9-methyl-8,9-dihydro-1H-naphtho[2,1-g]isochromene-1,4(11H)-dione
IBX (172 mg, 0.61 mmol) was added to a solution of 57 (142 mg, 0.20 mmol) in DMSO (10 mL). After 21 h at room temperature, saturated aqueous Na2S2O3 (5 mL) and saturated aqueous NaHCO3 (5 mL) were added, and the reaction was stirred for 10 min at room temperature. Brine (50 mL) was added, and the mixture was extracted with Et2O (3 × 50 mL). The combined organic phases were washed with brine (2 × 50 mL) and dried (Na2SO4). The solvent was removed under reduced pressure to give 142 mg (100%) of 2-(2-(tert-butyldimethylsilyloxy)-3,6-dimethoxybenzoyl)-7,12-bis(methoxymethoxy)-9-methyl-8,9-dihydro-1H-naphtho[2,1-g]isochromene-1,4(11H)-dione as a red oil; 1H NMR (400 MHz, CDCl3) δ 8.32 (d, J = 8.8 Hz, 1 H), 7.98 (d, J = 8.8 Hz, 1 H), 6.91 (s, 1 H), 6.86 (d, J = 9.2 Hz, 1 H), 6.46 (d, J = 9.2 Hz), 5.33 (d, J = 16.4 Hz, 1 H), 5.09 (d, J = 5.8 Hz, 1 H), 5.05 (d, J = 5.8 Hz, 1 H), 4.90 (d, J = 16.4 Hz, 1 H), 4.86 (d, J = 6.8 Hz, 1 H), 4.83 (d, J = 6.8 Hz, 1 H), 3.77 (s, 3 H), 3.66 (s, 3 H), 3.64 (s, 3 H), 3.45 (s, 3 H), 3.11 (dd, J = 17.3, 2.4 Hz, 1 H), 2.68 (dd, J = 17.3, 11.0 Hz, 1 H), 1.40 (d, J = 6.0 Hz, 3 H), 0.80 (s, 9 H), 0.13 (s, 6 H); 13C NMR (100 MHz, CDCl3) δ 191.0, 186.0, 184.2, 151.6, 147.8, 147.7, 146.2, 144.7, 143.5, 135.0, 133.9, 132.1, 131.4, 130.6, 129.8, 128.0, 121.6, 120.7, 120.6, 113.4, 103.4, 100.4, 99.9, 70.2, 65.8, 57.8, 57.6, 56.2, 55.3, 31.6, 25.7, 21.7, 18.6, −4.0, −4.1; IR (neat) 2934, 1697, 1663, 1599, 1486, 1256, 1099 cm−1; mass spectrum (ESI) m/z 715.2557 [C39H39O11Si (M+1) requires 715.2545], 747, 715 (base).
4,7-Dimethoxy-7′,12′-bis(methoxymethoxy)-9′-methyl-8′,9′-dihydro-3H-spiro[benzofuran-2,2′-naphtho[2,1-g]isochromene]-1′,3,4′(3′H,11′H)-trione (58)
Et3N·HF (51 μL, 0.31 mmol) was added to a solution of 2-(2-(tert- butyldimethylsilyloxy)-3,6-dimethoxybenzoyl)-7,12-bis(methoxymethoxy)-9-methyl-8,9-dihydro-1H-naphtho[2,1-g]isochromene-1,4(11H)-dione (51 mg, 0.07 mmol) in CH3CN at room temperature. The reaction was stirred for 20 min, then diluted with EtOAc (15 mL). The mixture was washed with brine (3 × 10 mL). The organic layers were separated, dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash chromatography eluting with EtOAc/Hexane (1: 2 to 1:1) to give 36 mg (86%) of 58 as an amorphous yellow solid; 1H NMR (400 MHz, CDCl3, 1:1 mixture of diastereomers) δ 8.34 (d, J = 8.8 Hz, 0.5 H), 8.33 (d, J = 9.2 Hz, 0.5 H), 8.06 (d, J = 8.8 Hz, 0.5 H), 8.05 (d, J = 9.2 Hz, 0.5 H), 7.14 (d, J = 8.9 Hz, 0.5 H), 7.13 (d, J = 8.9 Hz, 0.5 H), 6.41 (app d, J = 8.9 Hz, 1 H), 5.25 (d, J = 16.4 Hz, 0.5 H), 5.16 (d, J = 16.4 Hz, 0.5 H), 5.12 (d, J = 5.6 Hz, 0.5 H), 5.08 (d, J = 6.0 Hz, 0.5 H), 5.06 (d, J = 6.0 Hz, 0.5 H), 5.05 (d, J = 5.6 Hz, 0.5 H), 4.87 (d, J = 16.4 Hz, 0.5 H), 4.76 (d, J = 16.4 Hz, 0.5 H), 4.50 (d, J = 6.0 Hz, 0.5 H), 4.46 (d, J = 6.0 Hz, 0.5 H), 4.42 (d, J = 6.4 Hz, 0.5 H), 4.41 (d, J = 6.4 Hz, 0.5 H), 3.93 (s, 1.5 H), 3.91 (s, 1.5 H), 3.83 (app s, 3 H), 3.78-3.69 (m, 1 H), 3.64 (s, 1.5 H), 3.63 (s, 1.5 H), 3.57 (d, J = 17.6 Hz, 0.5 H), 3.55 (d, J = 17.6 Hz, 0.5 H), 3.50 (d, J = 17.6 Hz, 0.5 H), 3.48 (d, J = 17.6 Hz, 0.5 H), 3.15 (dd, J = 17.2, 2.8 Hz, 0.5 H), 3.12 (s, 1.5 H), 3.08 (s, 1.5 H), 3.07 (dd, J = 17.2, 10.8 Hz, 0.5 H), 2.71 (dd, J = 17.2, 10.8 Hz, 0.5 H), 1.38 (d, J = 6.0 Hz, 1.5 H), 1.37 (d, J = 6.0 Hz, 1.5 H); 13C NMR (100 MHz, CDCl3, mixture of diastereomers) δ 191.6, 191.3, 190.5, 190.4, 187.7, 187.1, 161.4, 161.2, 151.8, 148.0, 147.8, 147.2, 147.0, 140.0, 139.9, 136.4, 136.1, 134.3, 134.0, 131.1, 130.4, 130.3, 129.9, 129.8, 128.4, 128.2, 122.6, 122.5, 122.5, 122.3, 121.5, 109.9, 109.7, 103.5, 103.4, 100.4, 100.0, 93.5, 93.3, 70.3, 70.1, 65.5, 57.8, 57.3, 57.1, 57.02, 56.98, 56.1, 46.8, 46.7, 31.6, 31.5, 21.7, 21.6; IR (neat) 2934, 1704, 1598, 1515, 1272 cm−1; mass spectrum (ESI) m/z 601.1680 [C31H30O11 (M+23) requires 601.1680].
10,13-Dimethoxy-5,16-bis(methoxymethoxy)-3-methyl-3,4-dihydropyrano[4′,3′:6,7]naphtho[1,2-b]xanthene-8,14,15(1H)-trione (45)
A solution of spirocycle 58 (10 mg, 0.02 mmol) in PhNO2 (2.5 mL) was heated for 15 min at 150 °C, 30 min at 200 °C, and 45 min at 215 °C. The reaction was cooled to room temperature, and the majority of the solvent was removed by distillation under high vacuum at 54–56 °C. The residual mixture was dissolved in CH2Cl2 (15 mL), and the solution was extracted with 1 M KOH (2 × 10 mL). The combined aqueous phases were extracted with CH2Cl2 (3 × 10 mL) and dried (Na2SO4). The solvent was removed under reduced pressure, and the residue was purified by flash column chromatography eluting with hexanes/EtOAc (1:3→0:1) to give 6.6 mg (67%) of 45 as a red oil; 1H NMR (400 MHz, CDCl3) δ 8.35 (d, J = 8.8 Hz, 1 H), 8.10 (d, J = 8.8 Hz, 1 H), 7.22 (d, J = 9.2 Hz, 1 H), 6.82 (d, J = 9.2 Hz, 1 H), 5.36 (d, J = 16.4 Hz, 1 H), 5.11 (d, J = 6.0 Hz, 1 H), 5.06 (d, J = 6.0 Hz, 1 H), 5.05 (d, J = 6.8 Hz, 1 H), 4.97 (d, J = 6.8 Hz, 1 H), 4.93 (d, J = 16.4 Hz, 1 H), 4.00 (s, 3 H), 3.94 (s, 3 H), 3.84-3.69 (m, 1 H), 3.65 (s, 3 H3.48 (s, 3 H), 3.13 (dd, J = 16.8, 2.8 Hz, 1 H), 2.69 (dd, J = 16.8, 11.2 Hz, 1 H), 1.42 (d, J = 6.0 Hz, 3 H); 13C NMR (125 MHz, CDCl3) δ 181.7, 178.7, 173.3, 153.2, 152.4, 148.0, 147.8, 146.6, 143.1, 135.4, 131.8, 130.1, 129.9, 128.0, 122.4, 121.5, 120.4, 117.4, 117.1, 107.4, 104.8, 100.5, 99.9, 70.1, 65.7, 57.9, 57.8, 57.1, 56.8, 31.7, 21.7; IR (neat) 2928, 1691, 1648, 1630, 1585, 1489, 1273, 1158, 1099 cm−1; mass spectrum (ESI) m/z 577.1704 [C31H29O11 (M+1) requires 577.1704].
2-((2-((tert-Butyldimethylsilyl)oxy)-3,6-dimethoxyphenyl)(hydroxy)methyl)-11-methoxy-7,12-bis(methoxymethoxy)-9-methyl-8,9-dihydro-1H-naphtho[2,1-g]isochromene-1,4(11H)-dione (61)
A mixture of 57 (174 mg, 0.250 mmol), BaCO3 (148.3 mg, 0.751 mmol), dry MeOH (30.4 μL, 0.751 mmol), and DDQ (170.5 mg, 0.751 mmol) in CH2Cl2 (5 mL) was stirred at room temperature for 18 h. Saturated NaHCO3 (20 mL) was added, and the reaction mixture was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with brine (30 mL), dried (Na2SO4), and concentrated under reduced pressure. The residue was purified by flash chromatography eluting with EtOAc/Hexane (1: 2) to give 154 mg (85%) of 61 as a red oil; 1H NMR (600 MHz, CDCl3, mixture of diastereomers) δ 8.25 (d, J = 8.8 Hz, 0.5 H), 8.22 (d, J = 8.8, 0.5 H), 8.00 (d, J = 8.8 Hz, 0.5 H), 7.997 (d, J = 8.7 Hz, 0.5 H), 6.77 (d, J = 9.0 Hz, 0.5 H), 6.76 (d, J = 9.0 Hz, 0.5 H), 6.59 (dd, J = 10.2, 1.8 Hz, 0.5 H), 6.58 (dd, J = 10.6, 2.0 Hz, 0.5 H), 6.464 (d, J = 9.0 Hz, 0.5 H), 6.462 (d, J = 9.0 Hz, 0.5 H), 6.31 (d, J = 1.8 Hz, 0.5 H), 6.28 (d, J = 1.9 Hz, 0.5 H), 5.95 (s, 0.5 H), 5.92 (s, 0.5 H), 5.08-5.03 (comp, 2.5 H), 4.91 (d, J = 5.9 Hz, 0.5 H), 4.76 (d, J = 6.3 Hz, 0.5 H), 4.73 (d, J = 5.9 Hz, 0.5 H), 4.38-4.32 (m, 1 H), 4.29 (d, J = 10.2 Hz, 0.5 H), 4.15 (d, J = 10.6 Hz, 0.5 H), 3.75 (s, 3 H), 3.73 (s, 1.5 H), 3.71 (s, 1.5 H), 3.63 (s, 1.5 H), 3.62 (s, 1.5 H), 3.56 (s, 1.5 H), 3.52 (s, 1.5 H), 3.28 (s, 1.5 H), 3.17-3.09 (comp, 2.5 H), 2.69-2.58 (m, 1 H), 1.411 (d, J = 6.2 Hz, 1.5 H), 1.408 (d, J = 6.2 Hz, 1.5 H), 0.924 (s, 4.5 H), 0.919 (s, 4.5 H), 0.22 (s, 1.5 H), 0.19 (s, 1.5 H), 0.154 (s, 1.5 H), 0.145 (s, 1.5 H). 13C NMR (150 MHz, CDCl3) δ 186.8, 186.3, 185.13, 185.08, 155.1, 154.5, 152.5, 152.4, 150.2, 149.9, 147.3, 147.2, 145.05, 144.98, 143.69, 143.66, 136.4, 135.6, 132.0, 131.98, 131.82, 131.78, 129.5, 129.4, 129.1, 129.0, 128.3, 128.0, 127.2, 126.6, 123.5, 122.5, 121.49, 121.48, 119.0, 118.6, 111.0, 103.12, 103.05, 100.8, 100.38, 100.35, 100.2, 95.52, 95.47, 65.3, 65.2, 61.6, 61.4, 57.79, 57.76, 57.7, 57.4, 55.7, 55.6, 55.23, 55.17, 54.9, 54.8, 31.0, 30.9, 26.02, 25.99, 21.5, 21.4, 18.9, 18.8, −3.7, −3.94, −3.95, −3.98. IR 3519, 2933, 2857, 1661, 1600, 1489, 1466, 1443, 1361, 1337, 1303, 1256, 1228, 1159, 1104, 1052, 1003 cm–1. Mass spectrum (ESI) m/z 747.2807 [C38H48O12NaSi (M+Na)+ requires 747.2813].
2-(2-((tert-Butyldimethylsilyl)oxy)-3,6-dimethoxybenzoyl)-11-methoxy-7,12-bis(methoxymethoxy)-9-methyl-8,9-dihydro-1H-naphtho[2,1-g]isochromene-1,4(11H)-dione (64)
A solution of 61 (58.0 mg, 0.080 mmol) and IBX (45.0 mg, 0.161 mmol) in DMSO (4.0 mL) was stirred at room temperature for 5 h. The reaction mixture was diluted with Et2O (40 mL) and then washed with saturated NaHCO3 (10 mL) and brine (3 × 20 mL). The organic layer was dried (Na2SO4), filtered, and concentrated under reduced pressure, and the residue was purified by flash chromatography eluting with EtOAc/hexane (1: 2) to give 41 mg (70%) of 64 as a red oil ; 1H NMR (600 MHz, CDCl3) δ 8.26 (d, J = 8.8 Hz, 1 H), 7.98 (d, J = 8.8 Hz, 1 H), 6.86-6.84 (comp, 2 H), 6.45 (d, J = 8.8 Hz, 1 H), 5.97 (s, 1 H), 5.08 (d, J = 5.9 Hz, 1 H), 5.06 (d, J = 5.9 Hz, 1 H), 5.05 (s, J = 5.9 Hz, 1 H), 4.83 (d, J = 5.9 Hz, 1 H), 4.38-4.32 (m, 1 H), 3.77 (s, 3 H), 3.64 (comp, 6 H), 3.54 (s, 3 H), 3.39 (s, 3 H) , 3.15 (dd, J = 17.6, 3.5 Hz, 1 H), 2.62 (dd, J = 17.6, 11.7 Hz, 1 H), 1.41 (d, J = 6.5 Hz, 3 H), 0.82 (s, 9 H), 0.14 (s, 6 H). 13C NMR (150 MHz, CDCl3) δ 191.0, 186.2, 183.8, 151.7, 150.6, 147.3, 145.4, 144.8, 143.6, 135.9, 135.7, 132.2, 131.7, 129.7, 128.4, 127.1, 123.3, 121.2, 120.6, 113.3, 103.3, 101.2, 100.4, 95.4, 61.4, 57.8, 57.6, 56.1, 55.4, 54.8, 31.0, 25.8, 21.5, 18.7, −3.96, −4.01. IR 2933, 2857, 1699, 1663, 1599, 1485, 1441, 1361, 1335, 1256, 1206, 1159, 1010, 1051, 1014 cm–1. Mass spectrum (ESI) m/z 745.2654 [C38H46O12NaSi (M+Na)+ requires 745.2651].
4,7,11′-Trimethoxy-7′,12′-bis(methoxymethoxy)-9′-methyl-8′,9′-dihydro-3H-spiro[benzofuran-2,2′-naphtho[2,1-g]isochromene]-1′,3,4′(3′H,11′H)-trione (65)
A solution of 64 (58.4 mg, 0.081 mmol) and Et3N•3HF (45.6 mg, 46.0 μL, 0.283 mmol) in CH3CN (4.0 mL) was stirred at room temperature for 20 min. EtOAc (40 mL) was added, and the solution was washed with brine (3 × 10 ml). The organic layer was dried (Na2SO4) and concentrated under reduced pressure, and the residue was purified by flash chromatography eluting with EtOAc/Hexane (1:1) to give 43 mg (87%) of 65 as a yellow oil; 1H NMR (600 MHz, CDCl3, mixture of diastereomers) δ 8.31 (d, J = 9.0 Hz, 0.52 H), 8.29 (d, J = 9.0 Hz, 0.48 H), 8.10 (d, J = 8.8 Hz, 0.52 H), 8.07 (d, J = 9.0 Hz, 0.48 H), 7.12 (d, J = 8.8 Hz, 0.48 H), 7.11 (d, J = 8.8 Hz, 0.52 H), 6.39 (d, J = 8.8 Hz, 0.52 H), 6.38 (d, J = 8.8 Hz, 0.48 H), 5.82 (s, 0.52 H), 5.79 (s, 0.48 H), 5.08-5.03 (comp, 2 H), 4.74-4.73 (comp, 1 H), 4.49 (d, J = 5.7, 0.48 H), 4.40 (d, J = 5.1 Hz, 0.52 H), 4.34-4.25 (m, 1 H), 3.93 (s, 1.6 H), 3.90 (s, 1.4 H), 3.82 (s, 1.4 H), 3.81 (s, 1.6 H), 3.622 (s, 1.4 H), 3.616 (s, 1.6 H) , 3.55-3.40 (comp, 5 H), 3.14 (dd, J = 17.2, 3.4 Hz, 0.52 H), 3.09 (dd, J = 17.3, 3.7 Hz, 0.48 H), 3.01 (s, 1.4 H), 2.89 (s, 1.6 H), 2.66 (dd, J = 17.3, 11.2 Hz, 0.48 H), 2.56 (dd, J = 17.2, 11.4 Hz, 0.52 H), 1.38 (d, J = 6.3 Hz, 1.6 H), 1.37 (d, J = 6.3 Hz, 1.4 H). 13C NMR (150 MHz, CDCl3) δ 191.39, 191.37, 190.6, 190.3, 187.55, 187.49, 161.5, 160.6, 152.0, 151.8, 149.4, 149.1, 147.44, 147.39, 140.0, 139.9, 136.6, 135.9, 135.4, 135.3, 132.13, 132.06, 129.7, 129.4, 128.8, 128.4, 127.8, 127.7, 123.4, 123.3, 122.31, 122.27, 122.2, 122.1, 110.3, 109.9, 103.4, 103.3, 101.5, 100.40, 100.38, 95.35, 95.27, 93.20, 93.18, 61.5, 61.3, 57.81, 57.78, 57.1, 57.0, 56.69, 56.67, 56.2, 56.1, 54.6, 54.5, 48.3, 46.8, 30.9, 30.8, 21.40, 21.37. IR 2933, 2831, 1708, 1600, 1515, 1441, 1359, 1340, 1273, 1208, 1162, 1127, 1045 cm–1. Mass spectrum (ESI) m/z 631.1787 [C32H32O12Na (M+Na)+ requires 631.1786].
11′-Hydroxy-4,7-dimethoxy-7′,12′-bis(methoxymethoxy)-9′-methyl-8′,9′-dihydro-3H-spiro[benzofuran-2,2′-naphtho[2,1-g]isochromene]-1′,3,4′(3′H,11′H)-trione
A solution of 65 (24.0 mg, 0.039 mmol) and p-TsOH (4.5 mg, 0.024 mmol) in a mixture of acetone (4.0 mL) and H2O (0.8 mL) was stirred at room temperature for 8 h. Et2O (40 mL) was added, and the solution was washed with H2O (3 × 15 mL) and brine (15 mL). The organic layer was dried (Na2SO4) and concentrated under reduced pressure, and the residue was purified by flash chromatography eluting with EtOAc/Hexane (2:1) to give 2.0 mg (8%) of recovered 65 and 21.0 mg (90%) of 11′-hydroxy-4,7-dimethoxy-7′,12′-bis(methoxymethoxy)-9′-methyl-8′,9′-dihydro-3H-spiro[benzofuran-2,2′-naphtho[2,1-g]isochromene]-1′,3,4′(3′H,11′H)-trione as an amorphous yellow solid; 1H NMR (600 MHz, CDCl3, mixture of diastereomers) δ 8.34 (d, J = 8.8 Hz, 0.52 H), 8.32 (d, J = 8.8 Hz, 0.48 H), 8.09 (d, J = 8.8 Hz, 0.52 H), 8.06 (d, J = 9.0 Hz, 0.48 H), 7.14 (d, J = 8.8 Hz, 0.52 H), 7.13 (d, J = 8.8 Hz, 0.48 H), 6.408 (d, J = 8.8 Hz, 0.52 H), 6.406 (d, J = 8.8 Hz, 0.48 H), 6.33 (d, J = 1.8 Hz, 0.52 H), 6.25 (s, 0.48 H), 5.09-5.04 (comp, 2 H), 4.67 (d, J = 6.2, 0.48 H), 4.57 (d, J = 5.6 Hz, 0.52 H), 4.52-4.43 (comp, 2 H), 4.17 (d, J = 1.8 Hz, 0.52 H), 3.92 (s, 1.6 H), 3.90 (s, 1.4 H), 3.823 (s, 1.4 H), 3.821 (s, 1.6 H), 3.63 (s, 1.4 H), 3.62 (s, 1.6 H) , 3.58-3.47 (comp, 2 H), 3.17-3.05 (comp, 4 H), 2.63 (dd, J = 17.3, 11.4 Hz, 0.48 H), 2.55 (dd, J = 17.2, 11.4 Hz, 0.52 H), 1.37 (d, J = 6.2 Hz, 3 H). 13C NMR (150 MHz, CDCl3) δ 191.6, 191.2, 190.4, 190.2, 187.5, 186.7, 161.23, 161.22, 151.84, 151.82, 149.1, 148.8, 147.83, 147.78, 140.0, 139.9, 136.7, 136.0, 134.9, 134.3, 132.1, 132.0, 130.8, 130.5, 129.6, 129.3, 128.5, 128.2, 122.9, 122.6, 122.4, 122.3, 122.2, 109.9, 109.8, 103.53, 103.49, 100.5, 100.45, 100.39, 100.1, 93.5, 93.2, 88.8, 88.3, 62.2, 62.0, 57.84, 57.81, 57.05, 57.00, 56.98, 56.95, 56.1, 47.2, 46.5, 31.1, 30.8, 21.44, 21.40. IR 3422, 2934, 1703, 1599, 1515, 1441, 1345, 1273, 1206, 1159, 1076, 1041 cm–1. Mass spectrum (ESI) m/z 617.1636 [C31H30O12Na (M+Na)+ requires 617.1630].
4,7-Dimethoxy-7′,12′-bis(methoxymethoxy)-9′-methyl-8′,9′-dihydro-3H-spiro[benzofuran-2,2′-naphtho[2,1-g]isochromene]-1′,3,4′,11′(3′H)-tetraone (67)
A suspension of 11′-hydroxy-4,7-dimethoxy-7′,12′-bis(methoxymethoxy)-9′-methyl-8′,9′-dihydro-3H-spiro[benzofuran-2,2′-naphtho[2,1-g]isochromene]-1′,3,4′(3′H,11′H)-trione (20.2 mg, 0.034 mmol), pyridinium dichromate (PDC) (38.3 mg, 0.102 mmol), and 3 Å molecular sieves (34 mg) in CH2Cl2 (3.4 mL) was stirred at room temperature for 3 h. The mixture was then filtered through a thin plug of silica gel, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography eluting with EtOAc/hexane (2: 1) to give 18.1 mg (90%) of 67 as an amorphous yellow solid; 1H NMR (600 MHz, CDCl3, mixture of diastereomers) δ 8.303 (d, J = 8.8 Hz, 0.46 H), 8.302 (d, J = 8.8 Hz, 0.54 H), 8.20 (d, J = 8.7 Hz, 0.54 H), 8.19 (d, J = 8.8 Hz, 0.46 H), 7.15 (d, J = 9.0 Hz, 0.54 H), 7.14 (d, J = 8.8 Hz, 0.46 H), 6.412 (d, J = 8.8 Hz, 0.54 H), 6.411 (d, J = 8.8 Hz, 0.46 H), 5.11-5.05 (comp, 2.46 H), 4.81 (d, J = 5.3, 0.54 H), 4.71 (d, J = 5.3 Hz, 0.54 H), 4.70 (d, J = 4.6 Hz, 0.46 H), 4.55-4.48 (m, 1 H), 3.94 (s, 1.6 H), 3.93 (s, 1.4 H), 3.824 (s, 1.6 H), 3.823 (s, 1.4 H), 3.61 (s, 1.4 H), 3.60 (s, 1.6 H) , 3.56-3.46 (comp, 2 H), 3.38 (dd, J = 16.4, 2.3 Hz, 0.54 H), 3.31 (dd, J = 16.4, 2.6 Hz, 0.46 H), 2.98 (s, 1.4 H), 2.97 (s, 1.6 H), 2.89 (dd, J = 16.4, 10.6 Hz, 0.46 H), 2.80 (dd, J = 16.4, 11.4 Hz, 0.54 H), 1.52 (d, J = 6.3 Hz, 1.6 H), 1.48 (d, J = 6.5 Hz, 1.4 H). 13C NMR (150 MHz, CDCl3) δ 191.3, 191.1, 190.03, 190.0, 188.7, 187.9, 162.6, 162.2, 161.4, 160.8, 153.5, 152.9, 152.0, 151.9, 145.5, 144.9, 140.1, 139.9, 137.4, 136.9, 136.45, 136.38, 134.9, 134.6, 131.1, 130.9, 127.2, 127.0, 125.3, 124.8, 124.7, 124.6, 122.7, 122.3, 117.4, 116.5, 110.1, 109.8, 103.50, 103.45, 100.8, 100.7, 100.5, 100.1, 93.3, 93.0, 74.4, 74.1, 58.0, 57.98, 57.4, 57.14, 57.08, 56.14, 56.07, 47.9, 47.3, 31.6, 31.5, 20.6, 20.5. IR 2926, 2853, 1711, 1600, 1515, 1444, 1352, 1274, 1210, 1189, 1159, 1134, 1077, 1044. Mass spectrum (ESI) m/z 615.1478 [C31H28O12Na (M+Na)+ requires 615.1473].
16-Hydroxy-10,13-dimethoxy-5-(methoxymethoxy)-3-methyl-3,4-dihydropyrano[4′,3′:6,7]naphtho[1,2-b]xanthene-1,8,14,15-tetraone (69)
A solution of 67 (10.0 mg, 0.017 mmol) in CH3CN (6 mL) was heated in a microwave oven (sealed tube) at 180 °C (300 watts) for 25 min. The reaction mixture was then stirred under O2 at room temperature overnight and then again heated in a sealed tube at 180 °C in a microwave oven for 15 min. The solution was concentrated under reduced pressure to give a red-colored residue. The residue was precipitated from a mixture of CHCl3/Hexane (0.1 mL/0.9 mL) 5-times to give 7.3 mg (79%) of 69 as a red powder. 1H NMR (600 MHz, CDCl3): δ 12.62 (br s, 1H), 8.28 (d, J = 8.6 Hz, 1 H), 8.26 (d, J = 8.6 Hz, 1 H), 7.21 (d, J = 9.2 Hz, 1 H), 6.82 (d, J = 8.9 Hz, 1 H), 5.04 (d, J = 6.3 Hz, 1 H), 5.03 (d, J = 6.3 Hz, 1 H), 4.74-4.69 (m, 1 H), 4.00 (s, 3 H), 3.93 (s, 3 H), 3.62 (s, 3 H), 3.40 (dd, J = 16.4, 3.0 Hz, 1 H), 2.92 (dd, J = 16.4, 11.2 Hz, 1 H), 1.58 (d, J = 6.3 Hz, 3 H. 13C NMR (150 MHz, CDCl3): δ 181.1, 178.3, 172.8, 170.1, 159.8, 153.5, 152.5, 146.5, 143.0, 141,4, 138.5, 136.7, 129.82, 129.78, 126.8, 124.8, 122.1, 119.9, 117.5, 117.1, 107.8, 104.6, 100.8, 75.6, 57.9, 57.0, 56.9, 30.3, 20.9. IR 1698, 1679, 1647, 1614, 1586, 1491, 1441, 1409, 1323, 1277, 1210, 1161, 1132, 1102, 1073, 1017 cm–1. Mass spectrum (ESI) m/z 569.1050 [C29H22O11Na (M+Na)+ requires 569.1054].
5,16-Dihydroxy-10,13-dimethoxy-3-methyl-3,4-dihydropyrano[4′,3′:6,7]naphtho[1,2-b]xanthene-1,8,14,15-tetraone and 8,15-dihydroxy-10,13-dimethoxy-3-methyl-3,4-dihydropyrano[4′,3′:6,7]naphtho[1,2-b]xanthene-1,5,14,16-tetraone (70a,b)
A mixture of 69 (4.0 mg, 0.0073 mmol) and freshly distilled TMSBr (9.7 μL, 11.2 mg, 0.073 mmol) in CH2Cl2 (1.2 mL) containing 4 Å molecular sieves (7.3 mg) was stirred at room temperature for 16 h, whereupon another portion of TMSBr (4.8 μL, 5.5 mg, 0.036 mmol) was added and stirring continued for another 5 h. CH2Cl2 (15 mL) was added, and the molecular sieves were removed by filtration. The filtrate was washed with H2O (3 × 10 mL) and brine (10 mL). The organic layer was dried (Na2SO4) and concentrated under reduced pressure to give a red-colored residue. The residue was then dissolved in CHCl3 (0.25 mL) and precipitated with hexane (0.75 mL), and this process was repeated three-times. A final reprecipitation from CHCl3 (0.5 mL) and hexane (0.5 mL) gave 2.0 mg (54%) of an mixture (~2:1) of two isomeric compounds 70a,b as an amorphous red solid. 1H NMR (600 MHz, CD3CN) δ 8.44 (d, J = 8.8 Hz, 0.67 H, major), 8.20 (d, J = 9.3 Hz, 0.33 H, minor), 8.17 (d, J = 8.8 Hz, 0.67 H, major), 8.12 (d, J = 9.3 Hz, 0.33 H, minor), 7.41 (d, J = 9.0 Hz, 0.33 H, minor), 7.38 (d, J = 9.1 Hz, 0.67 H, major), 6.96 (d, J = 9.1 Hz, 0.67 H, major), 6.87 (d, J = 9.1 Hz, 0.33 H, minor), 4.84-4.77 (comp, 1 H), 3.99 (s, 1 H, minor), 3.98 (s, 2 H, major), 3.93 (s, 1 H, minor), 3.89 (s, 2 H, major), 3.35-3.27 (comp, 1 H), 2.90-2.82 (comp, 1 H), 1.56 (d, J = 6.3 Hz, 1 H, minor), 1.55 (d, J = 6.3 Hz, 2 H, major). Mass spectrum: major isomer (ESI) m/z 525.0792 [C27H18O10Na+ (M+Na)+ requires 525.0792]; (ESI neg) m/z 501.0804 [C27H17O10– (M-H)– requires 501.0827]: Mass spectrum: minor isomer (ESI) m/z 525.0790 [C27H18O10Na+ (M+Na)+ requires 525.0792]; (ESI neg) m/z 501.0812 [C27H17O10– (M-H)– requires 501.0827].
Acknowledgments
We thank the National Institutes of Health (GM31077) and the Robert A. Welch Foundation (F-0652) for support. DK thanks the National Science Foundation for a graduate research fellowship, JY thanks the Cancer Prevention Research Institute of Texas for support as postdoctoral trainee, and DM thanks the NIH for a postdoctoral fellowship. We thank Dr. Richard Pederson (Materia, Inc.) for catalyst support, Mr. Shawn Blumberg (The University of Texas at Austin) for technical assistance, and Dr. Ian Riddington (The University of Texas at Austin) for helpful discussions of mass spec data. We would also like thank Vincent Lynch (The University of Texas at Austin) for obtaining X-ray data for compound 41.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.a Malet-Cascon L, Romero F, Espliego-Vazquez F, Gravalos D, Fernandez-Puentes JL. J. Antibiot. 2003;56:219–225. doi: 10.7164/antibiotics.56.219. [DOI] [PubMed] [Google Scholar]; b Rodriguez JC, Puentes JLF, Baz JP, Canedo LM. J. Antibiot. 2003;56:318–321. doi: 10.7164/antibiotics.56.318. [DOI] [PubMed] [Google Scholar]
- 2.a Carter GT, Nietsche JA, Williams DR, Borders DB. J. Antibiot. 1990;43:504–512. doi: 10.7164/antibiotics.43.504. [DOI] [PubMed] [Google Scholar]; b Hopp DC, Milanowski DJ, Rhea J, Jacobsen D, Rabenstein J, Smith C, Romari K, Clarke M, Francis L, Irigoyen M, Luche M, Carr GJ, Mocek U. J. Nat. Prod. 2008;71:2032–2035. doi: 10.1021/np800503z. [DOI] [PubMed] [Google Scholar]
- 3.a Ratnayake R, Lacey E, Tennant S, Gill JH, Capon R. J. Org. Lett. 2006;8:5267–5270. doi: 10.1021/ol062113e. [DOI] [PubMed] [Google Scholar]; b Ratnayake R, Lacey E, Tennant S, Gill JH, Capon R. J. Chem. Eur. J. 2007;13:1610–1690. doi: 10.1002/chem.200601236. [DOI] [PubMed] [Google Scholar]
- 4.a Nakagawa A, Iwai Y, Shimizu H, Omura S. J. Antibiot. 1986;39:1636–1638. doi: 10.7164/antibiotics.39.1636. [DOI] [PubMed] [Google Scholar]; b Omura S, Nakagawa A, Kushida K, Shimizu H, Lukacs G. J. Antibiot. 1987;40:301–308. doi: 10.7164/antibiotics.40.301. [DOI] [PubMed] [Google Scholar]; c Tanka H, Kawakita K, Suzuki H, Nakagawa PS, Omura S. J. Antibiot. 1989;42:431–439. doi: 10.7164/antibiotics.42.431. [DOI] [PubMed] [Google Scholar]
- 5.For syntheses of cervinomycins, see: Kelly TR, Jagoe CT, Li Q. J. Am. Chem. Soc. 1989;111:4522–4524. Rao AVR, Yadav JS, Reddy KK, Upender V. Tetrahedron Lett. 1991;32:5199–5202. Mehta G, Shah SR, Venkateswarlu Y. Tetrahedron. 1994;50:11729–11742.
- 6.For syntheses of the kibdelones, see: Sloman DL, Bacon JW, Porco JA., Jr. J. Am. Chem. Soc. 2011;133:9952–9955. doi: 10.1021/ja203642n. Butler JR, Wang C, Bian J, Ready JM. J. Am. Chem. Soc. 2011;133:9956–9959. doi: 10.1021/ja204040k.
- 7.For syntheses of other natural polycyclic xanthones, see: Masuo R, Ohmori K, Hintermann L, Yoshida S, Suzuki K. Angew. Chem. Int. Ed. 2009;48:3452–3465. doi: 10.1002/anie.200806338.
- 8.For a review, see: Winter DK, Sloman DL, Porco JA., Jr. Nat. Prod. Rep. 2013;30:382–391. doi: 10.1039/c3np20122h.
- 9.For leading references to prepare 1,4-dioxygenated xanthones: Ellis RC, Whalley WB, Ball K. Chem. Commun. 1967:803–804. Stout GH, Balkenhol W. J. Tetrahedron. 1969;25:1947–1960. Jain AC, Khanna VK, Seshadri TR. Indian J. Chem. 1970;8:667–669. Simoneau B, Brassard P. J. Chem. Soc., Perkin Trans. I. 1984:1507–1510. Bekaert A, Andrieux J, Plat M. Tetrahedron Lett. 1992;33:2805–2806. Elix JA, Gaul KL, Jiang H. Aust. J. Chem. 1993;46:95–110. Sun L, Liebeskind LS. J. Am. Chem. Soc. 1997;118:12473–12474. Hauser FM, Dorsch WA. Org. Lett. 2003;5:3753–3754.. doi: 10.1021/ol0354876. Masuo R, Ohmori K, Hintermann L, Suzuki K. Angew. Chem., Int. Ed. 2009;48:3462–3465. doi: 10.1002/anie.200806338.
- 10.a Karlsson JO, Nguyen NV, Foland LD, Moore HW. J. Am. Chem. Soc. 1985;107:3392–3393. [Google Scholar]; b Foland LD, Karlsson JO, Perri ST, Schwabe R, Xu SL, Patil S, Moore HW. J. Am. Chem. Soc. 1989;111:975–989. [Google Scholar]
- 11.a Nichols AL, Zhang P, Martin SF. Org. Lett. 2011;13:4696–4699. doi: 10.1021/ol201910v. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nichols AL, Zhang P, Martin SF. Tetrahedron. 2012;68:7591–7597. doi: 10.1016/j.tet.2012.05.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang J, Knueppel D, Cheng B, Mans D, Martin SF. Org. Lett. 2015;17:114–117. doi: 10.1021/ol503336t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.For a related transformation, see: Danheiser RL, Gee SK. J. Org. Chem. 1984;49:1672–1674. Danheiser RL, Brisbois RG, James J, Kowalczyk, Miller RF. J. Am. Chem. Soc. 1990;112:3093–3100.
- 14.a Knueppel D, Martin SF. Angew. Chem. Int. Ed. 2009;48:2569–2571. doi: 10.1002/anie.200806269. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Knueppel D, Martin SF. Tetrahedron. 2011;67:9765–9770. doi: 10.1016/j.tet.2011.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Serra S, Fuganti C. Synlett. 2003;13:2005–2008. [Google Scholar]
- 16.For related work: Serra S, Fuganti C. Synlett. 2002;10:1661–1664. Serra S, Fuganti C. Synlett. 2005;5:809–812.
- 17.Gayo LM, Winters MP, Moore HW. J. Org. Chem. 1992;57:6896–6899. [Google Scholar]
- 18.Grubbs RH. Angew. Chem., Int. Ed. 2006;45:3760–3765. doi: 10.1002/anie.200600680. [DOI] [PubMed] [Google Scholar]
- 19.Capecchi T, de Koning CB, Michael JP. J. Chem. Soc., Perkin Trans. 2000;1:2681–2688. [Google Scholar]
- 20.See Experimental section for additional details on how this substrate was prepared.
- 21.Corey EJ, Hopkins PB. Tetrahedron Lett. 1982;23:4871–4874. [Google Scholar]
- 22.a Leinweber D, Butenschoen H. Tetrahedron Lett. 1997;38:6385–6386. [Google Scholar]; b Leinweber D, Schnebel M, Wartchow R, Wey HG, Butenschoen H. Eur. J. Org. Chem. 2002:2385–2390. [Google Scholar]
- 23.Hu Y, Li C, Kulkarni BA, Strobel G, Lobkovsky E, Torczynski RM, Porco JA. Org. Lett. 2001;3:1649–1652. doi: 10.1021/ol0159367. [DOI] [PubMed] [Google Scholar]
- 24.Tian Y, Chen C-Y, Yang C-C, Young AC, Jang S-H, Chen W-C, Jen AK-Y. Chem. Mater. 2008;20:1977–1987. [Google Scholar]
- 25.Tisdale EJ, Kochman DA, Theodorakis EA. Tetrahedron Lett. 2003;44:3281–3284. [Google Scholar]
- 26.Adapted from procedure to prepare enantiomerically pure (S)-50, see: Wagner JM, McElhinny CJ, Lewin AH, Carroll FI. Tetrahedron: Asymmetry. 2003;14:2119–2125.
- 27.Tanemura K, Suzuki T, Nishida Y, Satsumabayashi K, Horaguchi T. Chem. Lett. 2003;32:932–933. [Google Scholar]
- 28.For a related example, see: Sawant R, Waghmode SB. Tetrahedron. 2009;65:1599–1602.
- 29.Fkyerat A, Dubin G-M, Tabacchi R. Helv. Chim. Acta. 1999;82:1418–1422. [Google Scholar]
- 30.Smith AB, III, Basu K. J. Am. Chem. Soc. 2009;131:2348–2358. doi: 10.1021/ja8084669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kwon HK, Kee YE, Lee E. Org. Lett. 2008;10:2995–2996. doi: 10.1021/ol801020w. [DOI] [PubMed] [Google Scholar]
- 32.Salmond WG, Barata MA, Havens JL. J. Org. Chem. 1978;43:2057–2059. [Google Scholar]
- 33.Maier MW, Ritschel J. ARKIVOC. 2008:314–329. [Google Scholar]
- 34.Xu Y, Lebeau E, Gillard JW, Attardo G. Tetrahedron Lett. 1993;34:3841–3844. [Google Scholar]
- 35.For use of molecular sieves to accelerate oxidation, see: Herscovici J, Antonakis K. J. Chem. Soc., Chem. Commun. 1980:561–562.
- 36.Hanessian S, Delorme D, Dufresne Y. Tetrahedron Lett. 1984;24:2515–2518. [Google Scholar]
- 37.Li C, Johnson RP, Porco JA. J. Am. Chem. Soc. 2003;125:5095–5106. doi: 10.1021/ja021396c. [DOI] [PubMed] [Google Scholar]