

Abstract
The reductive cyclization of N-oxoacyl ortho-bromoanilides to form 3-hydroxy-2-oxindoles under the conditions of palladium catalyzed hydrogenation is described. This work may be viewed as a prelude to intermolecular hydrogen-mediated Grignard-type reductive couplings of organic halides with carbonyl compounds.
Keywords: hydrogenation, carbonyl addition, reductive coupling, hydroxyl oxindole, palladium
Graphical abstract

1. Introduction
Carbonyl addition is a cornerstone of chemical synthesis.1 The Grignard reaction,2 the magnesium mediated reductive coupling of organic halides and carbonyl compounds, persists as one of the most broadly utilized methods for carbonyl addition. Despite its broad scope, operational simplicity and cost-effective nature, the Grignard reaction requires organomagnesium reagents, which are highly basic and, hence, moisture sensitive, and which generate stoichiometric quantities of metallic byproducts, posing challenges for use on scale.3,4,5 Additionally, enantioselective variants of the Grignard addition have proven elusive. 6 These limitations are potentially addressed through the development of metal catalyzed organic halide-carbonyl reductive couplings, especially those employing non-metallic low molecular weight terminal reductants (Scheme 1). 7 The Nozaki-Hiyama-Kishi (NHK) reaction is perhaps the most notable metal catalyzed reaction of this type.8 While enantioselective variants of the NHK reaction have been developed,8 this process employs toxic chromium base catalysts and, as elemental manganese is used as terminal reductant, it does not circumvent generation of stoichiometric metallic byproducts.
Scheme 1.

Transition metal catalyzed reductive carbonyl addition.
We have developed the first “C-C bond forming hydrogenations” beyond hydroformylation, the largest volume application of homogeneous catalysis. 9 In these processes, π-unsaturated reactants are hydrogenated in the presence of carbonyl compounds or imines to furnish products of reductive coupling.10 As catalytic hydrogenation is used routinely for the reduction of organic halides to form the corresponding C-H compounds,11 we became attracted to the prospect of capturing the intervening organometallic species via carbonyl addition. Such hydrogen-mediated organic halide-carbonyl reductive couplings would bypass the generation of stoichiometric metallic byproducts and potentially provide a conduit to enantiomerically enriched adducts. The feasibility of hydrogen-mediated Grignard-type reactions is supported by reports of related halo-ketone reductive cyclizations under the conditions of transfer hydrogenation, wherein alcohols 12 or tertiary amines 13,14,15 are utilized as terminal reductants (Scheme 2, eq. 1–3).16,17 Here, we demonstrate palladium catalyzed hydrogenation of N-oxoacyl ortho-bromoanilides promotes reductive cyclization to form 3-hydroxy-2-oxindoles in good to excellent yield with complete suppression of conventional aryl halide hydrogenolysis pathways (Scheme 2, eq. 4).
Scheme 2.

Reductive cyclization of aromatic halo-ketones in the absence of stoichiometric metals
2. Results and Discussion
Initial experiments focused on the reductive cyclization of α-ketoamides 1a (X = Br) under hydrogenation conditions using Pd2(dba)3•CHCl3 as precatalyst in combination with various phosphine ligands (eq. 5). Gaseous hydrogen was introduced simply using balloons. Several phosphine ligands were tested for this transformation, for example, 1,1′-bis(di-tert-butylphosphino)ferrocene (DtBPF, 55% yield), XPhos (64% yield), and 1,2-bis(dicyclohexylphosphino)ethane (DCyPE, 68% yield). The palladium catalyst modified by the chelating phosphine ligand 1,1′-bis(di-iso-propylphosphino)ferrocene (DiPPF) provided the best results, enabling formation of 3-hydroxy-2-oxindole 2a in 95% yield after isolation by silica gel flash column chromatography. The use of Cs2CO3 as base is important, as substantially diminished isolated yields were observed in reactions conducted using Na2CO3 (trace conversion), K2CO3 (47% yield) or K3PO4 (20% yield) under otherwise identical conditions, which may, in part, be due to solubility. Under the indicated conditions (eq. 5), the corresponding ortho-chloro ketoamide 1a (X = Cl) provided 2a in 30% yield due to a combination of incomplete conversion and hydrogenolysis of the chloride. The triflyl derivative of ketoamide 1a (X = OTf) did not convert to oxindoles 2a under these conditions due to hydrolysis to form the phenol.14
 |
(5) |
Using the following conditions, Pd2(dba)3•CHCl3 (2.5 mol%), DiPPF (5 mol%), Cs2CO3 (200 mol%) in toluene (0.05 M) at 80 °C in the presence of H2 (1 atm) the reductive cyclization of α-ketoamides 1a–1f was explored (Table 2). The hydroxy oxindoles 2a-2f were obtained in moderate to excellent yields. Aryl 2a-2c, alkyl 2d-2e, and heteroaryl 2f groups at the C3 position of the resulting oxindoles 2a-2f were tolerated. Reactants 1a-1c that incorporate aryl substituents gave uniformly better results compared to reactants incorporating alkyl groups (1d-1e) or heteroaryl groups (2f).18
Table 2.
Reductive cyclization of α-ketoamides 1a 1f under the conditions of palladium catalyzed hydrogenation.a

|
Yields are of material isolated by silica gel chromatography.
K2CO3 (200 mol%).18
To further probe the scope of this process, compounds 1g and 1h, derived from 2-amino-3-bromopyridine and 4-amino-3-bromopyridine, respectively, were subjected to standard conditions for reductive cyclization (eq. 6, 7). Compound 1g was transformed to 6-aza-3-hydroxy-2-oxindole 2g in good yield (eq. 6). For compound 1h, only trace quantities of the 4-aza-3-hydroxy-2-oxindole 2h were observed under standard conditions. Elevated temperatures (130 °C) were required to enforce conversion to the 4-aza-3-hydroxy-2-oxindole 2h, which was isolated in 30% yield along with dehalogenated material (eq. 7).18 The diminished reactivity of 1h may be due to coordination of the less hindered pyridyl nitrogen to the palladium catalyst.
 |
(6) |
 |
(7) |
To probe the feasibility of engaging less activated carbonyl partners, compounds 1i and 1j were subjected to standard conditions for reductive cyclization (eq. 8, 9). Compound 1i delivered 3-phenyl indole 2i due to dehydration-aromatization of the tertiary alcohol derived upon reductive cyclization (eq. 8).18 The relatively low isolated yield of 2i stems from competing dehalogenation of 1i. For compound 1j, reductive cyclization to form 2j occurred in 32% isolated yield (eq. 9).18 Dehydration-aromatization to form the corresponding benzofuran was not observed. However, dehalogenation of 1j again contributed to a relatively low isolated yield of 2j.
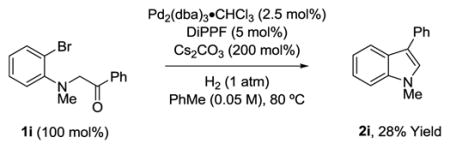 |
(8) |
 |
(9) |
The collective data suggest a catalytic mechanism involving oxidative addition of a palladium(0) complex to ortho-haloarenes 1a-1f to form the square planer σ-arylpalladium(II) complex A (eq. 10). Insertion of the tethered carbonyl followed by hydrogen activation delivers products of reductive cyclization 2a-2f. Use of chiral chelating phosphine ligands, Josiphos or Walphos type ligands, DuPhos, and BINAP, did not result in enantiomeric enrichment (>1% ee), suggesting loss of chelating ligand in advance of carbonyl insertion. Competing dehalogenation of 1a-1f presumably stems from the reaction of complex A with elemental hydrogen via pathways involving σ-bond metathesis-C-H reductive elimination.11 The identification of second generation catalysts that promote rapid carbonyl insertion with respect to hydrogen activation is currently under investigation in our laboratory.
 |
(10) |
3. Conclusions
In summary, we report that palladium catalyzed hydrogenation of ortho-bromoaryl α-ketoamides 1a-1f results in reductive cyclization to form 3-substituted-3-hydroxy-2-oxindoles 2a-2f in good to excellent isolated yields. Additionally, as illustrated in the conversion of 2-amino-3-bromopyridine derived α-ketoamides 1g and 4-amino-3-bromopyridine derived α-ketoamides 1h to compounds 2g and 2h, respectively, this method also provides access to aza-3-hydroxy-2-oxindoles. These results may be viewed as proof-of-concept studies with respect to the long term objective of developing general intermolecular hydrogen-mediated Grignard type additions of organic halides and carbonyl partners. The observation of competing aryl halide hydrogenolysis in the present transformations foreshadows the principal challenge associated with this endeavor.
4. Experimental
4.1. General Experimental
Tetrahydrofuran (THF) and toluene (PhMe) were distilled from sodium/benzophenone, and dichloromethane (CH2Cl2) and mesitylene were distilled from calcium hydride under a nitrogen atmosphere. Unless otherwise stated, commercially obtained reagents were used as received. 1H and 13C NMR spectra were recorded at 400 and 100 MHz with a Varian Gemini spectrometer. Chemical shifts are reported as parts per million (ppm) from tetramethylsilane (TMS) or ppm relative to the deuteriochloroform (CDCl3), 7.26 ppm for 1H NMR and 77.16 ppm for 13C NMR, respectively. Melting points (°C) are uncorrected. Infrared spectra were recorded on a Thermo Nicolet 380 spectrometer. Analytical thin layer chromatography (TLC) was performed on TLC silica gel plates (250 μm) precoated with a fluorescent indicator. Standard flash column chromatography procedures were followed using 40–63 μm silica gel. Visualization was effected with p-anisaldehyde, potassium permanganate, and iodine stains.
4.2 General Experimental Procedure for Palladium Catalyzed Reductive Cyclization
To a flask charged with Pd2(dba)3•CHCl3 (2.6 mg, 0.0025 mmol, 2.5 mol%), DiPPF (2.1 mg, 0.005 mmol, 5 mol%), and Cs2CO3 (65.2 mg, 0.2 mmol, 200 mol%) under an argon atmosphere was added freshly distilled toluene (2 mL, 0.05 M). The mixture was allowed to stir at 80 °C for 1 h, during which time the color of the solution changed from purple to orange or brown. N-Acyl ortho-bromoanilides 1 (0.1 mmol, 100 mol%) was added in one portion to the flask, then an argon balloon was switched to H2 balloon, and the reaction mixture was allowed to stir at 80 °C for 20 h. The reaction mixture was evaporated and the residue was subjected to flash silica gel column chromatography to furnish the title compound 2.
4.2.1. 3-Hydroxy-1-methyl-3-phenylindolin-2-one (2a)
According to general procedure for palladium catalyzed reductive cyclization with 1a (31.8 mg, 0.1 mmol, 100 mol%), the title product 2a was obtained as a white solid in 95% yield (22.7 mg, 0.095 mmol) after flash column chromatography (hexanes/EtOAc = 1:1). 1H NMR (400 MHz, CDCl3): δ 7.36–7.18 (m, 7H), 7.01 (td, J = 7.6, 1.0 Hz, 1H), 6.84 (dt, J = 7.8, 0.8 Hz, 1H), 3.46 (br s, 1H), 3.18 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 177.4, 143.5, 140.0, 131.5, 129.8, 128.5, 128.2, 125.3, 124.9, 123.5, 108.6, 77.9, 26.5. Data is consistent with reported literature.12b
4.2.2. 3-(3,5-Difluorophenyl)-3-hydroxy-1-methylindolin-2-one (2b)
According to general procedure for palladium catalyzed reductive cyclization with 1b (35.4 mg, 0.1 mmol, 100 mol%), the title product 2b was obtained as a white solid in 95% yield (26.1 mg, 0.095 mmol) after flash column chromatography (hexanes/EtOAc = 1:1). mp: 45 °C; 1H NMR (400 MHz, CDCl3) δ 7.40 (td, J = 7.7, 1.3 Hz, 1H), 7.26 (ddd, J = 7.4, 1.4, 0.6 Hz, 1H), 7.12 (td, J = 7.6, 1.0 Hz, 1H), 6.96–6.93 (m, 1H), 6.93–6.87 (m, 2H), 6.74 (tt, J = 8.7, 2.3 Hz, 1H), 3.27 (s, 3H), 3.22 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 176.5, 163.9 (d, J = 12.6 Hz), 162.2 (d, J = 12.6 Hz), 144.1 (t, J = 8.6 Hz), 143.3, 130.7, 130.4, 129.1, 124.8, 123.9, 109.0, 108.7 (dd, J = 21.1, 5.6 Hz), 103.7 (t, J = 25.4 Hz), 77.4 (t, J = 2.2 Hz), 26.7; IR (neat) 3410, 1701, 1610, 1595, 1450, 1111 cm−1; HRMS (ESI+) calcd. for C15H11F2NO2Na [M+Na]+ 298.0650, found 298.0649.
4.2.3. 3-(Benzo[d][1,3]dioxol-5-yl)-3-hydroxy-1-methylindolin- 2-one (2c)
According to general procedure for palladium catalyzed reductive cyclization with 1c (36.2 mg, 0.1 mmol, 100 mol%), the title product 2c was obtained as a colorless oil in 72% yield (19.8 mg, 0.072 mmol) after flash column chromatography (hexanes/EtOAc = 1:1). 1H NMR (400 MHz, CDCl3) δ 7.35 (td, J = 7.8, 1.3, 1H), 7.29 (ddd, J = 7.4, 1.3, 0.6 Hz, 1H), 7.10 (td, J = 7.5, 1.0 Hz, 1H), 6.91–6.89 (m, 2H), 6.83 (dd, J = 8.1, 1.8 Hz, 1H), 6.73 (dd, J = 8.1, 0.5 Hz, 1H), 5.93 (q, J = 1.4 Hz, 2H), 3.35 (s, 3H), 3.34 (s, 1H); 13C NMR (100 MHz, CDCl3) 177.2, 147.9, 147.7, 143.1, 133.8, 131.3, 129.9, 124.8, 123.5, 118.9, 108.7, 108.1, 106.3, 101.2, 29.6, 26.5. Data is consistent with reported literature.12b
4.2.4. 3-Hydroxy-1,3-dimethylindolin-2-one (2d)
According to general procedure for palladium catalyzed reductive cyclization with 1d (25.6 mg, 0.1 mmol, 100 mol%), the title product 2d was obtained as a white solid in 76% yield (13.5 mg, 0.076 mmol) after flash column chromatography (hexanes/EtOAc = 1:1). mp: 141–143 °C (lit: 141–143 °C); 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 7.5 Hz, 1H), 7.34 (td, J = 8.0 Hz, 1.5 Hz, 1H), 7.12 (t, J = 7.5 Hz, 1H), 6.86 (d, J = 8.0 Hz, 1H), 3.23 (s, 1H), 3.21 (s, 3H), 3.93 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.9, 143.0, 131.4, 129.6, 123.4, 123.0, 108.0, 74.0, 26.1, 25.0. Data is consistent with reported literature.19
4.2.5. 3-Cyclopropyl-3-hydroxy-1-methylindolin-2-one (2e)
According to general procedure for palladium catalyzed reductive cyclization with 1e (28.2 mg, 0.1 mmol, 100 mol%), the title product 2e was obtained as a white solid in 50% yield (10.2 mg, 0.050 mmol) after flash column chromatography (hexanes/EtOAc = 15:1 to 2:1). mp: 179–183 °C; 1H NMR (400 MHz, CDCl3) δ 7.37–7.30 (m, 2H), 7.11–7.04 (m, 1H), 6.83 (d, J = 7.8 Hz, 1H), 3.18 (s, 3H), 2.70 (br s, 1H), 1.39–1.31 (m, 1H), 0.66–0.53 (m, 2H), 0.48–0.34 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 177.6, 143.2, 129.5, 129.3, 123.8, 122.8, 108.2, 75.6, 29.6, 26.1, 17.9; IR (neat) 3331, 2922, 1699, 1615, 1467, 1260, 1090, 1021 cm−1; HRMS (ESI+) calcd. for C12H13NO2Na [M+Na]+ 226.0838, found 226.0836.
4.2.6. 3-Hydroxy-1-methyl-3-(thiophen-2-yl)indolin-2-one (2f)
According to general procedure for palladium catalyzed reductive cyclization with 1f (32.4 mg, 0.1 mmol, 100 mol%), the title product 2f was obtained as a yellow solid in 51% yield (12.5 mg, 0.051 mmol) after flash column chromatography (hexanes/EtOAc = 6:1 to 2:1). mp: 119–123 °C; 1H NMR (400 MHz, CDCl3) δ 7.54 (d, J = 7.4 Hz, 1H), 7.38 (dd, J = 7.6, 7.6 Hz, 1H), 7.31 (d, J = 4.9 Hz, 1H), 7.15 (dd, J = 7.4, 7.4 Hz, 1H), 7.01–6.96 (m, 1H), 6.96–6.91 (m, 1H), 6.89 (d, J = 7.8 Hz, 1H), 3.50 (br s, 1H), 3.23 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 176.2, 143.4, 143.3, 130.5, 126.9, 126.1, 125.1, 123.6, 109.0, 75.5, 26.7 (one carbon was not detected); IR (neat) 3346, 1702, 612, 1469, 1373, 1347, 1093, 1018 cm−1; HRMS (ESI+) calcd. for C13H11NO2SNa [M+Na]+ 268.0403, found 268.0405.
4.2.7. 3-Hydroxy-1-methyl-3-phenyl-1,3-dihydro-2H-pyrrolo[2,3- b]pyridin-2-one (2g)
According to general procedure for palladium catalyzed reductive cyclization with 1g (31.9 mg, 0.1 mmol, 100 mol%), the title product 2g was obtained as a colorless oil in 73% yield (17.6 mg, 0.073 mmol) after flash column chromatography (hexanes/EtOAc = 1:1). 1H NMR (400 MHz, CDCl3) δ 8.19 (dd, J = 5.3, 1.6 Hz, 1H), 7.47 (dd, J = 7.3, 1.6 Hz, 1H), 7.38–7.23 (m, 5H), 6.93 (dd, J = 7.3, 5.3, 1H), 3.29 (s, 3H), 3.18 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 177.2, 157.0, 148.7, 139.2, 132.4, 128.8, 127.7, 125.9, 125.2, 118.9, 77.6, 25.6; IR (neat) 3340, 1635, 1467, 1351 cm−1; HRMS (ESI+) calcd. for C14H12N2O2Na [M+Na]+ 263.0791, found 263.0792.
4.2.7. 3-Hydroxy-1-methyl-3-phenyl-1,3-dihydro-2H-pyrrolo[3,2- c]pyridin-2-one (2h)
According to general procedure for palladium catalyzed reductive cyclization with 1h (31.9 mg, 0.1 mmol, 100 mol%) in mesitylene at 130 °C, the title product 2h was obtained as a colorless oil in 30% yield (7.2 mg, 0.03 mmol) after flash column chromatography (100% EtOAc). 1H NMR (400 MHz, CDCl3) δ 8.45 (d, J = 5.4 Hz, 1H), 8.26 (d, J = 0.9 Hz, 1H), 7.42–7.31 (m, 5H), 6.83 (dd, J = 5.4, 0.8 Hz, 1H), 3.22 (s, 3H), 1.28 (br s, 1H); 13C NMR (100 MHz, CDCl3) δ 177.3, 151.4, 151.2, 145.1, 139.0, 131.1, 129.0, 127.5, 125.5, 123.4, 104.5, 26.7; IR (neat) 3350, 1630, 1466, 1359 cm−1; HRMS (ESI+) calcd. for C14H12N2O2Na [M+Na]+ 263.0791, found 263.0789.
4.2.8. 1-Methyl-3-phenyl-1H-indole (2i)
According to general procedure for palladium catalyzed reductive cyclization with 1i (30.4 mg, 0.1 mmol, 100 mol%), the title product 2i was obtained as a yellow oil in 28% yield (5.8 mg, 0.028 mmol) after flash column chromatography (hexanes/EtOAc = 3:1). 1H NMR (400 MHz, CDCl3) δ 7.88 (dt, J = 8.0, 1.0 Hz, 1H), 7.62–7.57 (m, 2H), 7.41–7.34 (m, 2H), 7.30 (dt, J = 8.3, 1.0 Hz, 1H), 7.24–7.17 (m, 3H), 7.12 (ddd, J = 8.0, 7.0, 1.1 Hz, 1H), 3.78 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 137.7, 136.6, 127.8, 127.9, 126.1, 125.6, 121.8, 120.0, 119.9, 117.0, 109.1, 32.9. Data is consistent with reported literature.20
4.2.9. 3-Phenyl-2,3-dihydrobenzofuran-3-ol (2j)
According to general procedure for palladium catalyzed reductive cyclization with 1j (29.1 mg, 0.1 mmol, 100 mol%), the title product 2j was obtained as a light yellow oil in 32% yield (6.8 mg, 0.032 mmol) after flash column chromatography (hexanes/EtOAc = 25:1). 1H NMR (400 MHz, CDCl3) δ 7.46– 7.41 (m, 2H), 7.33–7.27 (m, 2H), 7.27–7.20 (m, 2H), 7.05–7.02 (m, 1H), 6.93–6.83 (m, 2H), 4.64 (d, J = 10.2 Hz, 1H), 4.44 (d, J = 10.2 Hz, 1H), 2.23 (br s, 1H); 13C NMR (100 MHz, CDCl3) δ 160.8, 142.7, 132.3, 130.8, 128.4, 127.7, 126.2, 124.5, 121.6, 110.9, 86.3, 82.7. Data is consistent with that reported in the literature.21
Supplementary Material
Acknowledgments
The Robert A. Welch Foundation (F-0038) and the NIHNIGMS (RO1-GM069445) are acknowledged for partial support of this research.
Footnotes
1H and 13C NMR spectra for all new compounds are available. Supplementary data related to this article can be found at http://.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Knochel P, Molander GA. Comprehensive Organic Synthesis. 2. 1 and 2 Elsevier Ltd; 2014. [Google Scholar]; (b) Otera J. Modern Carbonyl Chemistry. Wiley-VCH; Weinheim, Germany: 2000. [Google Scholar]
- 2.Grignard V. Compt Rend. 1900;130:1322. [Google Scholar]
- 3.The two largest volume applications of homogenous metal catalysis, alkene hydroformylation and the carbonylation of methanol, the Monsanto or Cativa Processes, are byproduct-free C-C bond formations.
- 4.For selected reviews on hydroformylation, see: Kalck P, Peres Y, Jenck J. Adv Organomet Chem. 1991;32:121.Breit B, Seiche W. Synthesis. 2001:1.Van Keeuwen PWNM, Claver C. Rhodium Catalyzed Hydroformylation. Kluwer Academic Publishers; Dordrecht, Netherlands: 2002. Weissermel K, Arpe HJ. Industrial Organic Chemistry. Wiley-VCH; Weinheim, Germany: 2003. pp. 127–141.Franke R, Selent D, Börner A. Chem Rev. 2012;112:5675. doi: 10.1021/cr3001803.
- 5.For selected reviews on methanol carbonylation, the Monsanto or Cativa Processes, see: Maitlis PM, Haynes A, Sunley GJ, Howard MJ. J Chem Soc, Dalton Trans. 1996:2187. and references therein.Jones JH. Plantinum Met Rev. 2000;44:94.Haynes A. Top Organomet Chem. 2006;18:179.Kalck P, Serp P. Iridium Complexes in Organic Synthesis. Wiley-VCH; Weinheim: 2008. pp. 195–209.
- 6.Stoichiometric quantities of chiral modifiers have been employed in Grignard additions to enforce enantioselectivity: Weber B, Seebach D. Angew Chem Int Ed Engl. 1992;31:84.
- 7.For a recent review on the reductive coupling of organic halides to carbonyl compounds, see: Moragas T, Correa A, Martin R. Chem Eur J. 2014;20:8242. doi: 10.1002/chem.201402509.
- 8.For selected reviews on enantioselective carbonyl allylation via Nozaki-Hiyama-Kishi coupling, see: Avalos M, Babiano R, Cintas P, Jiménez JL, Palacios JC. Chem Soc Rev. 1999;28:169.Bandini M, Cozzi PG, Umani-Ronchi A. Chem Commun. 2002:919. doi: 10.1039/b109945k.Hargaden GC, Guiry PJ. Adv Synth Catal. 2007;349:2407.
- 9.For a review on C-C bond forming hydrogenation and transfer hydrogenation, see: Hassan A, Krische MJ. Org Process Res Devel. 2011;15:1236. doi: 10.1021/op200195m.
- 10.For selected examples of enantioselective C-C bond forming hydrogenation involving carbonyl and imine addition, see: Kong JR, Ngai MY, Krische MJ. J Am Chem Soc. 2006;128:718. doi: 10.1021/ja056474l.Rhee JU, Krische MJ. J Am Chem Soc. 2006;128:10674. doi: 10.1021/ja0637954.Skucas E, Kong JR, Krische MJ. J Am Chem Soc. 2007;129:7242. doi: 10.1021/ja0715896.Ngai MY, Barchuk A, Krische MJ. J Am Chem Soc. 2007;129:12644. doi: 10.1021/ja075438e.Bee C, Han SB, Hassan A, Iida H, Krische MJ. J Am Chem Soc. 2008;130:2746. doi: 10.1021/ja710862u.
- 11.For reviews on the hydrogenolysis of organic halides, see: Pinder AR. Synthesis. 1980:425.Hudlicky M. In: Reduction in Organic Chemistry in Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 8. Elsevier Ltd; 1991. pp. 895–922.Urbano FJ, Marinas JM. J Mol Cat A: Chem. 2001;173:329.Sheldon RA, van Bekkum H. Fine Chemicals through Heterogeneous Catalysis. Wiley-VCH; Weinheim, Germany: 2001. pp. 415–442.Alonso F, Beletskaya IP, Yus M. Chem Rev. 2002;102:4009. doi: 10.1021/cr0102967.Chelucci G, Baldino S, Pinna GA, Pinna G. Curr Org Chem. 2012;16:2921.
- 12.For palladium catalyzed reductive cyclization of aryl ortho-halo-ketones mediated by alcohol reductants, see: Quan LG, Lamrani M, Yamamoto Y. J Am Chem Soc. 2000;122:4827.Jia YX, Katayev D, Kündig EP. Chem Commun. 2010;46:130. doi: 10.1039/b917958e.
- 13.For palladium catalyzed reductive cyclization of aryl ortho-halo-ketones mediated by tertiary amines, see: Solé D, Vallverdú L, Peidró E, Bonjoch J. Chem Commun. 2001:1888. doi: 10.1039/b105686g.Solé D, Vallverdú L, Solans X, Font-Bardía M, Bonjoch J. J Am Chem Soc. 2003;125:1587. doi: 10.1021/ja029114w.
- 14.For a related palladium catalyzed reductive cyclization of N-α-(imino)acyl ortho-(triflyl)anilides to form 3-amino-2-oxindoles with triethylamine as reductant, see: Tolstoy P, Lee SXY, Sparr C, Ley SV. Org Lett. 2012;14:4810. doi: 10.1021/ol302119j.
- 15.For the palladium catalyzed reductive coupling of aryl iodides with anhydrides via hydrogen transfer from Hunig’s base, see: Cacchi S, Fabrizi G, Gavazza F, Goggiamani A. Org Lett. 2003;5:289. doi: 10.1021/ol027243b.
- 16.Reported reductive cyclization of halo-nitriles in DMF likely involve adventitious dimethylamine as terminal reductant: Pletnev AA, Larock RC. J Org Chem. 2002;67:9428. doi: 10.1021/jo0262006.Pletnev AA, Larock RC. Tetrahedron Lett. 2002;43:2133.
- 17.Couplings of ortho-halobenzaldehydes or ortho-halobenzonitriles with alkynes to form indenones and related products occur via hydrogen auto-transfer or transfer hydrogenation: Tao W, Silverberg LJ, Rheingold AL, Heck RF. Organometallics. 1989;8:2550.Larock RC, Doty MJ. J Org Chem. 1993;58:4579.Larock RC, Tian Q, Pletnev AA. J Am Chem Soc. 1999;121:3238.Quan LG, Gevorgyan V, Yamamoto Y. J Am Chem Soc. 1999;121:3545.Gevorgyan V, Quan LG, Yamamoto Y. Tetrahedron Lett. 1999;40:4089.
- 18.Use of K2CO3 did not improve the yields of compounds 2e, 2f or 2h–j.
- 19.Gorokhovik I, Neuville L, Jieping ZC. Org Lett. 2011;13:5536. doi: 10.1021/ol202263a. [DOI] [PubMed] [Google Scholar]
- 20.Chen S, Liao Y, Zhao F, Qi H, Liu S, Deng G-J. Org Lett. 2014;16:1618. doi: 10.1021/ol500231c. [DOI] [PubMed] [Google Scholar]
- 21.Liu G, Lu X. J Am Chem Soc. 2006;128:16504. doi: 10.1021/ja0672425. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.