

Abstract
The progression of an infection within a host determines the ability of a pathogen to transmit to new hosts and to maintain itself in the population. While the general connection between the infection dynamics within a host and the population-level transmission dynamics of pathogens is widely acknowledged, a comprehensive and quantitative understanding that would allow full integration of the two scales is still lacking. Here, we provide a brief discussion of both models and data that have attempted to provide quantitative mappings from within-host infection dynamics to transmission fitness. We present a conceptual framework and provide examples of studies that have taken first steps towards development of a quantitative framework that scales from within-host infections to population-level fitness of different pathogens. We hope to illustrate some general themes, summarize some of the recent advances and—maybe most importantly—discuss gaps in our ability to bridge these scales, and to stimulate future research on this important topic.
Keywords: multi-scale, infectious disease, computational models
1. Introduction
In this review, we argue that a detailed understanding of the within-host dynamics of infectious diseases is both scientifically important and timely. Specifically, we submit that the processes of pathogen invasion of the host, its subsequent spread, interplay with host immunity and the consequent pathogenesis impacts are central to understanding population-level transmission and mitigating the morbidity and mortality of infected hosts. To illustrate this claim, let us consider neuraminidase inhibitors (NAI), a class of therapeutic drugs used to treat influenza patients. While the clinical benefits of NAI in reducing the severity of complications in infected patients remain debated [1–4], NAI are known to shorten the symptomatic period and reduce virus load [5]. This, in turn, can potentially lead to reduced transmission of those treated with NAI, and therefore, make NAI a potentially important tool in outbreak mitigation or the curtailment of localized transmission [6–10]. However, even though we appreciate the need for quantifying the epidemiological impacts of NAI, obtaining suitable population-level information to do so remains difficult [11–15]. If instead there is a general theory on scaling from individual-level measurements of virus load and symptom severity to between-host transmission fitness, we could use more readily available within-host data to make quantitative predictions about the impact of NAI on outbreak mitigation.
Another example illustrating the importance of knowing quantitatively the link between within-host infection dynamics and transmission fitness comes from a recent study on avian influenza persistence [16]. Aiming to dissect the fitness consequences of differential sensitivity of virus subtypes to temperature, we considered three distinct ways in which transmission fitness might be linked to viral load. We found that the predicted population-level fitness of different low-pathogenic avian influenza strains strongly depended on the specific assumed link between viral load and transmission potential. In the absence of additional information on transmission and viral shedding, we were unable to make more precise predictions.
These are just two examples pointing to the importance of developing a framework that bridges within-host and between-host levels in a quantitative and predictive manner. Increasing awareness of the importance to integrate within- and between-host scales has led to the development of models that explicitly link the two scales [7,17–21]. These models, often referred to as ‘multi-scale’ models, have increased in popularity in recent years [22–26]. While there have been exciting advances made in this area, most studies linking within- and between-host scales are conceptual or theoretical with mainly qualitative and little quantitative support from data. Progress towards a predictive multi-scale framework will require a more precise, quantitative understanding of how infection dynamics, pathogen load, target cell depletion, immunology, symptomatology and other clinical features combine to shape pathogen transmission fitness at the population level.
In the following, we discuss some of the quantitative links that have been or need to be made in bridging the scales. To guide our discussion, we introduce a conceptual model, shown in figure 1. The main protagonists in any infection are the pathogenic organism and the immune response, which vary dynamically over the course of an infection. The interplay between these determines the time course of pathogen abundance in the host (pathogen load), and host symptoms, which in turn can interact with pathogen and immune response. Pathogen load, immune response and symptoms dictate (i) the host infectiousness profile and (ii) host behaviour as it relates to pathogen spread. In the following sections, we provide a collection of case studies that highlight some of the steps that have recently been made with regard to the quantitative bridging of individual host infection dynamics (pathogen, immune response and symptoms) to (i) host infectiousness and (ii) host behaviour and further on to transmission fitness.
Figure 1.
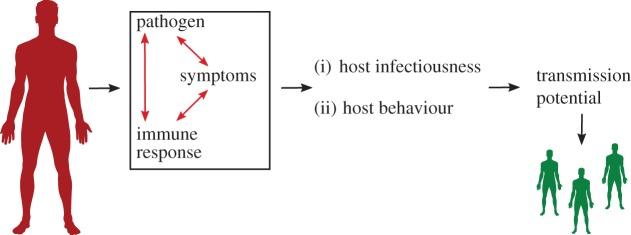
Schematic of the within-host infection and between host-transmission link. Inside an infected host, pathogen and immune response interact. These interactions dictate time-varying pathogen load, immune response and symptoms. Pathogen, immune response and symptoms impact (i) host infectiousness and (ii) host behaviour relating to pathogen spread. These components in turn influence pathogen transmission potential. (Online version in colour.)
2. Host infectiousness
To ensure non-extinction in a host population, a pathogen needs to replicate to levels within an infected host that are sufficient to generate ongoing chains of transmission to new hosts. It makes intuitive sense to assume that—all else being equal—the transmission potential of an infectious host increases with increasing pathogen load in the appropriate host tissues. For instance, high pathogen load in the respiratory tract may be expected to correspond to high infectiousness for a respiratory pathogen.
This—arguably simplest—assumption that transmission potential only depends on pathogen load has been used in a number of recent influenza modelling studies. However, the assumed functional association between viral load and transmission varied considerably. Some studies have considered transmission to be linked to the instantaneous viral load [27,28], whereas others have instead explored the total area under the curve (AUC) [29–31]. Among those models assuming transmission to scale with total virus load (AUC), alternative assumptions include transmission scaling with viral load on a logarithmic scale [16,32] or through a linear relationship [16].
For other infectious diseases, similar assumptions have been incorporated in mathematical models. For instance, studies of HIV and hepatitis C virus (HCV) assumed that virus load and possibly the number of infected cells are positively associated with transmission fitness [33–35]. In another model for HCV, it was assumed transmission fitness is proportional to the logarithm of the infected cell density [36] (as a proxy of virus load). Similar assumptions of the relation between virus load and transmission fitness have been made for generic, conceptual infection models [37–40].
While these models make plausible, pragmatic assumptions about the link between pathogen load and transmission rate, direct empirical support is not widely available. Possibly, one of the best studied pathogens in this regard is HIV. Data for HIV correlating the viral load in serum with probability of infection in a partner suggest a sigmoid relationship (figure 2; [42–44]). However, higher viral load also leads to more rapid progression to the terminal AIDS stage [43,45], therefore reducing the time during which transmission can occur (figure 2). The impact of increasing virus load on both increased instantaneous infectiousness and faster progression towards AIDS lead to the suggestion that overall lifetime transmission potential is maximized at intermediate viral loads [41,43].
Figure 2.
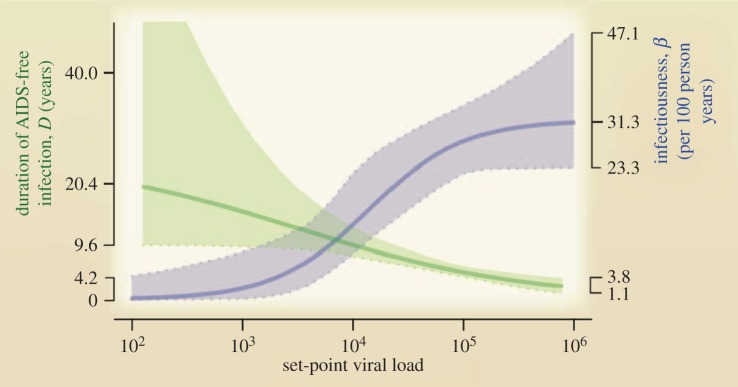
HIV transmission between discordant couples (blue) and duration of asymptomatic period (green) as function of set-point virus load for HIV. Reproduced from Fraser et al. [41]; see this study and references therein for more details. Reprinted with permission from AAAS. (Online version in colour.)
Figure 3 provides another example of a direct mapping from within-human pathogen load data to transmission for dengue infections. For each of the four dengue serotypes, the association between viraemia in dengue patients and the infectiousness of these patients to mosquitoes is shown (figure 3; [46]). In acute infections, such as dengue, pathogen levels do not reach a defined steady state at which pathogen levels are more or less constant for an extended period. Instead, the whole pathogen time-course during the infection likely determines overall transmission potential. It is likely that acute infections show a negative correlation between, for instance, peak pathogen load and the duration of infection. This can happen either because a higher peak leads to more rapid host death or a stronger ensuing immune response clears the infection more quickly. In figure 4, we present the duration of infection as a function of pathogen peak for several acute viral infections in animal hosts. In four of the five viruses, we observe a negative correlation between virus peak load and infection duration, with influenza A the exception. In general, such patterns will likely vary depending on the details of inoculum dose, host species, virus strain, etc. It will be important to determine how different components of infection dynamics such as duration, peak load, total area under the curve, etc., determine overall transmission potential. As far as we are aware, this has not yet been determined for dengue or any other acute human infection.
Figure 3.

Probability of a mosquito getting infected with dengue virus when exposed to an infected human, as a function of dengue virus load in the blood. Data are indicated by the symbols, lines show model fits. The figure is shown as originally published in Nguyen et al. [46]. See the original publication for more detailed descriptions and meaning of all features shown in the figure. (Online version in colour.)
Figure 4.
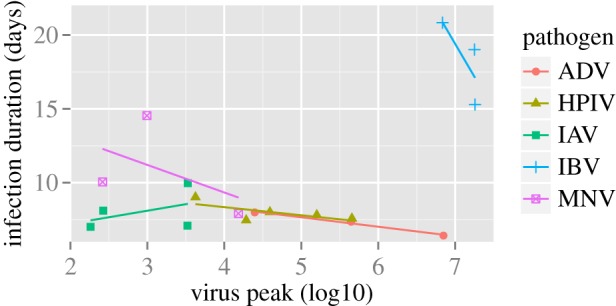
Duration of infection as function of pathogen peak load for several acute viral infections. Symbols show data, lines are linear fits to indicate trend. The infections shown are adenovirus (ADV) in cotton rats [47], human parainfluenza virus (HPIV) in cotton rats [48], influenza A virus (IAV) in mice [49], infectious bronchitis virus (IBV) in chickens [50] and murine norovirus (MNV) in mice [51]. Duration of infection is defined as the time at which the pathogen load drops below some threshold, e.g. the limit of detection or 1 infectious particle. See the original studies and also Li & Handel [52] for more details. (Online version in colour.)
The relationship between pathogen load and infectivity presented in figures 2 and 3 has also been reported in other infectious disease systems. For instance, transmission of malaria from humans to mosquitoes was found to map onto pathogen load similar to the mappings shown for dengue [53,54]. Similar patterns were found in feeding experiments measuring infection of sand flies with Leishmania donovani and mosquitoes with chikungunya [55–57], vertical transmission of hepatitis B virus between mothers and infants [58] and human T lymphotropic virus transmission between males and females [59] and mothers and newborns through breastfeeding [60]. Several studies of transmission in animal hosts have also shown a scaling of transmission fitness with pathogen load, e.g. Salmonella and Clostridium difficile transmission in mice [61,62] and Escherichia coli in cattle [63]. All these examples suggest that for some diseases and under some scenarios, infectiousness might be directly determined by pathogen load. The simple view that the infectiousness of an individual is dictated by pathogen load is appealing inasmuch that pathogen load is often relatively easily measurable. Under such conditions, the impact of symptoms on infectiousness may be safely ignored. If one further assumes that the contact behaviour of a host is not affected by pathogen load or associated symptoms, one obtains the simplest possible mapping from within-host dynamics to transmission, with host infectiousness and transmission potential related according to some functional form to pathogen load alone.
Often, however, host symptoms play a central role in efficient transmission. For instance, while one might expect that for HIV, symptoms in the infected person are not required for efficient transmission, there is some evidence that symptoms such as ulcers and other tissue injuries increase infectiousness of HIV-infected host [64], and that the stage of the infection, and likely changes in the status of the immune response during these different stages, also seem to have some impact on transmission [65]. Prominent pathogens which appear only to transmit during the symptomatic phase include SARS and Ebola [66,67]—though it is not fully clear if the symptoms are strictly required for transmission or merely coincide with virus load levels that are sufficient for transmission. For respiratory pathogens, symptoms that can facilitate transmission involve coughing and sneezing, for gastrointestinal pathogens the symptoms are often vomiting and diarrhoea, which have been shown to affect transmission [68,69].
One way to allow for the role of symptoms on transmission but still keep the focus on pathogen load is to try and express symptoms as a function of pathogen load. In a previous study, we assumed that host infectiousness was proportional to virus concentration multiplied by total amount of shedding [70]. Shedding, while presumably influenced by symptoms, was mapped back onto virus load. A sigmoid relation between virus load and shedding as measured by nasal discharge provided a reasonable fit (figure 5). This approach thereby accounted for the contribution of symptoms to shedding, but through mapping of symptoms back to virus load expressed infectiousness as function of pathogen load alone.
Figure 5.
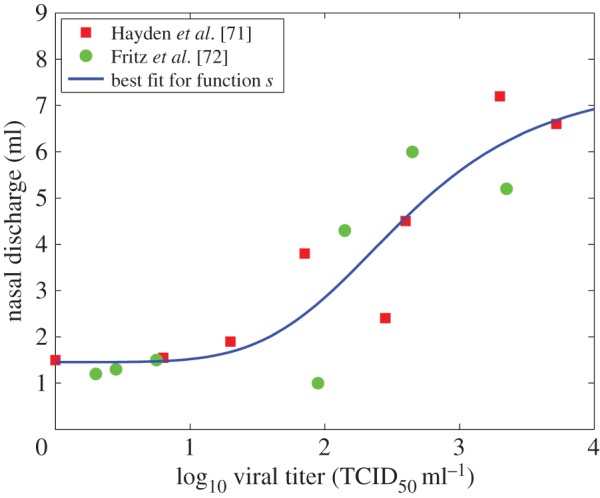
Nasal discharge as a function of viral load. Data are from Hayden et al. [71] (squares) and Fritz et al. [72] (circles) and measure viral load as determined by nasal wash, as well as total nasal discharge (i.e. snot) for 24 h time periods, produced by and collected from volunteers infected with influenza. Also shown is the best fit for a sigmoid function. Reproduced from Handel et al. [70], see the original publication for more details. (Online version in colour.)
While pathogen load and symptoms are closely correlated (see e.g. [73–75] for influenza), the ability to map symptoms back onto pathogen load will likely not work in general. Returning to influenza as our example, analyses of data from ferret infection studies showed that different influenza strains can generate similar viral loads but contrasting transmission potential [76,77]. Because the ferrets, in this study, were housed in cages with presumably little change in contact behaviour between different study groups, differences in transmission are not attributable to differences in host contact behaviour or viral load, but instead must be attributed to other features, such as qualitative differences in the virus [78] or differences in host infectiousness mediated by symptoms (e.g. frequency of sneezing). How best to associate symptoms to transmission potential remains an open question. For instance, while sneezing likely helps transmission in both humans and ferrets, there does not seem an easy and general relation between the two, with different mechanisms of transmission all contributing [79–82].
If it is not possible to map symptoms directly to pathogen load, one needs to specify a mapping between both symptoms and pathogen load (which both in turn are influenced by the immune response) and host infectiousness. This idea has been included in several influenza multi-scale modelling studies, which assumed that transmission/infectiousness was a product of virus load (not further defined) and a sigmoid function of interferon levels [83]. The latter was assumed to represent symptoms. A more recent modelling study assumed that transmission scaled with both virus load and pathogenicity by connecting these quantities through different (non-specified) linear and nonlinear functions [84].
Unfortunately, data that would allow a more detailed translation of pathogen load, immune response and symptoms into infectiousness is lacking for most pathogens. We could not find any data for a human pathogen that would allow such a direct, quantitative mapping. One detailed study that seems to be the most advanced effort in that direction was done for foot-and-mouth disease virus infection in cattle [85]. By carefully measuring multiple pathogen, immune response and symptom variables of infected animals over the course of the infection, and further exposing uninfected animals at different times during the infection and recording if transmission occurred, it was possible to devise a model that mapped within-host infection quantities to transmission potential. This study showed that, in addition to virus load, factors such as lesions, temperature and interferon, among others, impacted transmission potential [85,86]. More studies of this nature are needed, and we return to that point in the discussion.
To summarize this section, we conclude that while the simple assumption that infectiousness depends solely on pathogen load might be intuitively appealing and justifiable in some cases, in many situations, pathogen load alone is likely to be a poor predictor of infectiousness. The effect of symptoms will need to be included to properly characterize infectiousness. A major obstacle in taking this step for any pathogen remains the absence of suitable data that would permit the development of a more comprehensive, quantitative understanding. Even when one might reasonably assume that symptoms can be ignored, empirically supported explicit functional relationships between pathogen load and host infectiousness are still not available for many important pathogens. A major future challenge remains the determination of whether pathogen load alone is sufficiently predictive of instantaneous infectiousness. If yes, one needs to determine which organs are the most useful sampling sites that predict infectiousness, and then try to determine a quantitative mapping between pathogen load and infectiousness (e.g. linear or log scale, sigmoid or other). If pathogen load alone does not prove to be a good predictor of infectiousness, it would be important to identify those symptoms that influence infectiousness. One then either needs to measure those symptoms directly, or if possible, determine a mapping between pathogen load, appropriate components of the immune response, and symptoms, and measure those latter quantities. For instance, if one were to determine that sneezing is an important component of infectiousness for influenza, one could either directly measure sneezing frequency [80] or determine immune response correlates (e.g. histamine levels) and measure these.
3. Host behaviour
The behaviour of an infected host as it relates to the potential of transmission is the second component after infectiousness that determines overall transmission potential. Host behaviour is often influenced by symptoms, which in turn are determined by pathogen and immune response dynamics. (We do not further discuss behaviour changes specific to humans that are not related to the biology of the infection process, e.g. use of condoms by HIV-infected individuals and similar actions.)
The simplest assumption is that host behaviour is independent of the within-host infection process. This might often be a reasonable approximation for diseases that cause few or mild symptoms. Many sexually transmitted diseases might fall into this category for the majority of infected hosts, as might be mild infections with pathogens such as rhinovirus. If instead a pathogen causes significant symptoms, it often affects transmission potential in a complicated way. As discussed above, symptoms are often beneficial to the pathogen if they tend to increase infectiousness. However, beyond a certain point, there is likely a trade-off between enhanced infectiousness and reductions in host behaviour that can lead to transmission. The general analysis of such trade-offs has been under heavy theoretical development over the past few decades, commonly known as ‘virulence research’. We refer interested readers to reviews on this topic [87,88] and references therein for further details on this important topic. While the theory for such trade-offs is pretty well studied, the evidence from data is limited, especially for human pathogens.
Some data supporting the idea that changes in host behaviour limit the transmission potential for some diseases comes from a line of investigation by Ewald and co-workers [89–92]. Those studies showed that pathogens which do not rely strongly on host health and mobility tend to induce more severe symptoms (i.e. are more virulent) compared with pathogens that need the host to be reasonably healthy and mobile to support further spread.
Unfortunately, the existing evidence mostly comes from population-level analyses of aggregated data. Studies that try to quantify the relation between host infection and behaviour in individual hosts are much less common. Some examples come from animal infections, where some pathogens have been shown to actively manipulate host behaviour. For instance, toxoplasma is known to alter the behaviour of its rodent host, presumably to increase contact with the next host stage, the feline host [93]. Similar host altering behaviour to benefit the pathogen have been described for other pathogens [94]. While it is acknowledged that disease status can alter behaviour in humans as well (e.g. [95] and references therein), studies allowing quantification of the impact of infection on behaviour for human infections are rare. For the human infection examples presented in §2, the data were collected in experimental settings with little opportunity to observe and measure altered host behaviour. For instance, for the dengue data, the experimental set-up for measuring human infectivity to biting mosquitoes eliminated any impact of potential symptom-mediated behaviour change. In a less controlled experimental setting, such behaviour changes might impact overall transmission potential.
A study that provides some information on the link between infection status and host behaviour was done for influenza during the 2009 pandemic using a survey-based recording method [96,97]. Results showed that sick individuals had around one-fourth the number of daily contacts compared with healthy individuals, leading to an estimated reduction of transmission (as measured by the reproductive number) of the same amount [96,97]. This study also suggested that the number of symptoms correlated inversely with the number of contacts [96], possibly, because increased symptoms might make it more likely that an individual stays at home (self-quarantines). The latter finding supports the assumption of a previous modelling study [70]. In that study, we made the ad hoc assumption that there is an inverse correlation between contact rate, w, and symptoms, S, according to w ∼ 1/(1 + S).
Clearly, more detailed data would be useful to better parametrize and define the relation between symptoms and contact behaviour, not only for influenza but also for most other diseases. Based on the contact studies for influenza, it seems possible that the right kind of data linking symptoms with contact behaviour and therefore transmission potential could be obtained for a number of infectious diseases.
The findings on contact patterns shown in Eames et al. and Kerckhove et al. [96,97], combined with studies showing that prolonged viral shedding correlated with more severe disease [98,99], also suggest that a virulence–transmission trade-off exists for influenza, akin to the one for HIV mentioned above. Additional data would be needed to confirm and further quantify this potential trade-off.
Finally, while we have focused on potential contact behaviour changes in the infected host, it is worth pointing out that behaviour changes might also occur among susceptible hosts, who adjust their behaviour based on the perceived sickness of the host. For instance, if someone sneezes or coughs repeatedly, others might keep an increased distance. This symptom-induced behaviour change could also reduce transmission potential. For further discussions of this component, as well as more general discussions about the role of behaviour on the spread of infectious diseases, we refer the reader to Funk et al. and Manfredi & D'Onofrio [100–102] and references therein.
To summarize this section, we conclude that the impact of pathogen load, immune response and symptoms on host contact behaviour, and its subsequent impact on transmission, seems to be the least well studied part of the components linking the within-host and between-host scales and requires urgent future attention. Especially needed are data from either experimental or observational settings that could allow one to determine the mapping from infection dynamics to host contact behaviour and transmission potential.
4. Discussion
It is widely acknowledged that there is heterogeneity in the transmission potential of infected hosts [103–105]. Understanding how within-host factors of an infected individual contribute to transmission is important in targeting intervention strategies at high transmission hosts [106]. If, for some pathogen, increased transmission is mainly a function of host behaviour, a different strategy is called for compared with a situation where increased transmission is mainly associated with specific types of symptoms or high pathogen load. Beyond intervention strategies, linking the within-host and between-host scales will be important in obtaining a more complete and predictive understanding of host–pathogen ecology and evolution.
The past few decades have seen important advances in this regard. However, most of these advances have been theoretical, the much-needed comparison of the theory with data is often missing. Here, we sketched out some of the components linking the within-host and between-host scales, and provided some empirical examples that have demonstrated different aspects of how these scales could be bridged. It is obvious to us that this endeavour is still in its infancy. Even for the better studied of the components outlined above, namely host infectiousness, we appear to be in the early phase of a quantitative link. Less is known about the host behaviour component, especially for human pathogens.
While further theoretical advances are useful and necessary, the most beneficial studies are likely those that provide a tight integration of models with data. For instance, to estimate in detail the relation between infectiousness and within-host infection dynamics, one could perform experiments similar to the one described previously by Charleston et al. [85]. In figure 6, we show an experimental set-up that would allow one to determine the relation between pathogen load, immune response, symptoms and how it relates to transmission potential. By frequently measuring as many infection-related quantities as possible (e.g. pathogen load, various immune response components, symptoms) in the infected host, and further at frequent intervals exposing a number of naive hosts to the infected hosts and measuring transmission, one could obtain a detailed understanding how different within-host components affect transmission.
Figure 6.
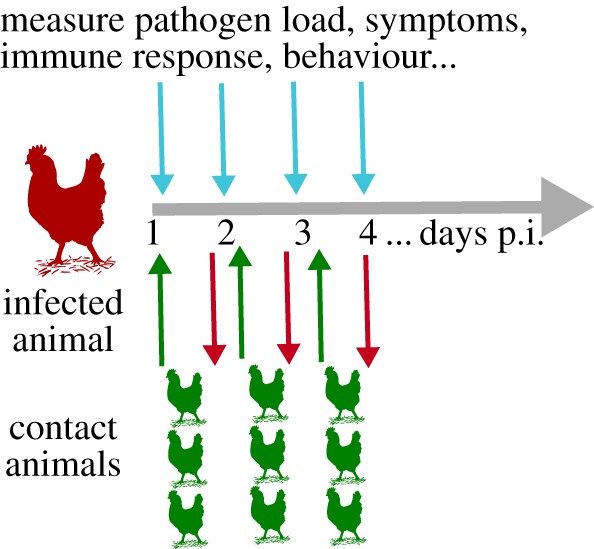
Experimental set-up to determine infectiousness as function of within-host infection dynamics. The infected host is repeatedly sampled to determine as many infection-related quantities as possible (e.g. pathogen load, various immune response components, symptoms). In addition, sets of susceptible hosts are exposed to the infected host at various intervals to determine transmission. This could allow one to obtain a quantitative mapping between quantities such as pathogen load and symptoms and transmission potential [85]. It might even be possible to set up the experiment in such a way that potential contact behaviour changes in the infected host or the contacts can be measured. (Online version in colour.)
While conceptually fairly straightforward, there are significant logistic challenges owing to the potentially frequent replacement of contacts. While the general feasibility of transmission experiments have for instance been demonstrated for influenza between humans [107], it would require a large number of human volunteers to enable frequent replacement of contacts. The same holds true for animal experiments. This might only be feasible for certain pathogen–host combinations. We expect small mammals (e.g. ferrets [108]) and birds (e.g. chicken [109]) to be potentially suitable hosts. Given the likely expense of such studies, it is important to design them in the most efficient way possible. For instance, the experimental set-up should be chosen such that there is variation in the number of contact hosts that get infected. If either all or none get infected every time, little information is gained. Further, the optimal frequency of contact animal replacement needs to be determined. Theoretical models have been devised to help plan small-scale transmission experiments [110–112]. These theoretical developments focused on experiments with the usual set-up where infected and contact animals were brought into contact for the duration of the infection to assess overall transmission potential, i.e. the estimation of a quantity such as R0. Similar methods could be devised for scenarios that require frequent adding and removal of contact animals. It would be important to estimate the optimal number of ‘rotations', the number of contact animals and infected animals per rotation, and the number of required replicates.
The collection of data allowing one to better estimate the second component of transmission potential, namely host behaviour, seems harder. Experimental studies often do not allow hosts to alter their behaviour in a meaningful way, because the enclosures in which animals (or humans) are kept during such experiments are very circumscribed. It might be possible for some hosts, e.g. chickens, to use large enough enclosures to potentially see changes in behaviour related to the infection. It is more likely that such data can come from careful observational studies. This is especially true for human pathogens. Setting up such studies and analysing the data in a way that will allow one to draw quantitative conclusions is likely a formidable challenge.
To further add to the task ahead of us, we point out that our ‘general’ conceptual overview provided here is still not very general. We have focused only on the question of scaling from within-host to between-host levels from a single pathogen genotype point of view, without considering changes in genotype, i.e. we did not consider explicitly evolutionary processes. This ignores the possibility of competitive dynamics between genotypes, which can be especially important for pathogens with high mutation rates and those that lead to long-term infection. The question of genetic and antigenic diversity, evolution and its relation to transmission has been addressed theoretically [36,44], but again experimental information is sparse [113]. The multi-genotype view also encompasses competition between unrelated pathogens, an area that has been explored somewhat in models [114] but for which data will be even harder to obtain.
We have also not discussed how to include a distinct transmission stage in the process of scaling from individual infection to population-level transmission. For some pathogens, e.g. HIV, the transmission between hosts is essentially direct, and therefore, one does not need to consider a distinct transmission stage. For other pathogens, such as influenza or cholera, an environmental stage may be important [115–117]. If there is no trade-off between infection dynamics within a host and survival in the environment, the pathogen can optimize both stages [32]. However, it is quite likely that trade-offs between within-host infection dynamics and environmental stage occur at least for some pathogens or in certain situations [92], though again, there is a general lack of experimental data on that topic. One notable exception is a study on environmental survival and growth in phages, where a trade-off between the environmental persistence with replication efficiency in the bacterial host was demonstrated [118]. If such trade-offs occur, the ability to persist versus replicate inside the host affects the overall fitness [119].
In sum, we are still in the early stages of what should be an extremely interesting and fruitful endeavour, building a theoretical framework that bridges within- and between-host scales and is thoroughly grounded in data. To do so, both future model development and collection of additional data, most usefully done in an integrated fashion, will be necessary.
Acknowledgements
P.R. was supported by the RAPIDD programme of the Science and Technology Directorate, Department of Homeland Security, and by MIDAS, National Institute of General Medical Sciences U54-GM111274 and U01-GM110744. The opinions expressed herein are those of the author(s) and do not necessarily reflect the views of any of the funding agencies. Discussions and the invitation to submit this article, arose from a NESCent working group and the NSF supported Research Coordination Network on Infectious Disease Evolution across Scales.
Competing interests
We declare we have no competing interests.
Funding
We received no funding for this study.
References
- 1.Ebell MH, Call M, Shinholser JA. 2013. Effectiveness of oseltamivir in adults: a meta-analysis of published and unpublished clinical trials. Fam. Pract. 30, 125–133. ( 10.1093/fampra/cms059) [DOI] [PubMed] [Google Scholar]
- 2.Jefferson T, et al. 2014. Neuraminidase inhibitors for preventing and treating influenza in healthy adults and children. The Cochrane Database Syst. Rev. 4, CD008965 ( 10.1590/1516-3180.20141324t2) [DOI] [PubMed] [Google Scholar]
- 3.Dobson J, Whitley RJ, Pocock S, Monto AS. 2015. Oseltamivir treatment for influenza in adults: a meta-analysis of randomised controlled trials. Lancet 385, 1729–1737. ( 10.1016/S0140-6736(14)62449-1) [DOI] [PubMed] [Google Scholar]
- 4.Muthuri SG, et al. 2014. Effectiveness of neuraminidase inhibitors in reducing mortality in patients admitted to hospital with influenza A H1n1pdm09 virus infection: a meta-analysis of individual participant data. Lancet Respir. Med. 2, 395–404. ( 10.1016/S2213-2600(14)70041-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayden FG, Treanor JJ, Betts RF, Lobo M, Esinhart JD, Hussey EK. 1996. Safety and efficacy of the neuraminidase inhibitor gg167 in experimental human influenza. JAMA 275, 295–299. ( 10.1001/jama.1996.03530280047035) [DOI] [PubMed] [Google Scholar]
- 6.Longini IM, Jr, Halloran EM, Nizam A, Yang Y. 2004. Containing pandemic influenza with antiviral agents. Am. J. Epidemiol. 159, 623–633. ( 10.1093/aje/kwh092) [DOI] [PubMed] [Google Scholar]
- 7.Ferguson NM, et al. 2005. Strategies for containing an emerging influenza pandemic in southeast Asia. Nature 437, 209–214. ( 10.1038/nature04017) [DOI] [PubMed] [Google Scholar]
- 8.Handel A, Longini IM, Jr, Antia R. 2009. Intervention strategies for an influenza pandemic taking into account secondary bacterial infections. Epidemics 1, 185–195. ( 10.1016/j.epidem.2009.09.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mizumoto K, Nishiura H, Yamamoto T. 2013. Effectiveness of antiviral prophylaxis coupled with contact tracing in reducing the transmission of the influenza A (H1N1–2009): a systematic review. Theor. Biol. Med. Model. 10, 4 ( 10.1186/1742-4682-10-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Handel A, Ebell MH. 2015. Neuraminidase inhibitors for influenza: fully evaluating benefits and harms. Lancet Respir. Med. 3, e7–e8. ( 10.1016/S2213-2600(15)00066-1) [DOI] [PubMed] [Google Scholar]
- 11.Halloran ME, Hayden FG, Yang Y, Longini IM, Jr, Monto AS. 2007. Antiviral effects on influenza viral transmission and pathogenicity: observations from household-based trials. Am. J. Epidemiol. 165, 212–221. ( 10.1093/aje/kwj362) [DOI] [PubMed] [Google Scholar]
- 12.Yang Y, Halloran EM, Longini IM., Jr 2009. A Bayesian model for evaluating influenza antiviral efficacy in household studies with asymptomatic infections. Biostatistics 10, 390–403. ( 10.1093/biostatistics/kxn045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishiura H, Oshitani H. 2011. Household transmission of influenza (H1n1-2009) in Japan: age-specificity and reduction of household transmission risk by zanamivir treatment. J. Int. Med. Res. 39, 619–628. ( 10.1177/147323001103900231) [DOI] [PubMed] [Google Scholar]
- 14.Carrat F, et al. 2012. Effect of oseltamivir, zanamivir or oseltamivir-zanamivir combination treatments on transmission of influenza in households. Antiviral Ther. 17, 1085–1090. ( 10.3851/IMP2128) [DOI] [PubMed] [Google Scholar]
- 15.Pebody RG. 2011. Use of antiviral drugs to reduce household transmission of pandemic (H1N1) 2009, United Kingdom. Emerg. Infect. Dis. 17, 990–999. ( 10.3201/eid/1706.101161) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Handel A, Brown J, Stallknecht D, Rohani P. 2013. A multi-scale analysis of influenza A virus fitness trade-offs due to temperature-dependent virus persistence. PLoS Comput. Biol. 9, e1002989 ( 10.1371/journal.pcbi.1002989) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Antia R, Levin BR, May RM. 1994. Within-host population dynamics and the evolution and maintenance of microparasite virulence. Am. Nat. 144, 457–472. ( 10.1086/285686) [DOI] [Google Scholar]
- 18.Read JM, Keeling MJ. 2006. Disease evolution across a range of spatio-temporal scales. Theor. Popul. Biol. 70, 201–213. ( 10.1016/j.tpb.2006.04.006) [DOI] [PubMed] [Google Scholar]
- 19.Longini IM, Jr, et al. 2005. Containing pandemic influenza at the source. Science 309, 1083–1087. ( 10.1126/science.1115717) [DOI] [PubMed] [Google Scholar]
- 20.Cen X, Feng Z, Zhao Y. 2014. Emerging disease dynamics in a model coupling within-host and between-host systems. J. Theor. Biol. 361, 141–151. ( 10.1016/j.jtbi.2014.07.030) [DOI] [PubMed] [Google Scholar]
- 21.Lukens S, et al. 2014. A large-scale immuno-epidemiological simulation of influenza A epidemics. BMC Public Health 14, 1019 ( 10.1186/1471-2458-14-1019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mideo N, Alizon S, Day T. 2008. Linking within- and between-host dynamics in the evolutionary epidemiology of infectious diseases. Trends Ecol. Evol. 23, 511–517. ( 10.1016/j.tree.2008.05.009) [DOI] [PubMed] [Google Scholar]
- 23.Alizon S, Luciani F, Regoes RR. 2011. Epidemiological and clinical consequences of within-host evolution. Trends Microbiol. 19, 24–32. ( 10.1016/j.tim.2010.09.005) [DOI] [PubMed] [Google Scholar]
- 24.Day T, Alizon S, Mideo N. 2011. Bridging scales in the evolution of infectious disease life histories: theory. Evolution 65, 3448–3461. ( 10.1111/j.1558-5646.2011.01394.x) [DOI] [PubMed] [Google Scholar]
- 25.Mideo N, Nelson WA, Reece SE, Bell AS, Read AF, Day T. 2011. Bridging scales in the evolution of infectious disease life histories: application. Evolution 65, 3298–3310. ( 10.1111/j.1558-5646.2011.01382.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murillo LN, Murillo MS, Perelson AS. 2013. Towards multiscale modeling of influenza infection. J. Theor. Biol. 332, 267–290. ( 10.1016/j.jtbi.2013.03.024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen SC, Chio CP, Jou LJ, Liao CM. 2009. Viral kinetics and exhaled droplet size affect indoor transmission dynamics of influenza infection. Indoor Air 19, 401–413. ( 10.1111/j.1600-0668.2009.00603.x) [DOI] [PubMed] [Google Scholar]
- 28.Halloran SK, Wexler AS, Ristenpart WD. 2012. A comprehensive breath plume model for disease transmission via expiratory aerosols. PLoS ONE 7, e37088 ( 10.1371/journal.pone.0037088) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liao C-M, Yang S-C, Chio C-P, Chen S-C. 2010. Understanding influenza virus-specific epidemiological properties by analysis of experimental human infections. Epidemiol. Infect. 138, 825–835. ( 10.1017/S0950268809991178) [DOI] [PubMed] [Google Scholar]
- 30.Canini L, Carrat F. 2011. Population modeling of influenza A/H1N1 virus kinetics and symptom dynamics. J. Virol. 85, 2764–2770. ( 10.1128/JVI.01318-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Handel A, Akin V, Pilyugin SS, Zarnitsyna V, Antia R. 2014. How sticky should a virus be? The impact of virus binding and release on transmission fitness using influenza as an example. J. R. Soc. Interface 11, 20131083 ( 10.1098/rsif.2013.1083) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Handel A, Lebarbenchon C, Stallknecht D, Rohani P. 2014. Trade-offs between and within scales: environmental persistence and within-host fitness of avian influenza viruses. Proc. R. Soc. B 281, 20133051 ( 10.1098/rspb.2013.3051) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gilchrist MA, Coombs D. 2006. Evolution of virulence: interdependence, constraints, and selection using nested models. Theor. Popul. Biol. 69, 145–153. ( 10.1016/j.tpb.2005.07.002) [DOI] [PubMed] [Google Scholar]
- 34.Coombs D, Gilchrist MA, Ball CL. 2007. Evaluating the importance of within- and between-host selection pressures on the evolution of chronic pathogens. Theor. Popul. Biol. 72, 576–591. ( 10.1016/j.tpb.2007.08.005) [DOI] [PubMed] [Google Scholar]
- 35.Saenz RA, Bonhoeffer S. 2013. Nested model reveals potential amplification of an HIV epidemic due to drug resistance. Epidemics 5, 34–43. ( 10.1016/j.epidem.2012.11.002) [DOI] [PubMed] [Google Scholar]
- 36.Luciani F, Alizon S. 2009. The evolutionary dynamics of a rapidly mutating virus within and between hosts: the case of hepatitis C virus. PLoS Comput. Biol. 5, e1000565 ( 10.1371/journal.pcbi.1000565) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andr J-B, Ferdy J-B, Godelle B. 2003. Within-host parasite dynamics, emerging trade-off, and evolution of virulence with immune system. Evolution 57, 1489–1497. ( 10.1111/j.0014-3820.2003.tb00357.x) [DOI] [PubMed] [Google Scholar]
- 38.Alizon S, van Baalen M. 2005. Emergence of a convex trade-off between transmission and virulence. Am. Nat. 165, E155–E167. ( 10.1086/430053) [DOI] [PubMed] [Google Scholar]
- 39.Pepin KM, Volkov I, Banavar JR, Wilke CO, Grenfell BT. 2010. Phenotypic differences in viral immune escape explained by linking within-host dynamics to host-population immunity. J. Theor. Biol. 265, 501–510. ( 10.1016/j.jtbi.2010.05.036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steinmeyer SH, Wilke CO, Pepin KM. 2010. Methods of modelling viral disease dynamics across the within- and between-host scales: the impact of virus dose on host population immunity. Phil. Trans. R. Soc. B 365, 1931–1941. ( 10.1098/rstb.2010.0065) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fraser C, Lythgoe K, Leventhal GE, Shirreff G, Hollingsworth TD, Alizon S, Bonhoeffer S. 2014. Virulence and pathogenesis of hiv-1 infection: an evolutionary perspective. Science 343, 1243727 ( 10.1126/science.1243727) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quinn TC, et al. 2000. Viral load and heterosexual transmission of human immunodeficiency virus type 1. Rakai project study group. N. Engl. J. Med. 342, 921–929. ( 10.1056/NEJM200003303421303) [DOI] [PubMed] [Google Scholar]
- 43.Fraser C, Hollingsworth TD, Chapman R, Wolf F, Hanage WP. 2007. Variation in HIV-1 set-point viral load: epidemiological analysis and an evolutionary hypothesis. Proc. Natl Acad. Sci. USA 104, 17 441–17 446. ( 10.1073/pnas.0708559104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lange A, Ferguson NM. 2009. Antigenic diversity, transmission mechanisms, and the evolution of pathogens. PLoS Comput. Biol. 5, e1000536 ( 10.1371/journal.pcbi.1000536) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mellors JW, Rinaldo CR, Jr, Gupta P, White RM, Todd JA, Kingsley LA. 1996. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science 272, 1167–1170. ( 10.1126/science.272.5265.1167) [DOI] [PubMed] [Google Scholar]
- 46.Nguyen NM, et al. 2013. Host and viral features of human dengue cases shape the population of infected and infectious Aedes aegypti mosquitoes. Proc. Natl Acad. Sci. USA 110, 9072–9077. ( 10.1073/pnas.1303395110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prince GA, Porter DD, Jenson AB, Horswood RL, Chanock RM, Ginsberg HS. 1993. Pathogenesis of adenovirus type 5 pneumonia in cotton rats (Sigmodon hispidus). J. Virol. 67, 101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ottolini MG, Porter DD, Hemming VG, Hensen SA, Sami IR, Prince GA. 1996. Semi-permissive replication and functional aspects of the immune response in a cotton rat model of human parainfluenza virus type 3 infection. J. Gen. Virol. 77, 1739–1743. ( 10.1099/0022-1317-77-8-1739) [DOI] [PubMed] [Google Scholar]
- 49.Ginsberg HS, Horsfall FL. 1952. Quantitative aspects of the multiplication of influenza A virus in the mouse lung: relation between the degree of viral multiplication and the extent of pneumonia. J. Exp. Med. 95, 135–145. ( 10.1084/jem.95.2.135) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Callison SA, Hilt DA, Boynton TO, Sample BF, Robison R, Swayne DE, Jackwood MW. 2006. Development and evaluation of a real-time Taqman RT-PCR assay for the detection of infectious bronchitis virus from infected chickens. J. Virol. Methods 138, 60–65. ( 10.1016/j.jviromet.2006.07.018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu G, Kahan SM, Jia Y, Karst SM. 2009. Primary high-dose murine norovirus 1 infection fails to protect from secondary challenge with homologous virus. J. Virol. 83, 6963–6968. ( 10.1128/JVI.00284-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Y, Handel A. 2014. Modeling inoculum dose dependent patterns of acute virus infections. J. Theor. Biol. 347, 63–73. ( 10.1016/j.jtbi.2014.01.008) [DOI] [PubMed] [Google Scholar]
- 53.Jeffery GM, Eyles DE. 1955. Infectivity to mosquitoes of Plasmodium falciparum as related to gametocyte density and duration of infection. Am. J. Trop. Med. Hyg. 4, 781–789. [DOI] [PubMed] [Google Scholar]
- 54.Eckhoff P. 2012. P. falciparum infection durations and infectiousness are shaped by antigenic variation and innate and adaptive host immunity in a mathematical model. PLoS ONE 7, e44950 ( 10.1371/journal.pone.0044950) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pesko K, Westbrook CJ, Mores CN, Lounibos LP, Reiskind MH. 2009. Effects of infectious virus dose and bloodmeal delivery method on susceptibility of Aedes aegypti and Aedes albopictus to chikungunya virus. J. Med. Entomol. 46, 395–399. ( 10.1603/033.046.0228) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seblova V, et al. 2013. Phlebotomus orientalis sand flies from two geographically distant Ethiopian localities: biology, genetic analyses and susceptibility to Leishmania donovani. PLoS Negl. Trop. Dis. 7, e2187 ( 10.1371/journal.pntd.0002187) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miller E, Warburg A, Novikov I, Hailu A, Volf P, Seblova V, Huppert A. 2014. Quantifying the contribution of hosts with different parasite concentrations to the transmission of visceral leishmaniasis in Ethiopia. PLoS Negl. Trop. Dis. 8, e3288 ( 10.1371/journal.pntd.0003288) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wen W-H, et al. 2013. Mother-to-infant transmission of hepatitis B virus infection: significance of maternal viral load and strategies for intervention. J. Hepatol. 59, 24–30. ( 10.1016/j.jhep.2013.02.015) [DOI] [PubMed] [Google Scholar]
- 59.Kaplan JE, et al. 1996. Male-to-female transmission of human T-cell lymphotropic virus types I and II: association with viral load. The retrovirus epidemiology donor study group. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 12, 193–201. ( 10.1097/00042560-199606010-00014) [DOI] [PubMed] [Google Scholar]
- 60.Li H-C, Biggar RJ, Miley WJ, Maloney EM, Cranston B, Hanchard B, Hisada M. 2004. Provirus load in breast milk and risk of mother-to-child transmission of human T lymphotropic virus type I. J. Infect. Dis. 190, 1275–1278. ( 10.1086/423941) [DOI] [PubMed] [Google Scholar]
- 61.Lawley TD, Bouley DM, Hoy YE, Gerke C, Relman DA, Monack DM. 2008. Host transmission of Salmonella enterica serovar typhimurium is controlled by virulence factors and indigenous intestinal microbiota. Infect. Immun. 76, 403–416. ( 10.1128/IAI.01189-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lawley TD, et al. 2009. Antibiotic treatment of Clostridium difficile carrier mice triggers a supershedder state, spore-mediated transmission, and severe disease in immunocompromised hosts. Infect. Immun. 77, 3661–3669. ( 10.1128/IAI.00558-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Matthews L, et al. 2006. Heterogeneous shedding of Escherichia coli O157 in cattle and its implications for control. Proc. Natl Acad. Sci. USA 103, 547–552. ( 10.1073/pnas.0503776103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gray RH, et al. 2001. Probability of HIV-1 transmission per coital act in monogamous, heterosexual, HIV-1-discordant couples in Rakai, Uganda. Lancet 357, 1149–1153. ( 10.1016/S0140-6736(00)04331-2) [DOI] [PubMed] [Google Scholar]
- 65.Hollingsworth TD, Anderson RM, Fraser C. 2008. HIV-1 transmission, by stage of infection. J. Infect. Dis. 198, 687–693. ( 10.1086/590501) [DOI] [PubMed] [Google Scholar]
- 66.Fraser C, Riley S, Anderson RM, Ferguson NM. 2004. Factors that make an infectious disease outbreak controllable. Proc. Natl Acad. Sci. USA 101, 6146–6151. ( 10.1073/pnas.0307506101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Feldmann H, Geisbert TW. 2011. Ebola haemorrhagic fever. Lancet 377, 849–862. ( 10.1016/S0140-6736(10)60667-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marks PJ, Vipond IB, Regan FM, Wedgwood K, Fey RE, Caul EO. 2003. A school outbreak of Norwalk-like virus: evidence for airborne transmission. Epidemiol. Infect. 131, 727–736. ( 10.1017/S0950268803008689) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.O'Neill PD, Marks PJ. 2005. Bayesian model choice and infection route modelling in an outbreak of norovirus. Stat. Med. 24, 2011–2024. ( 10.1002/sim.2090) [DOI] [PubMed] [Google Scholar]
- 70.Handel A, Longini IM, Antia R. 2007. Neuraminidase inhibitor resistance in influenza: assessing the danger of its generation and spread. PLoS Comput. Biol. 3, e240 ( 10.1371/journal.pcbi.0030240) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hayden FG, Fritz R, Lobo MC, Alvord W, Strober W, Straus SE. 1998. Local and systemic cytokine responses during experimental human influenza A virus infection. Relation to symptom formation and host defense. J. Clin. Invest. 101, 643–649. ( 10.1172/JCI1355) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fritz RS, Hayden FG, Calfee DP, Cass LM, Peng AW, Alvord WG, Strober W, Straus SE. 1999. Nasal cytokine and chemokine responses in experimental influenza A virus infection: results of a placebo-controlled trial of intravenous zanamivir treatment. J. Infect. Dis. 180, 586–593. ( 10.1086/314938) [DOI] [PubMed] [Google Scholar]
- 73.Carrat F, Vergu E, Ferguson NM, Lemaitre M, Cauchemez S, Leach S, Valleron A-J. 2008. Time lines of infection and disease in human influenza: a review of volunteer challenge studies. Am. J. Epidemiol. 167, 775–785. ( 10.1093/aje/kwm375) [DOI] [PubMed] [Google Scholar]
- 74.Lau LLH, et al. 2010. Viral shedding and clinical illness in naturally acquired influenza virus infections. J. Infect. Dis. 201, 1509–1516. ( 10.1086/652241) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bischoff WE, Swett K, Leng I, Peters TR. 2013. Exposure to influenza virus aerosols during routine patient care. J. Infect. Dis. 207, 1037–1046. ( 10.1093/infdis/jis773) [DOI] [PubMed] [Google Scholar]
- 76.Tumpey TM, et al. 2007. A two-amino acid change in the hemagglutinin of the 1918 influenza virus abolishes transmission. Science 315, 655–659. ( 10.1126/science.1136212) [DOI] [PubMed] [Google Scholar]
- 77.Zaraket H, et al. 2015. Mammalian adaptation of influenza A(H7N9) virus is limited by a narrow genetic bottleneck. Nat. Commun. 6, 6553 ( 10.1038/ncomms7553) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Blumenkrantz D, Roberts KL, Shelton H, Lycett S, Barclay WS. 2013. The short stalk length of highly pathogenic avian influenza H5N1 virus neuraminidase limits transmission of pandemic H1N1 virus in ferrets. J. Virol. 87, 10 539–10 551. ( 10.1128/JVI.00967-13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Roberts KL, Shelton H, Scull M, Pickles R, Barclay WS. 2011. Lack of transmission of a human influenza virus with avian receptor specificity between ferrets is not due to decreased virus shedding but rather a lower infectivity in vivo. J. Gen. Virol. 92, 1822–1831. ( 10.1099/vir.0.031203-0) [DOI] [PubMed] [Google Scholar]
- 80.Roberts KL, Shelton H, Stilwell P, Barclay WS. 2012. Transmission of a 2009 H1N1 pandemic influenza virus occurs before fever is detected, in the ferret model. PLoS ONE 7, e43303 ( 10.1371/journal.pone.0043303) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cowling BJ, et al. 2013. Aerosol transmission is an important mode of influenza A virus spread. Nat. Commun. 4, 1935 ( 10.1038/ncomms2922) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tellier R. 2009. Aerosol transmission of influenza A virus: a review of new studies. J. R. Soc. Interface 6(Suppl. 6), S783–S790. ( 10.1098/rsif.2009.0302.focus) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Saenz RA, et al. 2010. Dynamics of influenza virus infection and pathology. J. Virol. 84, 3974–3983. ( 10.1128/JVI.02078-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Reperant LA, Kuiken T, Grenfell BT, Osterhaus ADME, Dobson AP. 2012. Linking influenza virus tissue tropism to population-level reproductive fitness. PLoS ONE 7, e43115 ( 10.1371/journal.pone.0043115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Charleston B, et al. 2011. Relationship between clinical signs and transmission of an infectious disease and the implications for control. Science 332, 726–729. ( 10.1126/science.1199884) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chase-Topping ME, et al. 2013. Understanding foot-and-mouth disease virus transmission biology: identification of the indicators of infectiousness. Vet. Res. 44, 46 ( 10.1186/1297-9716-44-46) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lipsitch M, Moxon ER. 1997. Virulence and transmissibility of pathogens: what is the relationship? Trends Microbiol. 5, 31–37. ( 10.1016/S0966-842X(97)81772-6) [DOI] [PubMed] [Google Scholar]
- 88.Alizon S, Hurford A, Mideo N, Van Baalen M. 2009. Virulence evolution and the trade-off hypothesis: history, current state of affairs and the future. J. Evol. Biol. 22, 245–259. ( 10.1111/j.1420-9101.2008.01658.x) [DOI] [PubMed] [Google Scholar]
- 89.Ewald PW. 1987. Transmission modes and evolution of the parasitism-mutualism continuum. Ann. N.Y. Acad. Sci. 503, 295–306. ( 10.1111/j.1749-6632.1987.tb40616.x) [DOI] [PubMed] [Google Scholar]
- 90.Ewald PW. 1991. Waterborne transmission and the evolution of virulence among gastrointestinal bacteria. Epidemiol. Infect. 106, 83–119. ( 10.1017/S0950268800056478) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ewald PW. 1995. The evolution of virulence: a unifying link between parasitology and ecology. J. Parasitol. 81, 659–669. ( 10.2307/3283951) [DOI] [PubMed] [Google Scholar]
- 92.Walther BA, Ewald PW. 2004. Pathogen survival in the external environment and the evolution of virulence. Biol. Rev. Camb. Philos. Soc. 79, 849–869. ( 10.1017/S1464793104006475) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Afonso C, Paixão VB, Costa RM. 2012. Chronic toxoplasma infection modifies the structure and the risk of host behavior. PLoS ONE 7, e32489 ( 10.1371/journal.pone.0032489) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dobson AP. 1988. The population biology of parasite-induced changes in host behavior. Q. Rev. Biol. 63, 139–165. ( 10.1086/415837) [DOI] [PubMed] [Google Scholar]
- 95.Lloyd-Smith JO, Getz WM, Westerhoff HV. 2004. Frequency-dependent incidence in models of sexually transmitted diseases: portrayal of pair-based transmission and effects of illness on contact behaviour. Proc. R. Soc. Lond. B 271, 625–634. ( 10.1098/rspb.2003.2632) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Eames KTD, Tilston NL, White PJ, Adams E, Edmunds WJ. 2010. The impact of illness and the impact of school closure on social contact patterns. Health Technol. Assess. 14, 267–312. [DOI] [PubMed] [Google Scholar]
- 97.Kerckhove KV, Hens N, Edmunds WJ, Eames KTD. 2013. The impact of illness on social networks: implications for transmission and control of influenza. Am. J. Epidemiol. 178, 1655–1662. ( 10.1093/aje/kwt196) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kelvin KW, et al. 2010. Delayed clearance of viral load and marked cytokine activation in severe cases of pandemic H1N1 2009 influenza virus infection. Clin. Infect. Dis. 50, 850–859. ( 10.1086/650581) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fielding JE, Kelly HA, Mercer GN, Glass K. 2014. Systematic review of influenza A(H1N1)pdm09 virus shedding: duration is affected by severity, but not age. Influenza Other Respir. Viruses 8, 142–150. ( 10.1111/irv.12216) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Funk S, Salathé M, Jansen VAA. 2010. Modelling the influence of human behaviour on the spread of infectious diseases: a review. J. R. Soc. Interface 7, 1247–1256. ( 10.1098/rsif.2010.0142) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Manfredi P, D'Onofrio A. 2013. Modeling the interplay between human behavior and the spread of infectious diseases. Berlin, Germany: Springer. [Google Scholar]
- 102.Funk S, Bansal S, Bauch CT, Eames KTD, Edmunds JW, Galvani AP, Klepac P. 2014. Nine challenges in incorporating the dynamics of behaviour in infectious diseases models. Epidemics 10, 21–25. ( 10.1016/j.epidem.2014.09.005) [DOI] [PubMed] [Google Scholar]
- 103.Woolhouse ME, et al. 1997. Heterogeneities in the transmission of infectious agents: implications for the design of control programs. Proc. Natl Acad. Sci. USA 94, 338–342. ( 10.1073/pnas.94.1.338) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lloyd-Smith JO, Schreiber SJ, Kopp PE, Getz WM. 2005. Superspreading and the effect of individual variation on disease emergence. Nature 438, 355–359. ( 10.1038/nature04153) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Paull SH. 2012. From superspreaders to disease hotspots: linking transmission across hosts and space. Front. Ecol. Environ. 10, 75–82. ( 10.1890/110111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lau LLH, et al. 2013. Heterogeneity in viral shedding among individuals with medically attended influenza A virus infection. J. Infect. Dis. 207, 1281–1285. ( 10.1093/infdis/jit034) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Killingley B, et al. 2012. Use of a human influenza challenge model to assess person-to-person transmission: proof-of-concept study. J. Infect. Dis. 205, 35–43. ( 10.1093/infdis/jir701) [DOI] [PubMed] [Google Scholar]
- 108.Belser JA, Katz JM, Tumpey TM. 2011. The ferret as a model organism to study influenza A virus infection. Dis. Model. Mech. 4, 575–579. ( 10.1242/dmm.007823) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Spekreijse D, Bouma A, Stegeman JA, Koch G, de Jong MCM. 2011. The effect of inoculation dose of a highly pathogenic avian influenza virus strain H5N1 on the infectiousness of chickens. Vet. Microbiol. 147, 59–66 ( 10.1016/j.vetmic.2010.06.012) [DOI] [PubMed] [Google Scholar]
- 110.Velthuis AGJ, Bouma A, Katsma WEA, Nodelijk G, De Jong MCM. 2007. Design and analysis of small-scale transmission experiments with animals. Epidemiol. Infect. 135, 202–217. ( 10.1017/S095026880600673X) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Velthuis AGJ, De Jong MCM, De Bree J. 2007. Comparing methods to quantify experimental transmission of infectious agents. Math. Biosci. 210, 157–176. ( 10.1016/j.mbs.2007.04.009) [DOI] [PubMed] [Google Scholar]
- 112.Nishiura H, Yen H-L, Cowling BJ. 2013. Sample size considerations for one-to-one animal transmission studies of the influenza A viruses. PLoS ONE 8, e55358 ( 10.1371/journal.pone.0055358) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mideo N, Day T. 2008. On the evolution of reproductive restraint in malaria. Proc. R. Soc. B 275, 1217–1224. ( 10.1098/rspb.2007.1545) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shrestha S, Foxman B, Dawid S, Aiello AE, Davis BM, Berus J, Rohani P. 2013. Time and dose-dependent risk of pneumococcal pneumonia following influenza: a model for within-host interaction between influenza and Streptococcus pneumoniae. J. R. Soc. Interface 10, 20130233 ( 10.1098/rsif.2013.0233) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.King AA, Ionides EL, Pascual M, Bouma MJ. 2008. Inapparent infections and cholera dynamics. Nature 454, 877–880. ( 10.1038/nature07084) [DOI] [PubMed] [Google Scholar]
- 116.Rohani P, Breban R, Stallknecht DE, Drake JM. 2009. Environmental transmission of low pathogenicity avian influenza viruses and its implications for pathogen invasion. Proc. Natl Acad. Sci. USA 106, 10 365–10 369. ( 10.1073/pnas.0809026106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Roche B, Drake JM, Brown J, Stallknecht DE, Bedford T, Rohani P. 2014. Adaptive evolution and environmental durability jointly structure phylodynamic patterns in avian influenza viruses. PLoS Biol. 12, e1001931 ( 10.1371/journal.pbio.1001931) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Paepe MD, Taddei F. 2006. Viruses’ life history: towards a mechanistic basis of a trade-off between survival and reproduction among phages. PLoS Biol. 4, e193 ( 10.1371/journal.pbio.0040193) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Handel A, Bennett MR. 2008. Surviving the bottleneck: transmission mutants and the evolution of microbial populations. Genetics 180, 2193–2200. ( 10.1534/genetics.108.093013) [DOI] [PMC free article] [PubMed] [Google Scholar]