

Synthetic hydrogels have been increasingly engineered to provide and recapitulate the complex and dynamic environment of extra-cellular matrix (ECM) surrounding cells in vivo. Of significant interest is the design of chemical strategies not only to fabricate hydrogels that can function as cell culture platforms for both two-dimensional (2d) and three-dimensional (3d) cell culture systems, but also to incorporate biologically active features that mimic several critical aspects of the ECM.[1]–[4] One class of synthetic hydrogels, based on poly(ethylene glycol) (PEG), has emerged as a diverse synthetic ECM mimic,[5] as PEGs are hydrophilic in nature, commercially available in a range of molecular weights and structures, and possess materials properties similar to many soft tissues. Further, to complement their biophysical properties, the relative bio-inertness of PEG hydrogels renders opportunities to incorporate various biochemical cues and study their effects on host cells, while minimizing confounding non-specific interactions from serum proteins. Hence, there is a growing interest in strategies to functionalize PEG hydrogels with bioactive moieties, including small molecules, peptides, and proteins, aimed at directing and manipulating cellular functions ranging from adhesion, proliferation, differentiation, motility, etc.
But beyond the synthesis of biofunctional materials, recent efforts are focused on recapitulating the dynamic nature of ECM, which often requires multidimensional control over the presentation of the biochemical signals. For example, studies have demonstrated incorporation of peptides motifs that facilitate cell adhesion[6]–[9] (e.g., fibronectin derived RGD, PHSRN), allow cell directed degradability[5][10]–[12] (e.g., Matrix metalloproteanase (MMP) degradable GPQGI↓WGQ), and growth factor sequestering[13][14] (e.g., bFGF binding KRTGQYKL) in PEG hydrogels, but these strategies leave the experimenter as an observer. Thus, chemical strategies that allow external manipulation of the ECM environment in real time could provide insight as to how cells process queues from their surrounding environment, and this motivation had led to the development of new material approaches that allow sequential or reversible introduction of biological signals. Such strategies could offer new opportunities to tailor a cell’s local microenvironment in a biologically relevant fashion. In particular, bio-orthogonal click reactions have gained prominence owing to their efficiency, fidelity, facile incorporation of functional groups into widely used synthetic biomaterials, along with their relatively mild reaction conditions.[15]–[17] Further, photochemically controlled click reactions[18]–[20] allow confinement of the reaction of interest only to defined locations where light is delivered. Photolithographic techniques use photomasks and collimated light to confine and pattern reactions in two-dimensions, while focused light (single or multi- photon) enables control of the reaction within a locally defined ‘region of interest’ (ROI) in three-dimensions. While these chemical approaches have been successfully applied to cell-laden scaffolds to demonstrate localized control of cell spreading,[21] cell outgrowth,[22] control of stem cells fate,[23] and guided migration,[24],[25] complementary strategies that simultaneously achieve the addition and removal of biochemical signals would be beneficial. Since the majority of current strategies to functionalize bioscaffolds rely on irreversible, and thus ‘dead’, reactions,[21][23][26]–[30] the modified sites are consumed permanently and constrain stategies for sequential or multiple modifications. Hence, advances in cytocompatible chemical strategies that allow regeneration of reactive functional groups (i.e., reversible) afford powerful complementary tools in material based strategies to study cellular processes.
In this regard, a recent strategy exploiting the equilibrium switching of unreactive 3-(hydroxymethyl)-2-napthol and reactive o-2-napthoquinone-3-methides (NQM) was used to demonstrate attachment, removal and re-attachment of streptavidin to a thiol-derivitized glass slide.[31] This approach used a base catalyzed Michael addition for conjugation and 350 nm UV light to cleave the NQM functionalized chemical moieties to thiol-functionalized glass. Alternatively, Addition-Fragmentation-Chain transfer (AFCT) functional groups have been used as a unique paradigm to synthesize polymer network with excellent stress relaxation characteristics, as well as to create solution free physical patterns within the polymer substrates.[32]–[34] Key to achieving both of the functionalities in the polymer network is an AFCT capable allyl sulfide functional group.
Inspired from concepts and strategies outlined above, we hypothesized that allyl sulfide functional groups would be excellent candidates to precisely and simultaneously control the introduction, exchange, and/or removal of biochemical epitopes in hydrogel networks, while simultaneously regenerating the reactive functionality. This idea was to create a pseudo-living biomaterial system for the introduction of biological epitopes using cytocompatible thiol-ene click reactions. Thiol-ene click reactions are increasingly used for biological applications due to their robustness, simplicity, and bio-orthogonality to form hydrogel culture platforms,[35]–[37] as well as for patterning in biochemical cues in the presence of cells.[21][35][38] Towards achieving reversible photopatterning of biochemical ligands, here we report the synthesis of allyl sulfide functionalized hydrogel networks and characterize the reversible biochemical patterning using a model bioactive peptide motif, RGD, to control cell attachment and morphology.
The reversible exchange strategy reported in this study was inspired from the regenerative nature of the chain transfer agent used in RAFT based polymerizations.[39] Key to this reversible exchange of biochemical epitopes is addition fragmentation chain transfer (AFCT) capable allyl sulfide functional groups that enable reversible addition and removal of thiol-containing compounds. Scheme 1 shows the reported mechanism demonstrating the β-scission of the allyl sulfide. Upon attack of a thiyl radical on the double bond of allyl sulfide (Scheme 1a), the reaction results in a symmetric intermediate (Scheme 1b). This intermediate is unstable and undergoes β-scission resulting in addition of an attacking species and regeneration of a new double bond (Scheme 1c). We refer to this aspect of regeneration of the double bond as ‘living’ in this system. The regenerated double bond is susceptible to further attack by another thiyl radical, thus allowing exchange of any thiol containing biochemical moieties of interest.
Scheme 1.
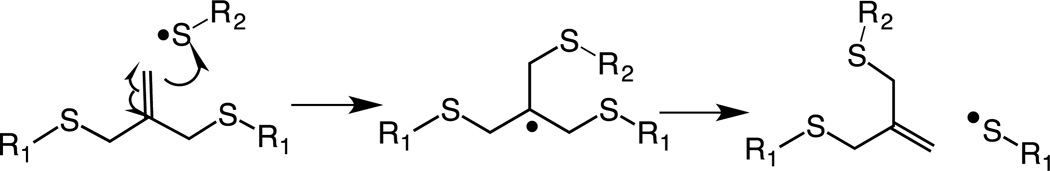
Mechanism of addition fragmentation chain transfer of an allyl sulfide functional group upon attack by a thiyl radical.
Hydrogels containing allyl sulfide functional groups were synthesized by a Cu-catalyzed click reaction between 4-arm PEG tetra azide and di-functional alkyne crosslinkers that proceeds quantitatively via a rapid, non-radical mechanism to form step growth networks.[38][40] Structures of the macromers used to form the hydrogel networks are shown in Figure 1A. Here, orthogonal Cu click reactions were chosen to synthesize the hydrogel networks to preserve the integrity of the double bond on the allyl sulfide functional group (i.e., the double bond is dormant during the formation of the hydrogel). Attack of a thiyl radical on the allyl sulfide functional group results in scission of cross-links and subsequent network degradation. As a result, the monomer formulations were adjusted to form step-growth networks containing 22% of the allyl sulfide crosslinker 2 (corresponding to 6.4 mM of allyl sulfide functional group in the formed hydrogel) and 78% of 3. Flory-Stockmayer theory[41] predicts that for the chosen formulation conditions, ~43% of crosslinks must be broken to reach the reverse gelation point, i.e. for transition from solid to liquid. Therefore, the chosen formulation does not undergo bulk degradation upon patterning, as a maximum of 22% of the crosslinks are cleaved if all the allyl sulfide functional groups are reacted.
Figure 1.

A) Structures of monomers used to form the hydrogel networks. 1: 4-arm-poly(ethylene glycol)-tetra azide. 2: 2-methylene-propane-1,3-bis(thioethyl 4-pentynoate). 3: Benzoic acid-3,5-bis(4-pentynoate). B) Mechanism of patterning fluorescently tagged CRGDS to the allyl sulphide functionalized hydrogel. C) Hollow cage pattern formed by exposing specific regions in –x and –y directions to 720 nm 2-photon light and rastering the focal point through z-direction. Thiol-ene reaction is initiated by I2959 initiator resulting in tethering of AF555-CRGDS only in the exposed regions. D) Amount of the peptide tethered to the network can be regulated by controlling the laser power and laser scan speed. E) Corresponding fluorescent patterns formed by controlling the laser scan speed and laser power.
The patterning process was initiated by homolytic photolysis of the photoinitiator 2-hydroxy-1-[4-(2-hydroxyehtoxy) phenyl]-2-methyl 1-propanone (I2959) using a 720 nm 2-photon laser light resulting in production of a thiyl radical that attacks the double bond on allyl sulfide. The cell adhesive RGDS peptide was used as model biochemical ligand to demonstrate photopatterning in the hydrogel networks. The RGDS peptide sequence was synthesized to contain a cysteine at the N-terminus end, to allow for photoconjugration via a thiol-ene mechanism, and 6-aminohexanionc acid to conjugate Alexa Fluor 555 (denoted as AF555AhxRGDSC) to visually confirm and study the addition of the patterning agent to the allyl sulfide functional group. Hydrogels were swollen in a solution containing photoinitiator and AF555AhxRGDSC for at least 2 hours and selected regions in -x and -y directions were exposed to 720 nm laser light. The focal point of the laser was rastered in the z-direction using a confocal microscope. The reaction mechanism of addition of AF555AhxRGDSC to allyl sulfide networks via photopatterning is shown in Figure 1B. Confinement of the reaction to the exposed regions was confirmed in three dimensions by the resultant open cage fluorescent pattern imaged in Figure 1C. Since the photo-initiated thiol-ene reaction can be regulated by the amount of light delivered to the system, we studied the biochemical functionalization of the hydrogels by varying the laser power and laser scan speed. Figure 1D,E shows the concentration of RGD patterned into the gel as a function of laser power at different laser scan speeds. The concentration of RGD patterned into the network was obtained from a calibration curve obtained by measuring the fluorescence in gels swollen in known concentrations of the patterning agent. The laser power was chosen not to exceed values that were reported to be safe in the presence of cells.[42] Results show that the amount of RGD patterned to the networks can be controlled by controlling the imaging conditions, and the concentration of RGD reacted into the networks varied from 0 to 1.2 mM, a range which has been previously shown to be biologically relevant.[9][43] Figure 1E shows the fluorescent patterns of RGD formed by varying the laser power and imaging speed demonstrating that the amount of biochemical functionalization of the hydrogels can be readily tailored using the laser properties and imaging conditions.
Next, we demonstrate that the ‘living’ alkene functionality of the allyl sulfide functional groups allows for sequential exchange of new biochemical moieties by using Alex fluor 555 and Alex fluor 488 conjugated AhxRGDSC peptides (AF555AhxRGDSC and AF488AhxRGDSCm respectively). AF488AhxRGDSC was synthesized similar to the AF555AhxRGDSC, and these peptide motifs allow fluorescence dependent monitoring of the thiol-ene exchange reaction. The mechanism and schematic process for the exchange of the patterning compounds in these hydrogel networks are shown in Figure 2A, B, respectively. Figure 2C–E demonstrates formation of a 250 µm long cube patterned using 10 mM of AF555AhxCRGDS, 4.4 mM I2959 at a laser scan speed which corresponds to ~9.41 µsec/µm2 pixel dwell time. To demonstrate the exchange reaction, we repeated the patterning process by the swelling the patterned gel with 10 mM AF488AhxRGDSC and 4.4 mM I2959. The buffalo shaped region inside the square red pattern was exposed to 720 nm focused laser light, facilitating the exchange reaction. Photo-exchange of the orginal peptide (AF555AhxRGDSC) with the new one (AF488AhxRGDSC) was confirmed by fluorescent images showing the disappearance of AF555 fluorescence and appearance of AF488 fluorescence in the exposed regions (Figure 2F–H). No change in AF555 fluorescence in the unexposed regions of the red square pattern (Figure 2F) confirmed the confinement of the photo-exchange only to the exposed regions.
Figure 2. Photoclick living strategy allows reversible exchange of biochemical ligands.

A) Mechanism of replacement of a thiol-containing compound on allyl sulphide functional group. B) Schematic of replacement of biochemical ligand. C–K) Demonstration of reversible exchange of thiol containing peptides. (C–E) 250µm square pattern of AF555AhxRGDSC. (F–H) Buffalo logo was formed by replacing AF555AhxRGDSC with AF488AhxRGDSC resulting in appearance of green fluorescence and disappearance of red fluorescence only in the exposed regions. (I–K) Demonstration of further replacement on living allyl sulfide: Letters ‘CU’ inside the buffalo logo were exposed to 720 nm light to photo-exchange with non-fluorescent AhxRGDSC peptide. Photo-exchange of peptides was confirmed by removal of green fluorescent (J) only at the exposed regions. Scale bar = 100 µm.
The living nature of the double bond allows performing further exchange reactions multiple times, theoretically infinitely, but is limited by irreversible termination of the intermediate.[44] To demonstrate this nature, we performed a second exchange step using a non-fluorescent AhxRGDSC peptide. User defined letters (CU) inside the green buffalo logo, obtained after the first exchange step, were exposed to laser light under similar conditions as the first exchange step (Figure 2I–K). Successful exchange of the patterned AF488AhxRGDSC peptide with the non-fluorescent peptide was confirmed by disappearance of green fluorescence in the exposed regions (Figure 2J). As expected, AF555 and AF488 fluorescence around the exposed regions after the second exchange step was not affected due to the exchange process (Figure I–K). Figure 2C–K clearly demonstrates the ‘living’ nature of alkene functionality on the allyl sulfide functional group that allows for the reversible exchange of the peptides. Since the exchange step allowed for complete exchange of the initially coupled moieties, at least within the detection limit of the fluorophore, exchange reactions could be extended to remove desired biochemical signals presented to the cells. Hence, the living characteristic of this hydrogel system allows, depending on the strategy, to achieve photo-coupling, photo-removal and photo-exchange of biochemical ligands with spatial and temporal control.
Since the extent of the exchange reaction depends on the production of initiator radicals responsible for initiating the thiol-ene reaction, we next characterized the kinetics of the exchange process by regulating the laser power and the pixel dwell time of laser light and monitoring the quantity of initially patterned peptide that remains attached after the exchange process. Figure 3A shows the amount of AF555AhxRGDSC peptide remaining after the exchange reaction in the presence of 10 mM AF488AhxRGDSC peptide and 8.8 mM I2959 with increasing laser intensity at different laser scan speeds of the laser. At a given pixel dwell time, an increase in laser power results in higher dosage per pixel, and hence, higher concentration of radicals produced resulting in the lower concentration of AF555RGDSC peptide as shown in Figure 3A. Increasing the pixel dwell time at any delivered laser intensity also results in a higher dosage to the reaction volume, and hence, lower concentration of AF555RGDSC peptide as shown in Figure 3A. Dependence of the amount of the peptide exchanged on light intensity was found to be linear in laser power at all the pixel dwell times studied. Figure 3B shows the dependence of addition of the AF488AhxRGDSC peptide to the network after the exchange reaction on laser power at different pixel dwell time. As expected, at a given pixel dwell time, the amount of peptide attached to the network increased with increasing the laser power, scaling linearly as was observed with Figure 3A. Also, at a given laser intensity, increasing the pixel dwell time increases the amount of peptide attached to the network at all laser power studied consistent with the results of Figure 3A. Collectively, our results demonstrate a high degree of control over the amount of the peptide exchanged by regulating the light delivered to pre-defined reaction volumes.
Figure 3. Kinetics of exchange of biochemical ligands in AFCT capable hydrogels.

A) Amount of AF555AhxRGDSC remained attached to the network after the photo-exchange reaction as a function of dosage at different laser scanning speeds. B) Amount of AF488AhxRGDSC photo-coupled to the network due to the exchange as a function of dosage at different pixel dwel times. C–E) Simultaneous generation of opposing gradient patterns of two different biomacromolecules. (C) AF555, (D) AF488RGDS and (E) combined. Gradient pattern is formed by exposing a uniform 250 µm square pattern to radially increasing laser power at pixel dwell time 36.67 µ/sec2
Furthermore, the ‘living’ aspect of this strategy offers unique opportunities to construct opposing patterns of two different biomacromolecules. As an example, we demonstrate synthesis of rectangular, opposing gradients in signals using the two AF555 and AF488 conjugated RGDSC peptides described above. Figure 3C–E shows the resultant rectangular radial gradient formed from exchanging reactions between AF555RGDSC and AF488RGDSC at a laser scan speed of 2. Specific polygon regions on a 250 µm long cube pattern were exposed to radially decreasing (measured from center to outside) laser power resulting in opposing patterns as shown in Figure 3C,D. The concentrations of the resultant AF555RGDSC and AF488RGDSC peptides are shown in Figure 3A,B respectively. Images clearly demonstrate the flexibility in constructing opposing gradient patterns by controlling the parameters of confocal microscopy. The patterning process occurs at typically used imaging speeds, and the whole patterning and exchange reactions occur on the order of seconds to minutes. Also, since the patterning strategy used in this work for the exchange reactions constructed ROIs using Zeiss software to form the patterns, advanced patterning procedures reported by Culver et al.[45] can be directly implemented to construct numerous relevant biomimetic patterns.
Finally, this chemistry allows manipulation of the local environment of cells, as it uses reaction conditions, wavelengths, and imaging conditions that have all been shown to be cytocompatible.[26][46] Towards the ability to manipulate the local cellular microenvironment, we demonstrate that the exchange reactions can be conducted in the presence of primary human mesenchymal stem cells in situ. Figure 4A,B shows that hMSCs (red, Figure 4B) attach on a rectangular pattern of RGD (green, Figure 4A). After one day of culture on these shapes, the RGD peptide was exchanged with a biologically inactive RDG peptide (non-fluorescent) at specific regions. Exposing specific regions of the pattern under conditions that allow exchange of the RGD (fluorescent) with RDG (non-fluorescent) peptide should result in disappearance of fluorescence only in the exposed regions. Figure 4C shows the RGD pattern and hMSC morpohology after exchanging with RDG. Figure 4C demonstrates that the exchange process does not adversely affect the local cells, as evidenced by no change in cell staining. Also, since the process uses 2-photon patterning via a confocal microscope, the shape of the pattern can be easily modified accordingly.
Figure 4. ‘Living’ strategy allows cytocompatible manipulation of biochemical environment of the gel.

A) 0.5 mM of rectangular RGD pattern was formed and seeded with hMSCs. B) hMSC stained with cell tracker red attached to the rectangular pattern. C) A smaller region was exposed to 720 nm light to allow for exchange of fluorescent-RGD with non-fluorescent RDG peptide. The photopatterning did not affect the cellular staining. Scale bar = 25 µm.
In this study we have demonstrate a emergent application for the RAFT agent allyl sulfide as a chemical tool that can be easily incorporated into hydrogels to provide a reactive handle for reversible addition and exchange of biochemical moieties under cytocompatible conditions. The strategy reported here provides an excellent opportunity for biomedical researchers to temporally and spatially manipulate the local microenvironment of cells, reversibly and multiple times using single and multi-photon wavelength light. We expect that such simple chemical strategies will help researchers to design experiments to understand and gather information to better understand how cells respond to signals from their surrounding environment.
Experimental
Complete experimental procedures for peptide and macromolecular precursor synthesis, hydrogel formation, biochemical patterning, cell culture and immunostaining are provided in the Supporting Information.
Supplementary Material
Acknowledgements
The authors acknowledge financial support from NSF (DMR 1006711) and the Howard Hughes Medical Institute.
Contributor Information
Navakanth R Gandavarapu, Department of Chemical and Biological Engineering and the BioFrontiers Institute, University of Colorado Boulder, Boulder, CO 80303 USA.
Malar A. Azagarsamy, Department of Chemical and Biological Engineering and the Howard Hughes Medical Institute, University of Colorado Boulder, Boulder, CO 80303 USA
Kristi S. Anseth, Email: Kristi.Anseth@colorado.edu, Department of Chemical and Biological Engineering, the BioFrontiers Institute, and the Howard Hughes Medical Institute, University of Colorado Boulder, Boulder, CO 80303 USA.
References
- 1.Zhu J. Biomaterials. 2010;31:4639. doi: 10.1016/j.biomaterials.2010.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peppas NA, Hilt JZ, Khademhosseini A, Langer R. Adv. Mater. 2006;18:1345. [Google Scholar]
- 3.Slaughter BV, Khurshid SS, Fisher OZ, Khademhosseini A, Peppas NA. Adv. Mater. 2009;21:3307. doi: 10.1002/adma.200802106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drury JL, Mooney DJ. Synth. Biomim. Polym. 2003;24:4337. doi: 10.1016/s0142-9612(03)00340-5. [DOI] [PubMed] [Google Scholar]
- 5.Lutolf MP, Hubbell JA. Nat. Biotechnol. 2005;23:47. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 6.Fittkau MH, Zilla P, Bezuidenhout D, Lutolf MP, Human P, Hubbell JA, Davies N. Biomaterials. 2005;26:167. doi: 10.1016/j.biomaterials.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 7.Schmidt DR, Kao WJ. J. Biomed. Mater. Res. A. 2007;83:617. doi: 10.1002/jbm.a.31270. [DOI] [PubMed] [Google Scholar]
- 8.Burdick JA, Anseth KS. Biomaterials. 2002;23:4315. doi: 10.1016/s0142-9612(02)00176-x. [DOI] [PubMed] [Google Scholar]
- 9.Liu Tsang V, Chen AA, Cho LM, Jadin KD, Sah RL, DeLong S, West JL, Bhatia SN. FASEB J. 2007;21:790. doi: 10.1096/fj.06-7117com. [DOI] [PubMed] [Google Scholar]
- 10.Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. Proc. Natl. Acad. Sci. 2003;100:5413. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee S-H, Moon JJ, Miller JS, West JL. Biomaterials. 2007;28:3163. doi: 10.1016/j.biomaterials.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 12.Aimetti AA, Machen AJ, Anseth KS. Biomaterials. 2009;30:6048. doi: 10.1016/j.biomaterials.2009.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin C-C, Anseth KS. Adv. Funct. Mater. 2009;19:2325. doi: 10.1002/adfm.200900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Impellitteri NA, Toepke MW, Lan Levengood SK, Murphy WL. Biomaterials. 2012;33:3475. doi: 10.1016/j.biomaterials.2012.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nimmo CM, Shoichet MS. Bioconjug. Chem. 2011;22:2199. doi: 10.1021/bc200281k. [DOI] [PubMed] [Google Scholar]
- 16.van Dijk M, Rijkers DTS, Liskamp RMJ, van Nostrum CF, Hennink WE. Bioconjug. Chem. 2009;20:2001. doi: 10.1021/bc900087a. [DOI] [PubMed] [Google Scholar]
- 17.Binder WH, Sachsenhofer R. Macromol. Rapid Commun. 2007;28:15. [Google Scholar]
- 18.Adzima BJ, Tao Y, Kloxin CJ, DeForest CA, Anseth KS, Bowman CN. Nat. Chem. 2011;3:256. doi: 10.1038/nchem.980. [DOI] [PubMed] [Google Scholar]
- 19.Hoyle CE, Bowman CN. Angew. Chem. Int. Ed. 2010;49:1540. doi: 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- 20.DeForest CA, Polizzotti BD, Anseth KS. Nat. Mater. 2009;8:659. doi: 10.1038/nmat2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeForest CA, Polizzotti BD, Anseth KS. Nat. Mater. 2009;8:659. doi: 10.1038/nmat2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seidlits SK, Schmidt CE, Shear JB. Adv. Funct. Mater. 2009;19:3543. [Google Scholar]
- 23.Khetan S, Burdick JA. Biomaterials. 2010;31:8228. doi: 10.1016/j.biomaterials.2010.07.035. [DOI] [PubMed] [Google Scholar]
- 24.Hahn MS, Miller JS, West JL. Adv. Mater. 2006;18:2679. [Google Scholar]
- 25.Aizawa Y, Wylie R, Shoichet M. Adv. Mater. 2010;22:4831. doi: 10.1002/adma.201001855. [DOI] [PubMed] [Google Scholar]
- 26.DeForest CA, Anseth KS. Angew. Chem. Int. Ed. 2012;51:1816. doi: 10.1002/anie.201106463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeForest CA, Sims EA, Anseth KS. Chem. Mater. 2010;22:4783. doi: 10.1021/cm101391y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DeForest CA, Anseth KS. Nat. Chem. 2011;3:925. doi: 10.1038/nchem.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wylie RG, Ahsan S, Aizawa Y, Maxwell KL, Morshead CM, Shoichet MS. Nat. Mater. 2011;10:799. doi: 10.1038/nmat3101. [DOI] [PubMed] [Google Scholar]
- 30.Luo Y, Shoichet MS. Nat. Mater. 2004;3:249. doi: 10.1038/nmat1092. [DOI] [PubMed] [Google Scholar]
- 31.Arumugam S, Popik VV. J. Am. Chem. Soc. 2012;134:8408. doi: 10.1021/ja302970x. [DOI] [PubMed] [Google Scholar]
- 32.Scott TF, Schneider AD, Cook WD, Bowman CN. Science. 2005;308:1615. doi: 10.1126/science.1110505. [DOI] [PubMed] [Google Scholar]
- 33.Kloxin CJ, Scott TF, Bowman CN. Macromolecules. 2009;42:2551. doi: 10.1021/ma802771b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kloxin CJ, Scott TF, Park HY, Bowman CN. Adv. Mater. 2011;23:1977. doi: 10.1002/adma.201100323. [DOI] [PubMed] [Google Scholar]
- 35.Fairbanks BD, Schwartz MP, Halevi AE, Nuttelman CR, Bowman CN, Anseth KS. Adv. Mater. 2009;21:5005. doi: 10.1002/adma.200901808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anderson SB, Lin C-C, Kuntzler DV, Anseth KS. Biomaterials. 2011;32:3564. doi: 10.1016/j.biomaterials.2011.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin C-C, Raza A, Shih H. Biomaterials. 2011;32:9685. doi: 10.1016/j.biomaterials.2011.08.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Polizzotti BD, Fairbanks BD, Anseth KS. Biomacromolecules. 2008;9:1084. doi: 10.1021/bm7012636. [DOI] [PubMed] [Google Scholar]
- 39.Chiefari J, (Bill) Chong YK, Ercole F, Krstina J, Jeffery J, Le TPT, Mayadunne RTA, Meijs GF, Moad CL, Moad G, Rizzardo E, Thang SH. Macromolecules. 1998;31:5559. [Google Scholar]
- 40.Malkoch M, Vestberg R, Gupta N, Mespouille L, Dubois P, Mason AF, Hedrick JL, Liao Q, Frank CW, Kingsbury K, Hawker CJ. Chem. Commun. 2006:2774. doi: 10.1039/b603438a. [DOI] [PubMed] [Google Scholar]
- 41.Stockmayer WH. J Chem. Phys. 1944;12:125. [Google Scholar]
- 42.Tibbitt MW, Kloxin AM, Dyamenahalli KU, Anseth KS. Soft. Matter. 2010;6:5100. doi: 10.1039/C0SM00174K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoffmann JC, West JL. Soft. Matter. 2010;6:5056. [Google Scholar]
- 44.Kloxin CJ, Scott TF, Bowman CN. Macromolecules. 2009;42:2551. doi: 10.1021/ma802771b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Culver JC, Hoffmann JC, Poché RA, Slater JH, West JL, Dickinson ME. Adv. Mater. 2012;24:2344. doi: 10.1002/adma.201200395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kasko AM, Wong DY. Future Med. Chem. 2010;2:1669. doi: 10.4155/fmc.10.253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.