

Abstract
Pancreatic cancer is one of the most lethal human cancers, and radiotherapy is often implemented for locally advanced pancreatic ductal adenocarcinoma. Tumor cell repopulation is a major challenge in treating cancers after radiotherapy. In order to address the problem of tumor repopulation, our previous studies have demonstrated that dying cells stimulate the proliferation of living tumor cells after radiotherapy. In particular, dying cells undergoing apoptosis also activate survival or proliferation signals and release growth factors to surrounding living cells. In the present study, we used an in vitro model to examine the possible mechanisms for dying cell stimulated tumor repopulation in pancreatic cancer. In this model, a small number of living, luciferase‐labeled pancreatic cancer cells (reporter) were seeded onto a layer of a much larger number of irradiated, unlabeled pancreatic cancer cells and the growth of the living cells was measured over time as a gage of tumor repopulation. Our results indicate that irradiated, dying Panc1 feeder cells significantly stimulated the proliferation of living Panc1 reporter cells. Importantly, we identified that the percentage of apoptotic cells and the cleavage of caspases 3 and 7 and protein kinase Cδ (PKCδ) were increased in irradiated Panc1 cells. We presumed that caspases 3 and 7 and PKCδ as integral mediators in the process of dying pancreatic cancer cell stimulation of living tumor cell growth. In order to demonstrate the importance of caspases 3, 7 and PKCδ, we introduced dominant‐negative mutants of caspase 3 (DN_C3), caspase 7 (DN_C7), or PKCδ (DN_PKCδ) into Panc1 cells using lentiviral vectors. The stably transduced Panc1 cells were irradiated and used as feeders and we found a significant decrease in the growth of living Panc1 reporter cells when compared with irradiated wild‐type Panc1 cells as feeders. Moreover, the role of PKCδ in the growth stimulation of living tumor cells was further confirmed using a pan PKC inhibitor GF109203x and a specific PKCδ inhibitor, rottlerin. Additionally, we found significantly increased phosphorylation of Akt, p38 mitogen‐activated protein kinase (MAPK) and c‐Jun N‐terminal kinase/stress‐activated protein kinase (JNK1/2) in the irradiated Panc1 cells. Mechanistically, PKCδ cleavage was attenuated in both DN_C3 and DN_C7 transduced Panc1 cells, and both Akt and p38 MAPK phosphorylation were attenuated in DN_PKCδ transduced Panc1 cells following radiation. Thus, this report suggests a novel finding that cellular signaling caspase 3/7‐PKCδ‐Akt/p38 MAPK is crucial to the repopulation in Panc1 cells after radiotherapy.
Keywords: Pancreatic cancer, Apoptosis, Repopulation, Caspase, PKCδ, Akt/MAPK
Highlights
Dying pancreatic tumor cells (Panc1) could stimulate living tumor cell growth.
Inhibition of caspase 3/7 and PKCδ activity attenuated growth stimulation effect.
PKCδ cleavage was attenuated in caspase 3/7 mutant transduced cells.
Akt and p38 MAPK could be downstream effector of PKCδ.
C3/7‐PKCδ‐Akt/p38 MAPK is crucial to Panc1 cell repopulation after radiotherapy.
Abbreviations
- PKC
protein kinase C
- MAPK
mitogen-activated protein kinase
- JNK1/2
c-Jun N-terminal kinase/stress-activated protein kinase
- iPLA2
calcium-independent phospholipase A2
- PGE2
prostaglandin E2
- SHH
Sonic Hedgehog
- ERK1/2
extracellular regulated protein kinase 1/2
- PKB
protein kinase B
- GFP
green fluorescent protein
- VEGF
vascular endothelial growth factor
- PI3K-PDK1
phosphotidylinositol 3′ kinase/Phosphoinositide-dependent kinase 1
- LCAF
late course accelerated hyperfractionated irradiation
1. Introduction
Pancreatic cancer portends a poor prognosis, and therefore remains a treatment challenge with currently available cancer therapies. Radiation therapy is often used for locally advanced pancreatic cancer, but unfortunately, many pancreatic tumors recur despite radiotherapy. Repopulation of tumor cells after radiation occurs in many tumor types, and represents a major cause of tumor relapse after radiotherapy (Rodriguez‐Wallberg, 2012). In fact, the accelerated repopulation of tumors after radiotherapy is the rationale for using radiation schedules involving continuous low dose rate radiation instead of fractionated radiation in order to improve local tumor control and patient survival (Vassileva et al., 2008). Although many proliferation‐associated signaling pathways have been implicated in the repopulation of surviving tumor cells after radiotherapy (Schmidt‐Ullrich et al., 1997), the initiating signal of tumor repopulation after radiotherapy of pancreatic cancer remains largely unknown.
In breast cancer cells, our previous studies have demonstrated that irradiated, dying tumor cells significantly stimulate the repopulation of living tumor cells after radiotherapy. Therefore, we sought to study the role of dying cell stimulation of living tumor cell growth in pancreatic cancer. In particular, we previously observed that when a small number of living tumor cells were seeded onto a much larger number of irradiated, dying tumor cells, the dying cells stimulated the growth of living tumor cells (Huang et al., 2011; Li et al., 2010). We discovered the “Phoenix Rising” pathway of dying cell stimulation of living tumor cell growth, which involves the release of paracrine signals resulting in proliferation of the living tumor cells. The mechanism involves radiation‐induced activation of caspase 3, which leads to the activation of calcium‐independent phospholipase A2 (iPLA2) and subsequent release of prostaglandin E2 (PGE2) from apoptotic cells (Huang et al., 2011; Galluzzi et al., 2012). In breast cancer cells, PGE2 was thought to increase the growth of living tumor cells, but we wish to explore additional signaling pathway in pancreatic cancer in this study. Interestingly, we recently showed that dying cells stimulate living tumor cell growth through the activation of the Sonic Hedgehog (SHH) signaling pathway and this effect was observed in pancreatic tumor cells (Ma et al., 2013). It is generally considered that apoptosis is one of the main cell death mechanisms following exposure to irradiation and execution of apoptosis is closely linked to serial activation of a family of proteases called caspases via external or internal inducers; and irrespective of the actual route to caspase activation, both pathways will lead to the activation of the effector caspases, caspase 3, caspase 6 and caspase 7 which perform much of the proteolysis. Therefore, the proteolysis in irradiated, apoptotic tumor cells may affect the function of other proteins and involve in regulation of multiple proliferation‐associated pathways. However, many of these pathways have yet to be identified. Thus, in order to improve efficacy of tumor radiotherapy, we must further investigate the role of apoptosis and its possible downstream mechanisms leading to tumor repopulation.
A likely possible downstream mechanism of dying cell stimulated living tumor cell growth is via activation of protein kinase C (PKC). In this study, we aimed to determine the role of caspase‐mediated activation of PKC in pancreatic tumor cell repopulation after radiotherapy. PKC is known to function in cell proliferation, differentiation, metabolism, and apoptosis (Nakajima, 2008; Bluwstein et al., 2013). It has been previously demonstrated that several PKC subtypes, such as δ, θ and ε can be activated by caspase 3‐mediated cleavage, and these activated PKCs participate in the positive regulation of radiation‐induced apoptosis (Emoto et al., 1995; Datta et al., 1997; Basu et al., 2002; Koriyama et al., 1999; Nakajima, 2006). Among the PKC subtypes, the cleavage site (DMQD329/N) and function of PKCδ in the radiation‐induced apoptosis cascade has been most well‐studied (Nakajima, 2006; D'Costa and Denning, 2005; Kurokawa and Kornbluth, 2009). Previous studies from the HL60 cell line also revealed that radiation activates PKC, and that PKC inhibitors may result in increased radiosensitivity of cancer cells (Hallahan et al., 1992, 1991).
In addition to studying PKC activation, we wanted to elucidate the downstream activities of PKC in pancreatic tumor repopulation after radiotherapy. Specifically, mitogen‐activated protein kinase (MAPK) cascades have been shown to lie within protein kinase cascades, so we decided to examine the MAPK cascades in pancreatic tumor repopulation. The following three MAPK families in mammalian cells have been clearly described and will be included in this study: Extracellular regulated protein kinase 1/2 (ERK1/2), c‐Jun N‐terminal kinase/stress‐activated protein kinase (JNK/SAPK, or JNK1/2) and p38 kinase. Since MAPK pathways are closely related to cell proliferation, differentiation, development and apoptosis, caspase‐mediated PKC activation leading to MAPK activation may provide a possible mechanistic link between dying cell and living tumor cell growth (Zhang and Liu, 2002; Park et al., 2001).
In addition to MAPK, Akt (also known as protein kinase B (PKB)) is thought to be a key mediator located in downstream of protein kinase cascades. As previously documented, Akt plays a key role in regulating the cell cycle and proliferation (Dugourd et al., 2003; Stabile et al., 2003). Therefore, we also examined Akt activation and its relationship with PKC δ.
Thus, in this study, we created a pancreatic tumor repopulation model after radiotherapy in order to study the importance of apoptosis and the activation of PKCδ and consequently its downstream effectors in this process. In this model, we seeded a small number of living Panc1Fluc cells, which were stably transduced with a firefly luciferase and green fluorescent protein (GFP) fusion gene, onto a feeder layer of 10 Gy X‐ray radiated Panc1 cells. This model simulates the environment of a tumor in which a small number of surrounding, living tumor cells survive and repopulate the tumor after the majority of tumor cells undergo apoptosis after a cytotoxic therapy. Our study highlights the crucial role of caspase 3/7‐PKCδ signaling during Panc1 tumor cell repopulation, and suggests that Akt and p38 MAPK are likely involved in the downstream mechanism. Therefore, we propose that caspase 3/7‐PKCδ‐Akt/p38 MAPK signaling may be a novel target to improve the efficacy of cancer radiotherapy in pancreatic cancer.
2. Materials and methods
2.1. Cell culture
Panc1 cells, a human pancreatic carcinoma of ductal cell origin (Lieber et al., 1975), were purchased from the Chinese Academy of Science (Shanghai, China) and cultured in Dulbecco's Modified Eagle's Medium (DMEM) (Thermo Scientific Inc., Beijing, China) containing 10% fetal bovine serum (FBS) (Tianhang Biological Technology Co., Ltd., Hangzhou, China), 100 U/ml penicillin, and 100 μg/ml streptomycin at 37 °C, 5% CO2.
2.2. Irradiation and in vitro repopulation of cancer cells
Cells cultured in 10‐cm dishes were irradiated with 10 Gy of X‐rays (a lethal dose) using an Oncor linear accelerator (Siemens, Amberg, Germany) located in the department of radiation oncology at Shanghai Jiao Tong University affiliated first people's hospital. The dose rate of the machine is about 3.6 Gy/min. Irradiated cells were immediately trypsinized and seeded into 24‐well plates as feeders at a density of 1.0 × 105 cells per well in DMEM containing 2% FBS. 1000 living Panc1 cells labeled with firefly luciferase and GFP fusion gene (Fluc) were added to each well at indicated time points after seeding the feeders. The medium was replaced with fresh DMEM containing 2% FBS every 2 days for 14 days. For all inhibitors, they were added into culture medium at indicated concentration. The media were replaced with fresh media containing inhibitor every other day until time to be analyzed.
2.3. Production of Panc1 cells stably expressing dominant‐negative caspase 3, caspase 7 and PKCδ
The cDNA encoded dominant‐negative caspase 3, caspase 7 and PKCδ has a single nucleotide mutation (C163A in caspase 3, C186A in caspase 7, D329A in PKCδ) which ablated caspase 3, caspase 7 and PKCδ cleavage activity. The lentiviral vectors expressing dominant‐negative caspase 3, caspase 7 and PKCδ cDNA were constructed using the pLEX system and packaged in 293T cells following manufacturer's instructions (Thermo Scientific Inc.). The expression of dominant‐negative caspase 3, caspase 7 and PKCδ are driven by a CMV promoter. Panc1 cells that stably expressed dominant‐negative caspase 3, caspase 7 and PKCδ were obtained by lentivirus infection and selection with 1.5 μg/ml puromycin for two weeks and were designated as Panc1DN_C3, Panc1DN_C7 and Panc1DN_PKCδ, respectively; and the pLEX without insert i.e. empty vector was used as control and designated as Panc1EV.
2.4. Photographing and bioluminescence imaging
The photos were taken using a confocal microscope from Leica Microsystems (Mannheim, Germany) located in the Experimental Research Center at Shanghai Jiao Tong University affiliated first people's hospital. The bioluminescence imaging was conducted using the NC100 instrument from Berthold Technologies (Bad Wildbad, Germany) located in School of Basic Medical Sciences, Shanghai Medical College, Fudan University, as previously described (Ma et al., 2013). In brief, for Fluc transduced Panc1 cells, luciferase signals were measured by adding D‐luciferin (Promega, Madison, WI, USA) in PBS at a final concentration of 0.15 mg/ml. Five minutes later, luciferase signals were measured and analyzed quantitatively using the manufacturer supplied software.
2.5. Clonogenic cell survival assay
Panc1 cells were trypsinized and prepared in single‐cell suspension, then seeded in 6‐cm dishes in different numbers in triplicates. Twenty‐four hours later, cells were irradiated with various doses of X‐ray (400, 600, 1000, 2000, 10,000 and 1,00,000 cells with 0 Gy, 2 Gy, 4 Gy, 6 Gy, 8 Gy and 10 Gy, respectively). Following 14 days incubation at 37 °C, colonies were washed twice with ice‐cold PBS and stained with crystal violet in 50% methanol and the number of colonies of 50 or more cells was counted. The surviving fraction was then calculated and analyzed following published instructions (Munshi et al., 2005).
2.6. Flow cytometric analysis
Cell apoptosis was determined using the PE Annexin V Apoptosis Detection Kit (Becton Dickinson, NJ, USA). In brief, Panc1 cells were grown in 6‐cm dishes to 80% confluence and then irradiated with 6 Gy or 10 Gy of X‐rays. After trypsinizion the cells were washed twice with ice‐cold PBS and then resuspended in Binding Buffer at a concentration of 1.0 × 106 cells/ml. 100 μl of the solution (1.0 × 105 cells) were transferred to a new culture tube and 5 μl of PE Annexin V and 5 μl of 7‐AAD were added into the solution. After incubation for 15 min at room temperature (25 °C) in the dark, an additional 400 μl of Binding Buffer was added prior to analysis using the Accuri C6 Flow Cytometer from Becton Dickinson located in the Experimental Research Center at Shanghai Jiao Tong University affiliated first people's hospital. The experiments were performed in triplicates.
2.7. Western blot analysis
Cells were washed twice with ice‐cold PBS and lysed using standard RIPA buffer containing proteinase inhibitors (Beyotime, Jiangsu, China). Following quantification, protein samples were heated to 95 °C for 5 min, then separated in an SDS‐polyacrylamide gel and transferred to PVDF membranes (Bio‐Rad, Hercules, CA, USA). The membranes were blocked with 5% non‐fat milk in TBS buffer for 1 h and then incubated with primary antibodies overnight at 4 °C. After washing with TBST buffer for 4*10 min, the membranes were incubated with HRP‐conjugated secondary antibody for 2 h at room temperature. The immune reaction was visualized using ECL Plus substrates (Roche, Basel, Switzerland).
2.8. Antibodies and key chemicals used in this study
Commercially available PKCδ, PKCθ and PKCε antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA); β‐actin, GAPDH, HA‐tag, caspase 3, caspase 7, pan‐Akt, phospho‐Akt(Ser473), pan‐p38 MAPK, phospho‐p38 MAPK, pan‐JNK1/2, phospho‐JNK1/2, pan‐ERK1/2 and phospho‐ERK1/2 antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA); and HRP‐conjugated secondary antibody was purchased from Bio‐Rad. PKC pan inhibitor GF109203x and PKCδ specific inhibitor rottlerin were purchased from Tocris Bioscience (Bristol, UK).
2.9. Statistical analysis
Data were analyzed using the Statistical Package for the Social Sciences Version 13.0 (SPSS Inc., Chicago, IL, USA). Data were presented as mean ± SEM (standard error of the mean). Mean comparisons were performed using one‐way analysis of variance (ANOVA) and covariance analysis. The p < 0.05 was considered statistically significant.
3. Results
3.1. Dying pancreatic tumor cells strongly stimulate living tumor cell growth
In order to mimic tumor cell repopulation in vivo after radiotherapy, in which the majority of tumor cells are eliminated by radiation and a small number of tumor cells survive and repopulate the tumor, we seeded a small number of living, luciferase and GFP‐labeled tumor cells (reporter cells), onto a larger number of lethally irradiated, unlabeled tumor cells (feeder cells) in vitro. We measured bioluminescence over time as a gage of tumor repopulation by living reporter cell growth.
First, we optimized the radiation dose and cell numbers for our tumor repopulation model conditions. In order to select the radiation dose to eliminate the majority of the tumor cells, we performed a clonogenic assay. As shown in Figure 1a, after two weeks, the survival fraction of 6 Gy irradiated Panc1 cells were approximately 3.22 ± 1.19%. For the 8 Gy and 10 Gy irradiated Panc1 cells, there were no colonies present that were qualified for counting. We then demonstrated that the bioluminescence signals from firefly luciferase‐labeled tumor cells linearly correlated (R 2 = 0.9982) with living Panc1Fluc cell numbers (upper panel of Figure 1b). The cells in each well were photographed and representative pictures are shown in lower panel of Figure 1b. Therefore, we used photon values to represent the number of living reporter cells in our in vitro tumor repopulation model.
Figure 1.

Dying tumor cells stimulate the proliferation of living tumor cells. (a) Survival fraction of Panc1 cells irradiated with 2 Gy, 4 Gy, 6 Gy, 8 Gy and 10 Gy tested by clonogenic assay. Error bars represent the standard error of the mean (SEM). N = 3. (b) Linearity of measured bioluminescence activity vs. Panc1Fluc cell numbers. Upper panel: Luciferase activity (photons/sec) plotted against cell numbers (N = 6, R2 = 0.9982, two‐tailed ANOVA analysis). Lower panel: representative images for luciferase activity (hot map) and photos which showed correlation between fluorescence activity and Panc1Fluc cell numbers plated. (c) The diagram of luciferase activity shown as photons/sec (upper panel) and luciferase activity imaging (lower panel) of Panc1Fluc cells when cultured alone or onto a layer of irradiated and non‐irradiated Panc1 cells in 24‐well plates. N = 9, ∗∗p < 0.01 (Panc1Fluc on irradiated feeder cells vs. on non‐irradiated feeder cells or Panc1Fluc alone).
Next, we examined the growth stimulating effect of irradiated pancreatic tumor cells on living tumor cells. A large number of lethally irradiated (10 Gy) Panc1 (1 × 105/well) cells were seeded into 24‐well plates as feeders. On days 1, 2, 3, 4, and 5 post radiation, 1000 living Panc1Fluc cells were seeded onto irradiated cells and continually co‐incubated in DMEM with 2% FBS for 14 days and medium was changed every other day. The luminescence signal was measured and compared with reporter alone cells or reporter cells grown on untreated tumor cells. As shown in Figure 1c, all wells with irradiated feeder cells showed a significant increase in luciferase activity, which peaked on day 3 (about 19 times higher than reporter alone cells and 7 times higher than reporter cells grown on untreated tumor cells). Representative images from bioluminescence imaging are shown in the lower panel of Figure 1c. Therefore, our results indicate that irradiated, dying Panc1 cells stimulate the growth of living Panc1 cells. The dying cell stimulated living cell growth was also observed in other pancreatic cancer cell lines such as BxPC‐3, AsPC‐1 and SW1990 as well as the colon cancer cell line HT29 (data not shown).
3.2. Caspases 3 and 7 mediate the dying cell induced proliferation of living tumor cell growth
Given our results that dying Panc1 cells stimulate the growth of living Panc1 cells (Figure 1), we then examined the role of apoptosis and caspases 3 and 7 in the stimulation of living reporter cell growth. Before studying caspases, we first determined the manner of cell death in Panc1 after radiation by examining apoptosis via FACS and caspases 3 and 7 cleavage via Western blot. As shown in Figure 2a, FACS analysis after PE Annexin V/7‐AAD staining showed a significantly increased number of apoptotic cells after radiation and this trend was dose‐dependent, with more apoptotic cells in the 10 Gy group (25.30 ± 0.29%) than in the 6 Gy group (15.73 ± 1.39%). For western blotting, Panc1 cells were irradiated with 10 Gy and then harvested at 1, 2, 3, 4, 5, and 6 days after radiation to measure caspases 3 and 7 protein levels. Western blot analysis showed significant increases in cleaved caspases 3 and 7 protein levels after radiation, which appeared at day 2 and day 3 for caspase 3 and caspase 7 respectively and lasted at least to day 6 (end of this observation, Figure 2b). Based on these findings, we used 10 Gy X‐ray to induce apoptosis in Panc1 cells in the following experiments to study caspases.
Figure 2.
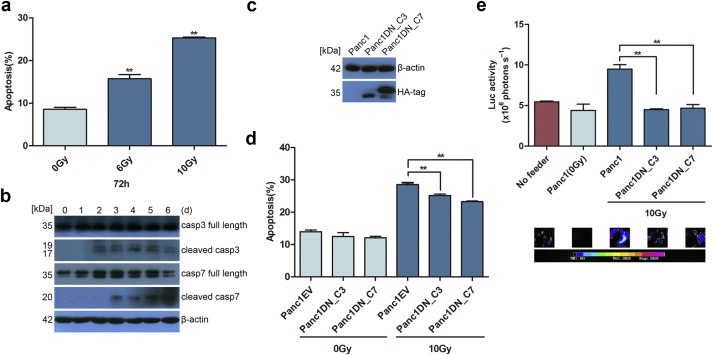
Activated caspases 3 or 7 in apoptotic cells stimulate living tumor cell growth. (a) Apoptotic cells detected by PE Annexin V/7‐AAD staining increased after radiation and correlated with X‐ray dose, N = 3, ∗∗p < 0.01. (b) Western blot showed increased caspases 3 and 7 cleavage after radiation. (c) Western blot showed that dominant‐negative caspase 3 or caspase 7 transduced Panc1 cells had HA‐tag expression, which was fused to dominant‐negative caspase 3 or caspase 7 cDNA. (d) The dominant‐negative caspases 3 or 7 transduced Panc1 cells showed significantly decreased apoptotic cells after 10 Gy radiation. N = 3, ∗∗p < 0.01. (e) Panc1DN_C3 and Panc1DN_C7 showed a significantly reduced growth stimulating effect on Panc1Fluc cells when compared with their parent Panc1 cells as feeder (luciferase activity in upper panel and imaging in lower panel). N = 9, ∗∗p < 0.01.
In order to demonstrate the importance of caspases 3 and 7 to the stimulatory effect of apoptotic cells on living tumor cells, we inhibited caspases 3 and 7 and measured living tumor cell growth. Inhibition of caspases 3 and 7 was achieved by introducing lentiviral vectors containing caspases 3 and 7 functional inactivated mutants i.e. dominant‐negative mutants into Panc1 cells. The dominant‐negative mutants (DN) have a single nucleotide mutation in the catalytic domains of caspases 3 or 7, which ablate caspases 3 or 7 cleavage. The expression of lentiviral vectors containing DN_C3 or DN_C7 was confirmed by HA‐tag (Figure 2c). Interestingly, the irradiated Panc1 cells expressing DN_C3 or DN_C7 showed significantly decreased apoptosis (Panc1EV: 28.53 ± 0.77%; Panc1DN_C3: 25.17 ± 0.59%; Panc1DN_C7: 23.23 ± 0.34%) as well as a decreased growth stimulating effect on living tumor cells compared with irradiated parent Panc1 cells or empty vector transduced cells (Figure 2d and e). These results were consistent with our previous study using caspase 3 shRNA as the method for inhibiting caspase 3 (Huang et al., 2011).
3.3. PKCδ plays a key role in the proliferation of living tumor cell stimulated by apoptotic tumor cells
As stated previously, activated caspases 3 and 7 may selectively cleave proteins, and in turn activate or inactivate the function of cleaved proteins, such as PKCδ. We irradiated Panc1 cells with 10 Gy X‐ray and harvested cells 1, 2, 3, 4, 5, and 6 days after radiation. Western blot assay showed a significantly increased protein levels of cleaved PKCδ, which peaked at day 3, but not in ε and θ protein levels after radiation. Also, we observed a significant decrease in the full length of PKCθ expression with the time, but we do not know its biological meaning (Figure 3a).
Figure 3.

Activated PKCδ stimulating living tumor cell growth. (a) Western blot showed that PKCδ but not ε or θ was cleaved in irradiated Panc1 cells peaked at day 3 and continued to day 6. (b) The pan PKC inhibitor GF109203x significantly inhibited Panc1Fluc cell repopulation in 0.2 μM and 0.6 μM concentration. N = 9, ∗∗p < 0.01. (c) The specific PKCδ inhibitor rottlerin significantly inhibited Panc1Fluc cell repopulation in 0.2 μM and 0.4 μM concentration respectively. N = 9, ∗∗p < 0.01. Panc1Fluc cell alone in same condition was used as control. The upper panel showed quantity of luciferase activity (photons/sec) and lower panel showed imaging of luciferase activity.
To further investigate the function of PKCδ during tumor repopulation, we tested the effects of the pan PKC inhibitor GF109203x (GF) and the specific PKCδ inhibitor rottlerin (Rot) on the stimulatory effect of apoptotic cells on living tumor cell growth. A small number (1000) of living Panc1Fluc cells were co‐cultured with a larger number of 10 Gy irradiated feeder cells and co‐incubated with or without different concentrations of GF or Rot for 14 days (the media containing inhibitor were replaced every other day). The living Panc1Fluc alone (1000 cells) was used as a control. As shown in Figure 3b and c, the 0.2 μM and 0.6 μM concentrations of GF as well as the 0.2 μM and 0.4 μM concentrations of Rot significantly inhibited the growth of living reporter cells co‐cultured with irradiated feeder cells compared to the controls without feeder cells. Further, this inhibitory effect on living tumor cell repopulation is dose‐dependent. Therefore, the inhibition of PKCδ significantly decreased the growth stimulating effect of dying Panc1 cells on living tumor cell repopulation after radiotherapy, thereby supporting a role for PKCδ in this process. Of mention, we also observed similar results in the colon cancer cell line HT29 (data not shown). Therefore, we believe that PKCδ plays a key role in the proliferation of living tumor cells stimulated by apoptotic tumor cells.
3.4. PKCδ acts downstream of caspase 3 during apoptotic tumor cell stimulated living tumor cell growth
In order to investigate whether or not caspases 3 or 7 act upstream of PKCδ during apoptotic cell stimulated living tumor cell growth, Panc1, Panc1DN_C3 and Panc1DN_C7 cells were either untreated or irradiated with 10 Gy X‐ray and harvested on day 3 after radiation and PKCδ protein levels were measured using Western blot analysis. Consistent with caspase‐mediated activation of PKCδ, western blots showed significant decreases in cleaved PKCδ protein levels in both irradiated Panc1DN_C3 and Panc1DN_C7 cells when compared with irradiated parent Panc1 cells (Figure 4a).
Figure 4.
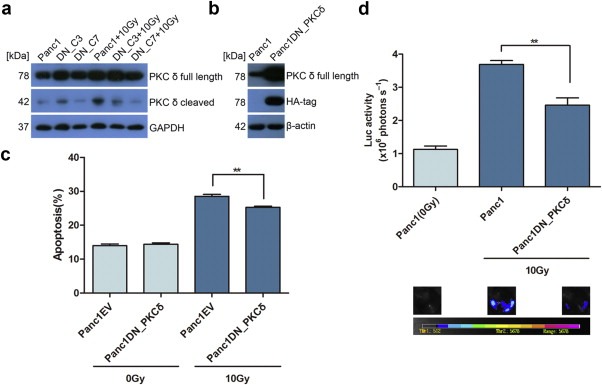
PKCδ activation correlated to caspases 3 and 7 activity and PKCδ inactivation inhibited living tumor cell repopulation. (a) Western blot showing decreased PKCδ cleavage in Panc1DN_C3 and Panc1DN_C7 cells after radiation in comparison with their parent cells. Data shown was from 3 days after radiation. (b) Western blot showing HA‐tag and full length PKCδ expression in dominant‐negative PKCδ transduced Panc1 cells. (c) The dominant‐negative PKCδ transduced Panc1 cells showed significantly decreased apoptotic cells after 10 Gy radiation. N = 3, ∗∗p < 0.01. (d) The dominant‐negative PKCδ transduced Panc1 cells showed significantly decreased growth stimulation effect on Panc1Fluc cells in comparison with parent Panc1 cells (luciferase activity in upper panel and imaging in lower panel). N = 9, ∗∗p < 0.01.
Since it has been shown that PKCδ contains a caspase 3 recognition sequence (DMQD329/N) in the hinge region that is cleaved specifically by caspase 3, we wanted to determine if Panc1 tumor repopulation after radiotherapy is dependent upon caspase 3 cleavage of PKCδ. We constructed a dominant‐negative mutant PKCδ (aspartic acid at 329aa to alanine, DN_PKCδ) that is resistant to caspase 3 cleavage (D'Costa and Denning, 2005; Kurokawa and Kornbluth, 2009). The DN_PKCδ stably transduced Panc1 cells were confirmed by full length PKCδ and HA‐tag expression using Western blot analysis (Figure 4b). As shown in Figure 4c, the irradiated Panc1 cells expressing DN_PKCδ showed significantly decreased apoptosis compared with irradiated empty vector transduced Panc1 cells (Panc1EV: 28.53 ± 0.77%; Panc1DN_PKCδ: 25.30 ± 0.46%). The repopulation assay was carried out as the method mentioned above. Interestingly, decreased living reporter cell growth was detected in groups which used irradiated Panc1DN_PKCδ cells as feeders when compared to the groups using irradiated parent Panc1 cells as feeders (Figure 4d). Thus, the caspase 3‐mediated cleavage of PKCδ may be involved in the dying cell stimulated living tumor cell growth after irradiation of Panc1 cells.
3.5. Possible signaling molecules involved in PKCδ‐mediated growth stimulatory effect on living tumor cells
Since we discovered the importance of caspase 3‐mediated PKCδ activation to the growth stimulating effect of dying Panc1 cells on living Panc1 cell growth, we wanted to further investigate the underlying signaling mechanism leading to living tumor cell growth. In order to do so, we examined the expression levels or phosphorylation status of several signaling molecules that have been shown to play key roles in the positive regulation of cell proliferation. Panc1 cells were irradiated with 10 Gy and cells were then harvested at 4, 12, 24, and 48 h after radiation. Western blot assays showed significant increases in phospho‐Akt (ser473) protein levels 4 h post radiation and in phospho‐p38 MAPK and phospho‐JNK1/2 protein levels 48 h post radiation. However, no significant increases in phospho‐ERK1/2 protein levels were observed after radiation (Figure 5a). To determine whether Akt, p38 MAPK and JNK1/2 are activated in a PKCδ‐dependent manner after radiation, parent Panc1 and Panc1DN_PKCδ cells were either untreated or irradiated with 10 Gy and harvested at 4 h (phospho‐Akt) and 48 h (phospho‐p38 MAPK and phospho‐JNK1/2) after radiation. Western blot analysis showed significant decreases of both phospho‐Akt (ser473) and phospho‐p38 MAPK protein levels in irradiated Panc1DN_PKCδ cells, compared to irradiated parent Panc1 cells (Figure 5b and c). There was no significant difference in phospho‐JNK1/2 protein levels between irradiated Panc1DN_PKCδ cells and their parent cells (Figure 5c). Interestingly, phospho‐Akt (ser473) but not phospho‐p38 MAPK protein levels were decreased in irradiated HT29DN_PKCδ cells compared to irradiated parent HT29 cells (data not shown). Thus, we hypothesized that Akt were more extensively involved in the downstream mechanism of the PKCδ‐mediated growth stimulatory effect on living tumor cells, while p38 MAPK might be more specific to Panc1 cells during repopulation.
Figure 5.

Activation of molecular signal in irradiated Panc1 cells and their relation with PKCδ. (a) Western blot showed phosphorylated Akt (Ser473), pan‐Akt, phosphorylated p38 MAPK, pan‐p38 MAPK, phosphorylated JNK1/2, pan‐JNK1/2, phosphorylated ERK1/2 and pan‐ERK1/2 expression levels in irradiated Panc1 cells 4, 12, 24 and 48 h after 10 Gy radiation. (b) Western blot showed decreased phosphorylated Akt (Ser473) in irradiated Panc1DN_PKCδ cells 4 h after radiation in comparison with their parent cells. (c) Western blot showed that phosphorylated p38 MAPK but not phosphorylated JNK1/2 was decreased in irradiated Panc1DN_PKCδ cells 48 h after radiation in comparison with their parent cells.
Given the repopulation model we used in this study represents a paracrine stimulatory effect induced by apoptotic cells, we deduced that the possible mechanism should associated with one or more downstream growth factors released by dying cells. In particular, studies have shown that vascular endothelial growth factor (VEGF) may activate Akt via PKCδ during angiogenesis (Gliki et al., 2002), and others have demonstrated that activated Akt could stimulate VEGF expression and increase tumor resistance to radiotherapy (Schuurbiers et al., 2009). Therefore, we examined VEGF expression in irradiated Panc1 cells and found significantly increased VEGF expression after radiation. Furthermore, radiation‐induced Akt activation and VEGF expression could be inhibited by the PI3K/Akt inhibitor LY294002 (data not shown), which suggested that VEGF expression may be resulted from Akt activation. Of note, Panc1 cells express extremely high levels of VEGF receptor i.e. Flk‐1 (data not shown). Taken together, these observations support that activated caspases 3 and 7 in apoptotic Panc1 cells cleave PKCδ, and then activated PKCδ leads to the phosphorylation of Akt and p38 MAPK directly or indirectly, subsequently causing release of growth factors such as VEGF, which in turn results in living tumor cell growth.
4. Discussion
In addition to surgery, aggressive tumors such as pancreatic cancer are often treated with cytotoxic therapies. However, pancreatic cancer often relapses after cytotoxic therapy, thereby suggesting that these therapies are often ineffective in eliminating all of the tumor cells. The data presented herein provide a novel explanation for the initiating events leading to pancreatic tumor repopulation after radiotherapy. Our data suggest that dying pancreatic tumor cells after radiotherapy stimulate the growth of living pancreatic tumor cells through a mechanism involving caspase 3/7‐PKCδ‐Akt/p38 MAPK signaling. To our knowledge, the role of these signaling events in pancreatic tumor repopulation has not been previously identified.
The concept of accelerated repopulation after cytotoxic therapy such as radiotherapy represents a major obstacle in modern cancer treatment (Hermens and Barendsen, 1969; Stephens et al., 1978). Several studies have attempted to understand the molecular mechanism of tumor repopulation after cytotoxic therapy, specifically regarding angiogenesis and inflammation. For example, radiation causes the upregulation of angiogenesis related factors such as hypoxia‐inducible factor‐1 (Moeller et al., 2004). The macrophages in tumors have been shown to facilitate tumor recovery after radiation (Li et al., 2007; Ahn and Brown, 2008). However, we believe that angiogenesis and inflammation are likely secondary events in tumor repopulation. Although previous studies have identified key mechanisms of tumor repopulation such as inflammation and angiogenesis, the initial events driving tumor repopulation have not been well‐characterized.
In this report, we expanded upon our previous findings of the “Phoenix Rising” pathway of tumor repopulation in the following ways: demonstrating the importance of caspase 7 in addition to caspase 3; and identifying PKC activation as a downstream mechanism of caspase‐mediated tumor repopulation. Specifically, caspase 7, an effector caspase similar to caspase 3, plays a key role during apoptotic tumor cell stimulated living tumor cell growth in pancreatic ductal adenocarcinoma. The transduction of DN_C3 or DN_C7 into Panc1 cells led to significantly decreased numbers of apoptotic cells detected by FACS after radiation and a reduced growth stimulating effect observed by bioluminescence imaging as compared with their parent cells or empty vector transduced cells. In addition to iPLA2 (the downstream mechanism of caspase‐mediated tumor repopulation in the “Phoenix Rising” pathway) we believe that there are several other targets of caspase 3 and caspase 7 in tumor repopulation, such as PKC. In this study we examined three subtypes of PKC by Western blot and found remarkably increased PKCδ cleavage in Panc1 cells after radiation. However, PKCδ cleavage was significantly decreased in the DN_C3 or DN_C7 transduced Panc1 cells after radiation, which confirmed a linkage between caspase 3/7 and PKCδ in apoptotic cells. The mechanism for reduced PKCδ cleavage in DN_C7 transduced Panc1 cells after radiation is unclear since there is no caspase 7 cleaving site been identified in PKCδ. One possibility is that caspase 7 indirectly activates PKCδ via caspase 3 since caspase 7 is located upstream of caspase 3 and can activate caspase 3.
Our results are consistent with the previous finding that the caspase 3‐mediated PKCδ cleavage is vital to the apoptotic cascade. For instance, in human keratinocytes, the overexpression of a mutant PKCδ, which cannot be cleaved by caspase 3, suppresses UV radiation‐induced apoptosis (D'Costa and Denning, 2005). Additionally, PKCδ is an essential component of the intrinsic and extrinsic apoptotic program, and it functions by phosphorylating many proapoptotic targets, including p53, Mcl‐1, and lamin B (Brodie and Blumberg, 2003; Reyland, 2009). However, several controversial reports regarding PKCδ and apoptosis have appeared recently. Cataldi et al. (2009) reported that the 1.5‐ and 6‐Gy doses of ionizing radiation‐induced apoptosis via PKCδ cleavage, but promote survival by Akt phosphorylation in surviving Jurkat T cells. In another study, Xia et al. (2009) reported that PKCδ functions as a critical anti‐apoptotic signal transducer in cells containing activated p21Ras by phosphotidylinositol 3′ kinase/Phosphoinositide‐dependent kinase 1 (PI3K‐PDK1). PKCδ then delivers the survival signal to Akt and results in the activation of Akt, thereby promoting cell survival. As to the relationship between PKCδ and p38 MAPK, several reports suggested that activated PKCδ could further activate p38 MAPK by phosphorylation (Ravi et al., 2008). In our study, both Akt and p38 MAPK activation were associated with PKCδ activation, while Akt activation occurred earlier than p38 MAPK activation. However, we did not detect any changes of phospho‐p38 MAPK protein levels in irradiated HT29DN_ PKCδ cells compared to irradiated parent HT29 cells (data not shown). Therefore, our findings suggest that Akt may be more broadly involved in dying cell stimulation of living tumor cell growth, while p38 is more specific to Panc1 cells during repopulation.
The advantage to the design of our study is that living tumor cells could be distinguished from dying cells so that the growth of living cells could be quantitatively monitored. Our results clearly indicated that presence of dying cells could stimulate living tumor cell growth. Therefore, we propose that dying cells can produce and secrete growth factor and stimulate living tumor cell proliferation via a paracrine manner. The results from this study showed that VEGF expression increased after radiation. Moreover, VEGF expression was inhibited by the PI3K/Akt inhibitor LY294002. However, the role of VEGF on tumor repopulation needs more investigation to further confirm.
Overall, this study demonstrated several meaningful findings. First, both apoptotic and survival signals were activated in irradiated, dying Panc1 cells. Second, dying Panc1 tumor cells could induce living Panc1 tumor cell growth. Third, a novel pathway which regulates tumor repopulation or resistance to radiotherapy might exist, i.e. activated caspases 7 and/or 3 in apoptotic Panc1 cells cleaves PKCδ, and subsequently activated PKCδ leads to the phosphorylation of Akt and p38 MAPK. Fourth, since activated Akt and p38 MAPK are known as extensively involved in regulation of cell proliferation, we speculate that activated Akt and p38 MAPK may result in release of growth factors and induce tumor cell growth (Figure 6). We therefore propose that the data from this study indicate a novel role for caspase 3‐mediated PKCδ activation in pancreatic tumor repopulation after radiotherapy. Although PKCδ activation has been reported to be related to fractionated‐radiation‐induced expansion of glioma‐initiating cells and resistance to cancer treatment (Kim et al., 2011), a direct role for PKCδ in stimulating tumor repopulation has not yet been described to our knowledge. Our observation that the pan PKC inhibitor (GF109203x) and the specific PKCδ inhibitor (rottlerin) strongly inhibited apoptotic cell induced tumor cell proliferation further highlighted the role of PKCδ during tumor repopulation. The pathway reported herein provides targets for interrupting tumor repopulation or resistance to radiotherapy. As a practical application of our findings, the use of inhibitors targeting either caspase 3/7, PKCδ, phospho‐Akt or p38 MAPK could be combined with radiation therapy in an effort to reduce tumor repopulation or resistance to radiotherapy and improve survival outcomes for patients with pancreatic cancer. Alternatively, late course accelerated hyperfractionated irradiation (LCAF) may also be used to decrease tumor repopulation stimulated by dying, irradiated cells. Taken together, these findings may improve the survival outcomes for patients with pancreatic cancer.
Figure 6.

A schematic representation of caspase 3/7‐PKCδ‐Akt/p38 MAPK signaling pathway in the radiation‐induced apoptotic cells stimulated live tumor cell growth in human pancreatic carcinoma (Panc1) in vitro. After radiation, apoptotic executioner caspase 7 and 3 are cleaved and activated. Activated caspase 7 and 3, eventually via caspase 3, cleave PKCδ to generate a catalytic fragment (CF) and then activate PKCδ. Activated PKCδ CF further phosphorylates and activates Akt or p38 MAPK directly or indirectly and then subsequently causes release of growth factors to stimulate the proliferation of neighboring live tumor cells.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgments
This study was supported by grants from National Natural Science Foundation (81120108017, 81172030) and National Basic Research Program of China (2010CB529902) (to Q Huang and L Tian) and in part by grants from National Cancer Institute, United States (CA131408, CA136748, CA155270) (to C‐Y Li).
Cheng Jin, Tian Ling, Ma Jingjing, Gong Yanping, Zhang Zhengxiang, Chen Zhiwei, Xu Bing, Xiong Hui, Li Chuanyuan, Huang Qian, (2015), Dying tumor cells stimulate proliferation of living tumor cells via caspase-dependent protein kinase Cδ activation in pancreatic ductal adenocarcinoma, Molecular Oncology, 9, doi: 10.1016/j.molonc.2014.07.024.
Contributor Information
Chuanyuan Li, Email: chuan.li@duke.edu.
Qian Huang, Email: qhuang2007@gmail.com.
References
- Ahn, G.O. , Brown, J.M. , 2008. Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: role of bone marrow-derived myelomonocytic cells. Cancer Cell. 13, 193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu, A. , Lu, D. , Sun, B. , Moor, A.N. , Akkaraju, G.R. , Huang, J. , 2002. Proteolytic activation of protein kinase C-epsilon by caspase-mediated processing and transduction of antiapoptotic signals. J. Biol. Chem. 277, 41850–41856. [DOI] [PubMed] [Google Scholar]
- Bluwstein, A. , Kumar, N. , Leger, K. , Traenkle, J. , Oostrum, J. , Rehrauer, H. , Baudis, M. , Hottiger, M.O. , 2013. PKC signaling prevents irradiation-induced apoptosis of primary human fibroblasts. Cell Death Dis. 4, e498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie, C. , Blumberg, P.M. , 2003. Regulation of cell apoptosis by protein kinase c delta. Apoptosis. 8, 19–27. [DOI] [PubMed] [Google Scholar]
- Cataldi, A. , Di Giacomo, V. , Rapino, M. , Zara, S. , Rana, R.A. , 2009. Ionizing radiation induces apoptotic signal through protein kinase Cdelta (delta) and survival signal through Akt and cyclic-nucleotide response element-binding protein (CREB) in Jurkat T cells. Biol. Bull. 217, 202–212. [DOI] [PubMed] [Google Scholar]
- Datta, R. , Kojima, H. , Yoshida, K. , Kufe, D. , 1997. Caspase-3-mediated cleavage of protein kinase C theta in induction of apoptosis. J. Biol. Chem. 272, 20317–20320. [DOI] [PubMed] [Google Scholar]
- D'Costa, A.M. , Denning, M.F. , 2005. A caspase-resistant mutant of PKC-delta protects keratinocytes from UV-induced apoptosis. Cell Death Differ. 12, 224–232. [DOI] [PubMed] [Google Scholar]
- Dugourd, C. , Gervais, M. , Corvol, P. , Monnot, C. , 2003. Akt is a major downstream target of PI3-kinase involved in angiotensin II-induced proliferation. Hypertension. 41, 882–890. [DOI] [PubMed] [Google Scholar]
- Emoto, Y. , Manome, Y. , Meinhardt, G. , Kisaki, H. , Kharbanda, S. , Robertson, M. , Ghayur, T. , Wong, W.W. , Kamen, R. , Weichselbaum, R. , 1995. Proteolytic activation of protein kinase C delta by an ICE-like protease in apoptotic cells. EMBO J. 14, 6148–6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi, L. , Kepp, O. , Kroemer, G. , 2012. Caspase-3 and prostaglandins signal for tumor regrowth in cancer therapy. Oncogene. 31, 2805–2808. [DOI] [PubMed] [Google Scholar]
- Gliki, G. , Wheeler-Jones, C. , Zachary, I. , 2002. Vascular endothelial growth factor induces protein kinase C (PKC)-dependent Akt/PKB activation and phosphatidylinositol 3'-kinase-mediates PKC delta phosphorylation: role of PKC in angiogenesis. Cell Biol. Int. 26, 751–759. [DOI] [PubMed] [Google Scholar]
- Hallahan, D.E. , Virudachalam, S. , Schwartz, J.L. , Panje, N. , Mustafi, R. , Weichselbaum, R.R. , 1992. Inhibition of protein kinases sensitizes human tumor cells to ionizing radiation. Radiat. Res. 129, 345–350. [PubMed] [Google Scholar]
- Hallahan, D.E. , Virudachalam, S. , Sherman, M.L. , Huberman, E. , Kufe, D.W. , Weichselbaum, R.R. , 1991. Tumor necrosis factor gene expression is mediated by protein kinase C following activation by ionizing radiation. Cancer Res. 51, 4565–4569. [PubMed] [Google Scholar]
- Hermens, A.F. , Barendsen, G.W. , 1969. Changes of cell proliferation characteristics in a rat rhabdomyosarcoma before and after x-irradiation. Eur. J. Cancer. 5, 173–189. [DOI] [PubMed] [Google Scholar]
- Huang, Q. , Li, F. , Liu, X. , Li, W. , Shi, W. , Liu, F.F. , O'Sullivan, B. , He, Z. , Peng, Y. , Tan, A.C. , Zhou, L. , Shen, J. , Han, G. , Wang, X.J. , Thorburn, J. , Thorburn, A. , Jimeno, A. , Raben, D. , Bedford, J.S. , Li, C.Y. , 2011. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 17, 860–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, M.J. , Kim, R.K. , Yoon, C.H. , An, S. , Hwang, S.G. , Suh, Y. , Park, M.J. , Chung, H.Y. , Kim, I.G. , Lee, S.J. , 2011. Importance of PKCdelta signaling in fractionated-radiation-induced expansion of glioma-initiating cells and resistance to cancer treatment. J. Cell Sci. 124, 3084–3094. [DOI] [PubMed] [Google Scholar]
- Koriyama, H. , Kouchi, Z. , Umeda, T. , Saido, T.C. , Momoi, T. , Ishiura, S. , Suzuki, K. , 1999. Proteolytic activation of protein kinase C delta and epsilon by caspase-3 in U937 cells during chemotherapeutic agent-induced apoptosis. Cell Signal. 11, 831–838. [DOI] [PubMed] [Google Scholar]
- Kurokawa, M. , Kornbluth, S. , 2009. Caspases and kinases in a death grip. Cell. 138, 838–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F. , Huang, Q. , Chen, J. , Peng, Y. , Roop, D.R. , Bedford, J.S. , Li, C.Y. , 2010. Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci. Signal. 3, ra13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F. , Sonveaux, P. , Rabbani, Z.N. , Liu, S. , Yan, B. , Huang, Q. , Vujaskovic, Z. , Dewhirst, M.W. , Li, C.Y. , 2007. Regulation of HIF-1alpha stability through S-nitrosylation. Mol. Cell. 26, 63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber, M. , Mazzetta, J. , Nelson-Rees, W. , Kaplan, M. , Todaro, G. , 1975. Establishment of a continuous tumor-cell line (panc-1) from a human carcinoma of the exocrine pancreas. Int. J. Cancer. 15, 741–747. [DOI] [PubMed] [Google Scholar]
- Ma, J. , Tian, L. , Cheng, J. , Chen, Z. , Xu, B. , Wang, L. , Li, C. , Huang, Q. , 2013. Sonic hedgehog signaling pathway supports cancer cell growth during cancer radiotherapy. PLoS One. 8, e65032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller, B.J. , Cao, Y. , Li, C.Y. , Dewhirst, M.W. , 2004. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell. 5, 429–441. [DOI] [PubMed] [Google Scholar]
- Munshi, A. , Hobbs, M. , Meyn, R.E. , 2005. Clonogenic cell survival assay. Methods Mol. Med. 110, 21–28. [DOI] [PubMed] [Google Scholar]
- Nakajima, T. , 2006. Signaling cascades in radiation-induced apoptosis: roles of protein kinase C in the apoptosis regulation. Med. Sci. Monit. 12, RA220–224. [PubMed] [Google Scholar]
- Nakajima, T. , 2008. Positive and negative regulation of radiation-induced apoptosis by protein kinase C. J. Rad. Res. 49, 1–8. [DOI] [PubMed] [Google Scholar]
- Park, J.S. , Qiao, L. , Su, Z.Z. , Hinman, D. , Willoughby, K. , McKinstry, R. , Yacoub, A. , Duigou, G.J. , Young, C.S. , Grant, S. , Hagan, M.P. , Ellis, E. , Fisher, P.B. , Dent, P. , 2001. Ionizing radiation modulates vascular endothelial growth factor (VEGF) expression through multiple mitogen activated protein kinase dependent pathways. Oncogene. 20, 3266–3280. [DOI] [PubMed] [Google Scholar]
- Ravi, D. , Muniyappa, H. , Das, K.C. , 2008. Caffeine inhibits UV-mediated NF-kappaB activation in A2058 melanoma cells: an ATM-PKCdelta-p38 MAPK-dependent mechanism. Mol. Cell Biochem. 308, 193–200. [DOI] [PubMed] [Google Scholar]
- Reyland, M.E. , 2009. Protein kinase C isoforms: multi-functional regulators of cell life and death. Front. Biosc. 14, 2386–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Wallberg, K.A. , 2012. Principles of cancer treatment: impact on reproduction. Adv. Exp. Med. Biol. 732, 1–8. [DOI] [PubMed] [Google Scholar]
- Schmidt-Ullrich, R.K. , Mikkelsen, R.B. , Dent, P. , Todd, D.G. , Valerie, K. , Kavanagh, B.D. , Contessa, J.N. , Rorrer, W.K. , Chen, P.B. , 1997. Radiation-induced proliferation of the human A431 squamous carcinoma cells is dependent on EGFR tyrosine phosphorylation. Oncogene. 15, 1191–1197. [DOI] [PubMed] [Google Scholar]
- Schuurbiers, O.C. , Kaanders, J.H. , van der Heijden, H.F. , Dekhuijzen, R.P. , Oyen, W.J. , Bussink, J. , 2009. The PI3-K/AKT-pathway and radiation resistance mechanisms in non-small cell lung cancer. J. Thorac. Oncol. 4, 761–767. [DOI] [PubMed] [Google Scholar]
- Stabile, E. , Zhou, Y.F. , Saji, M. , Castagna, M. , Shou, M. , Kinnaird, T.D. , Baffour, R. , Ringel, M.D. , Epstein, S.E. , Fuchs, S. , 2003. Akt controls vascular smooth muscle cell proliferation in vitro and in vivo by delaying G1/S exit. Circ. Res. 93, 1059–1065. [DOI] [PubMed] [Google Scholar]
- Stephens, T.C. , Currie, G.A. , Peacock, J.H. , 1978. Repopulation of gamma-irradiated Lewis lung carcinoma by malignant cells and host macrophage progenitors. Br. J. Cancer. 38, 573–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassileva, V. , Allen, C.J. , Piquette-Miller, M. , 2008. Effects of sustained and intermittent paclitaxel therapy on tumor repopulation in ovarian cancer. Mol. Cancer Ther. 7, 630–637. [DOI] [PubMed] [Google Scholar]
- Xia, S. , Chen, Z. , Forman, L.W. , Faller, D.V. , 2009. PKCdelta survival signaling in cells containing an activated p21Ras protein requires PDK1. Cell Signal. 21, 502–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, W. , Liu, H.T. , 2002. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 12, 9–18. [DOI] [PubMed] [Google Scholar]