

Developmentally regulated epigenetic modifications of upstream promoters are involved in photosynthesis and C4 metabolism.
Abstract
Histone modifications contribute to gene regulation in eukaryotes. We analyzed genome-wide histone H3 Lysine (Lys) 4 trimethylation and histone H3 Lys 9 acetylation (two modifications typically associated with active genes) in meristematic cells at the base and expanded cells in the blade of the maize (Zea mays) leaf. These data were compared with transcript levels of associated genes. For individual genes, regulations (fold changes) of histone modifications and transcript levels were much better correlated than absolute intensities. When focusing on regulated histone modification sites, we identified highly regulated secondary H3 Lys 9 acetylation peaks on upstream promoters (regulated secondary upstream peaks [R-SUPs]) on 10% of all genes. R-SUPs were more often found on genes that were up-regulated toward the blade than on down-regulated genes and specifically, photosynthetic genes. Among those genes, we identified six genes encoding enzymes of the C4 cycle and a significant enrichment of genes associated with the C4 trait derived from transcriptomic studies. On the DNA level, R-SUPs are frequently associated with ethylene-responsive elements. Based on these data, we suggest coevolution of epigenetic promoter elements during the establishment of C4 photosynthesis.
The transcription rate of genes is regulated by their promoters. In eukaryotes, genes are associated with proteins that condense the DNA molecule. The association of DNA and proteins is called chromatin. The basic repeat structure of chromatin is an octamer of two of the histone proteins H2A, H2B, H3, and H4 plus approximately 160 bp of DNA (Kouzarides, 2007). This structure not only forms a passive barrier to binding of other proteins to DNA but also, actively participates in the regulation of transcription. Posttranslational modifications of histones are important elements of this regulatory role (Lauria and Rossi, 2011). A wealth of different modifications was identified. Studies in different model organisms revealed that promoters and transcribed regions of active genes fundamentally differ in their histone modification profile compared with inactive genes (Berger, 2007; Zhang, 2008). Two major theories have been suggested of how histone modifications can regulate gene transcription. Acetylation of histones might neutralize the positive charge of lysines on the flexible histone N-terminal tails and by this, reduce the interaction with negatively charged DNA. This would enhance DNA accessibility for transcription factors and the RNA polymerase. Therefore, histone acetylation was mostly observed on actively transcribed genes (Dion et al., 2005; Wang et al., 2009). Other histone modifications do not neutralize the positive charge of Lys. The best studied of these modifications is Lys methylation. The three hydrogen atoms on the ε-amino group of lysines can be partially or fully replaced by methyl groups; thus, mono-, di-, and trimethylated forms of lysines exist. Different forms of Lys methylation have been associated with active or inactive genes (Wang et al., 2009; Roudier et al., 2011). The most obvious role for histone modifications in this scenario is the provision of binding sites for transcription factors. This has been exemplified by the binding of TATA box binding protein-associated factor3, a subunit of the general transcription initiator factor TFIID, to trimethylated Lys 4 on histone H3 (H3K4me3) to initiate transcription (Vermeulen et al., 2007; Lauberth et al., 2013) or the recognition of acetylated histones by bromodomain proteins (Hassan et al., 2007). The combination of all different histone modifications on a gene might form a histone code or epigenetic memory that collects and integrates information about developmental and environmental signals perceived by the organism (Turner, 2002; Song et al., 2012; To and Kim, 2014).
The leaf gradient of monocotyledonous plants is a useful tool to study developmental dynamics of biological processes. Monocot leaves develop from undifferentiated meristematic cells at the base to mature photosynthetic cells at the tip. Thus, different developmental stages can be isolated from the same leaf (Nelissen et al., 2012; Wang et al., 2014). Development of the maize (Zea mays) leaf is of special interest, because the photosynthetic tissue functionally differentiates during maturation into mesophyll (M) and bundle sheath (BS) cells: the two cell types required to perform C4 photosynthesis (Langdale, 1998; Majeran et al., 2005). Thus, the dynamics of transcriptomes, proteomes, metabolite levels, and photosynthesis on the maize leaf gradient have been studied (Li et al., 2010; Majeran et al., 2010; Pick et al., 2011; Tausta et al., 2014; Wang et al., 2014). Moreover, this type of analysis has also been transferred to dicotyledonous C4 plants, where leaves of different growth stages (Külahoglu et al., 2014) or different zones of an individual developed leaf were compared (Aubry et al., 2014). All of these studies aimed at identifying genes that were coregulated with photosynthetic genes and specifically, C4 genes.
We previously analyzed the function of histone modifications on C4 genes in maize. We defined specific modifications that were associated with light regulation (Offermann et al., 2006), differentiation of M and BS cells (Danker et al., 2008), circadian regulation (Horst et al., 2009), or metabolic regulation of C4 genes (Offermann et al., 2008). This code was, at least in part, conserved in other C4 grasses (Heimann et al., 2013; Horst et al., 2013). Most significantly, we found that acetylation of H3K9 (H3K9ac) on core promoters only occurred in the light but was independent of other stimuli. In contrast, H3K4me3 was associated with M and BS cell differentiation but independent of light (Offermann et al., 2008).
In this study, we compared transcript levels and histone modification levels between base and blade of maize leaves on a genome-wide level. Our data reveal a quantitative association between changes in transcription and changes in histone modifications of individual genes. The focus on regulated histone modifications in this study allowed the identification of regulated secondary upstream peaks (R-SUPs) on many developmentally regulated genes as a unique feature of H3K9ac. R-SUPs showed distinct properties compared with transcription initiation site (TIS) region peaks and were associated with ethylene-responsive elements (EREs) on the DNA level. Moreover, R-SUPs were significantly enriched on photosynthetic genes and found on some key genes encoding enzymes of the C4 cycle. Meta-analyses of R-SUP genes and lists of potential C4-associated genes derived from different transcriptomic studies revealed a substantial overlap.
RESULTS AND DISCUSSION
Assessment of Data Quality
We isolated RNA and chromatin from the base and the blade of the maize leaf (corresponding to segments −4 and +4 in Li et al., 2010). The abundance of mRNAs was measured by RNA sequencing (RNA-Seq), and the abundance of histone modifications was measured by chromatin immunoprecipitation sequencing (ChIP-Seq) after precipitation of chromatin with antibodies directed to H3K9ac and H3K4me3. Comparison of transcript abundance at the base and the blade revealed 7,634 transcripts that were significantly up- or down-regulated (false discovery rate [FDR] ≤ 0.05; fold change ≥ |2|) in our data set.
Quality of RNA data was analyzed by comparison with previously published data on transcription on the leaf gradient (Li et al., 2010). Pearson correlation coefficients of the two replicates generated here and the previous data set are given in Supplemental Figure S1; 88% of the regulated genes from our data set showed identical regulation in the data set in Li et al., 2010. To concentrate on robustly regulated genes, we further only considered genes that were identically regulated in both studies (2,865 up-regulated genes and 3,823 down-regulated genes when comparing base and blade).
For the comparative analyses and the statistical evaluation of quantitative ChIP-Seq data, we used DiffBind (Stark and Brown, 2011). This program was initially developed for the identification of differential transcription factor binding to DNA (Ross-Innes et al., 2012) but recently, also used in a study on genome-wide induced alterations in histone modifications in mice (Papait et al., 2013). DiffBind normalizes ChIP-Seq samples for differences in peak calling and the amount of mapped reads enabling a direct quantitative comparison of histone modifications from different biological situations (“Materials and Methods”). As a quality test, we determined the percentage of consensus peaks derived from expected or unexpected sample combinations (Table I). Expected combinations were merged from either all four samples (two replicates from the base and two replicates from the blade) or at least two associated samples (either the two replicates from the base or the two replicates from the blade). Unexpected combinations included detection of peaks in one of the base samples and one of the blade samples. The percentages of consensus peaks that were built from expected combinations were more than 95% for both tested modifications. For H3K9ac, 61% of all peaks were detected in both the blade and the base, whereas 10% were only detected at the leaf base and 8% were only in the blade. For H3K4me3, similar numbers were observed, but a higher number of peaks (75%) was detected both at the base and in the blade, indicating less regulation of H3K4me3 compared with H3K9ac. The percentages of consensus peaks that were derived from unexpected combinations were always lower than 5%. This indicated that peak calling was consistent in all samples and allowed for a comparative analysis of histone modifications at the leaf base and the blade.
Table I. Sources of merged consensus peaks of H3K9ac and H3K4me3 from DiffBind.
Histone modification peaks called in replicates (A and B) from the leaf base (Base) or the leaf blade (Blade) were analyzed with DiffBind. Number and percentage of consensus peaks merged from the indicated peak combinations of samples are shown.
| Combinations | Base A | Base B | Blade A | Blade B | H3K9ac Peaks |
H3K4me3 Peaks |
||
|---|---|---|---|---|---|---|---|---|
| No. | % | No. | % | |||||
| X | X | X | X | 26,742 | 61.1 | 24,661 | 74.9 | |
| X | X | 4,329 | 9.9 | 1,655 | 5.0 | |||
| X | X | X | 1,362 | 3.1 | 1,980 | 6.0 | ||
| X | X | X | 317 | 0.7 | 226 | 0.7 | ||
| X | X | 6,258 | 14.3 | 1,197 | 3.6 | |||
| X | X | X | 3,507 | 8.0 | 962 | 2.9 | ||
| X | X | X | 27 | 0.06 | 639 | 1.9 | ||
| Expected | 42,542 | 97.3 | 31,320 | 95.2 | ||||
| X | X | 940 | 2.1 | 846 | 2.6 | |||
| X | X | 236 | 0.5 | 91 | 0.3 | |||
| X | X | 10 | 0.02 | 608 | 1.8 | |||
| X | X | 6 | 0.01 | 43 | 0.1 | |||
| Unexpected | 1,192 | 2.7 | 1,588 | 4.8 | ||||
It has previously been described that H3K9ac and H3K4me3 were associated with actively transcribed genes. Modifications were mainly enriched around the TIS and with lower abundance in the transcribed region of maize genes (Wang et al., 2009; He et al., 2013). To investigate if our ChIP-Seq data from two different zones from the maize leaf confirm these findings, we tested whether histones on transcribed genes were methylated and/or acetylated (Fig. 1A). From the 26,055 genes that were expressed with reads per kilobase per million mapped reads (RPKM) > 0.1 in at least one of the investigated tissues, 20,841 (80%) showed H3K9ac modification, and 20,456 (79%) showed H3K4me3 modification. Both modifications were found on 19,560 (75%) transcribed genes. Thus, both modifications were highly associated with transcribed genes. When analyzing the distribution of H3K4me3 and H3K9ac signals, both modifications peaked between the TIS and position +1,000 bp of the transcribed region (Fig. 1B). This region was defined as the TIS region. The detected distributions were in accordance with other data sets on positioning of the investigated histone modifications on active genes in maize (Wang et al., 2009; He et al., 2013).
Figure 1.

Overlap between expressed and modified genes and distribution of H3K9ac and H3K4me3 peaks. A, Overlap between expressed genes and genes modified by H3K9ac and H3K4me3. Expressed genes showed a mean RPKM > 0.1 in one of the analyzed tissues. B, Distribution relative to the TIS. H3K9ac and H3K4me3 peaks were grouped into 250-bp bins according to their position. Percentages of all peaks are plotted on the y axes. Three distinct regions are indicated: upstream (U), TIS (T), and downstream (D). +, More than 4,500 bp downstream of the TIS.
A Dynamic Analysis of Histone Modifications Revealed a Unique Class of Upstream Acetylation Peaks
Whereas a static view on histone modifications on genes with low and high transcript levels provided important insights into their function, much less is known about the dynamics of histone modifications during gene activation or inactivation of individual genes. Our quantitative determination of transcript abundances and histone modifications in two different developmental zones allowed for the comparison of both parameters on the level of individual genes. Figure 2 shows a correlation analysis of transcript levels and histone modifications. Correlation between the absolute intensity of the tested histone modifications and the absolute transcript level of the corresponding gene (Fig. 2, Intensities) was low and only showed a slight tendency for more intense histone modification signals on higher transcribed genes. However, when plotting the fold change in histone modifications between leaf base and blade against the fold change in transcript levels from the same samples (Fig. 2, Fold changes), we observed significant correlation for both H3K9ac and H3K4me3. This was also exemplified by the much better Pearson correlation coefficients (P values) for fold changes compared with intensities (Fig. 2). Thus, although a rough correlation between the intensity of a histone modification peak and the abundance of the associated transcript in a given biological situation exists, changes in histone modification between two biological situations or developmental stages provide much better information about the activity of the gene. Our observations are supported by another study in maize, where transcript abundance and histone modification levels were compared between root and shoot tissues of maize. Best correlations were found for relative changes and not absolute values (He et al., 2013). In a recent study on histone modifications and leaf senescence in Arabidopsis (Arabidopsis thaliana), histone modifications increased during gene activation for a subset of genes, but other genes were already premarked with histone modifications before activation and did not show such increases (Brusslan et al., 2015). The very good correlation between fold changes in the tested histone modifications and fold changes in transcript levels in our study argues against widespread premarking of genes at the leaf base for later activation in the blade of the maize leaf.
Figure 2.

Correlation of H3K9ac or H3K4me3 signals and transcript levels for individual genes. Log2-transformed values of absolute signal intensities or fold changes between base and blade were plotted against each other. The values for H3K9ac and H3K4me3 are plotted on the y axes, and the values for transcription are plotted on the x axes. For absolute signal intensities, only the values from the tissue with the higher transcription are shown. P, Pearson correlation coefficient.
Our data analysis up to this point suggested that regulation of histone modifications between different biological situations was highly associated with regulation of transcript levels. We, therefore, focused our analyses on H3K9ac and H3K4me3 peaks that were regulated between leaf base and blade (FDR ≤ 0.05, peak fold change ≥ |2|; Fig. 3A, regulated peaks). A distribution of these peaks is shown in Figure 3A. For H3K4me3 (Fig. 3A, right), we observed a distribution that was very similar to the distribution observed for all peaks (Fig. 1B; i.e. most regulated peaks were found in the TIS region, the frequency slowly declined toward the downstream region, and only few regulated peaks were found in the upstream promoter region). However, for H3K9ac, we observed beside the TIS region and the downstream region a separate region of regulated acetylation peaks in the upstream promoter region. This region was distinct from the TIS region, because the number of regulated peaks declined between TIS and −500 bp and then increased again toward more upstream-located promoter regions. The highest number of these upstream acetylation peaks was found around position −1,000 bp relative to the TIS.
Figure 3.

Distribution of regulated H3K9ac and H3K4me3 peaks relative to the TIS and peak composition of genes with a regulated upstream peak. A, Regulated H3K9ac and H3K4me3 peaks (fold change ≥ |2|, FDR ≤ 0.05) were grouped into 250-bp bins according to their position. Percentages of all peaks were plotted on the y axes. +, More than 4,500 bp downstream of the TIS. B, Possible H3K9ac peak compositions of genes with a regulated H3K9ac upstream peak are indicated in left. The bars indicate absolute values. The fractions of the indicated composition from all genes with regulated upstream peaks are given as percentages. Three distinct regions are indicated: upstream (U), TIS (T), and downstream (D).
Whereas H3K9ac and H3K4me3 peaks in the TIS region and downstream region were already described in different species (Wang et al., 2009; Roudier et al., 2011; Du et al., 2013; He et al., 2013; Hussey et al., 2015), our observation of regulated upstream H3K9ac peaks on an genome-wide basis was, to our knowledge, unique. We first wanted to test whether regulated upstream acetylation was simply caused by false annotations of TIS. To exclude this possibility, we evaluated whether the 1,084 genes with regulated upstream H3K9ac peaks had additional H3K9ac peaks in the annotated TIS region or the downstream region (Fig. 3B). For comparison, data for the 251 upstream H3K4me3 peaks were included; 69% (53% + 16%) of all genes with a regulated upstream H3K9ac peak also had an H3K9ac peak in the annotated TIS region. On those genes, upstream H3K9ac peaks were distinct secondary peaks that were separate from the primary TIS region H3K9ac peak. For 31% (24% + 7%) of all tested genes, no TIS region acetylation peaks were detected, and it was unclear whether the TIS was correctly annotated. For H3K4me3, the distribution was different, and most genes with a regulated upstream H3K4me3 did not show a TIS region peak. We, therefore, concentrated on H3K9ac and analyzed R-SUPs in more detail. A list of the 748 (576 + 172) genes with R-SUPs is provided in Supplemental Table S1.
Distinct Properties of R-SUPs of H3K9ac and Associated Genes
We first tested whether R-SUPs differed in shape or the degree of regulation from the associated TIS region peaks on the same genes (Fig. 4A). Whereas both peak classes showed an average width of 600 to 700 bp, the peak intensity of R-SUPs was 3 times lower than the intensity of associated TIS region peaks. However, R-SUPs were clearly more regulated between leaf base and blade (factor 5.5× versus 3.1× for the TIS region peaks), suggesting that they might also be important for gene regulation.
Figure 4.

Characterization of R-SUPs and TIS region peaks from R-SUP-associated genes and comparison of ratios from induced to repressed H3K9ac peaks and genes from different groups. A, Mean widths, intensities and fold changes from R-SUPs and TIS region H3K9ac peaks of R-SUP-associated genes were calculated and plotted as indicated. The width is given as base pairs, and the intensities are given as normalized read counts. B, Ratios from induced to repressed H3K9ac peaks (left) and genes (right) from the indicated groups (x axes) were calculated and are plotted on the log10-transformed y axes. Genes and H3K9ac peaks are regulated with a fold change ≥ |2| and an FDR ≤ 0.05.
We furthermore tested if R-SUPs and associated genes showed the same ratio of induction and repression in the blade compared with the majority of H3K9ac peaks and genes (Fig. 4B). Interestingly, the ratio of induced and repressed H3K9ac peaks differed notably between R-SUPs and all H3K9ac peaks or all TIS region peaks. The ratio of induced to repressed H3K9ac peaks in the blade was only 0.7 for all H3K9ac peaks and 0.5 for all TIS region peaks, indicating that more H3K9ac peaks were repressed than induced when comparing leaf base and blade. However, the ratio for R-SUPs was 2.1, indicating that they were more often induced than repressed. Comparable ratios were observed for the transcriptional regulation of associated genes. More genes were down-regulated between leaf blade and base, but for R-SUP genes, we observed more up-regulation.
Histone modifications might facilitate access of transcription factors to DNA on promoters (Hassan et al., 2007; Vermeulen et al., 2007; Lauberth et al., 2013). We, therefore, analyzed whether specific DNA sequences were enriched under R-SUPs. We used the MEME and DREME algorithms of the MEME-ChIP Suite (Machanick and Bailey, 2011) for de novo identification of enriched sequences. These algorithms are insensitive for biases (e.g. caused by the C/G content of a genome, because the nucleotide compositions of the input sequences are used to generate a randomized reference sequence for the statistical enrichment analyses; Bailey and Elkan, 1994; Bailey, 2011). Both algorithms identified highly related sequences as the most overrepresented motifs (Table II). These motifs are most similar to EREs with a (A/C)GCCGCC core sequence (Hart et al., 1993). EREs are binding sites for ethylene response element binding factors (ERFs; also ERE binding proteins) and have so far been mainly associated with biological processes, such as pathogen response (Zhou et al., 1997) and abiotic stress (Gutterson and Reuber, 2004).
Table II. Enrichment of motifs within sequences from R-SUPs.
Genomic sequences covered by R-SUPs were subjected to MEME ChIP for de novo identification of motifs and subsequent alignment of discovered motifs to known binding sites from the JASPAR CORE Plantae database.
| Most Overrepresented Motif | Algorithm | E Value | Transcription Factor Family with the Most Similar Motif |
|---|---|---|---|
 |
MEME | 1.9e-13 | ERF1 |
 |
DREME | 3.3e-4 | ERF1 |
Functional Classification of R-SUP-Associated Genes
To analyze if the occurrence of R-SUPs can be associated with specific processes, we also tested biological processes as defined by MapMan bins (Thimm et al., 2004) for enrichment within R-SUP-associated genes (Fig. 5). To be more specific, we focused on those R-SUP genes that showed the typical expression and acetylation pattern defined in Figure 4B: induction of both transcription and R-SUP acetylation in the leaf blade compared with the base (201 genes; Supplemental Table S2). Six biological processes contained sufficient genes to reliably test for enrichment by χ2 test (expectation ≥ 5). Only the photosynthesis bin showed significant enrichment, with almost 2 times as many R-SUP genes as expected (Fig. 5). This was not caused by a general overrepresentation of photosynthesis genes among genes that were up-regulated in the leaf blade, because enrichment was calculated relative to all genes that were up-regulated in the leaf blade and not relative to all genes independent of their regulation.
Figure 5.
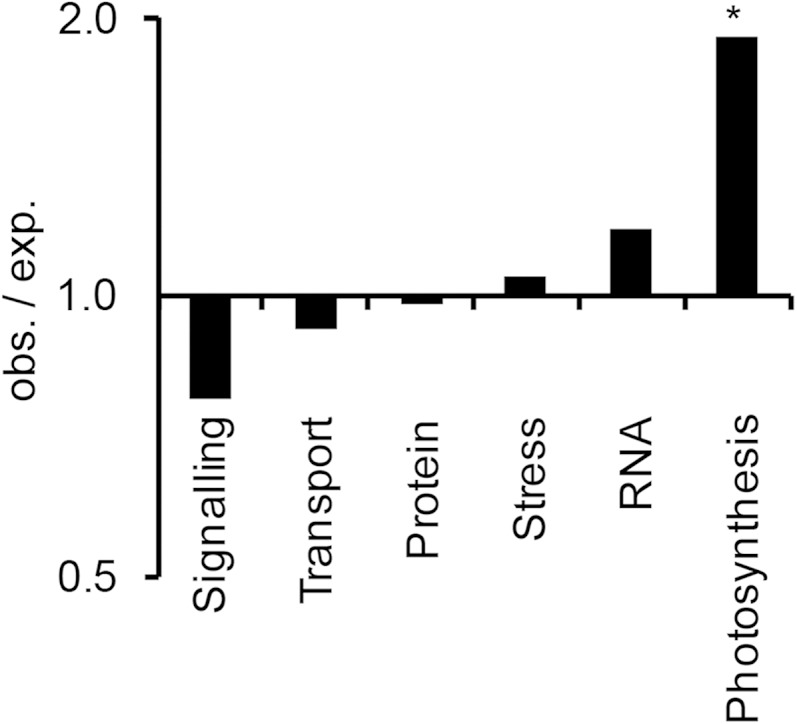
Enrichment analysis of R-SUP-associated genes with induction of both transcription and R-SUP acetylation in the leaf blade compared with the base within processes defined by MapMan bins. All genes that are transcriptionally induced and linked to an induced H3K9ac peak were chosen as reference for the analysis. The degree of enrichment is given as a ratio of expected (exp.) and observed (obs.) counts (log10-transformed y axis). The significances of overrepresentations were evaluated by χ2 test, and derived P values were adjusted using the Benjamini-Hochberg correction. Processes were analyzed only if at least five counts were expected in the tested group. *, FDR ≤ 0.05.
R-SUP-associated genes in the photosynthesis bin are listed in Supplemental Table S2. We found genes from all different subprocesses of photosynthesis, such as light reaction and the Calvin cycle. However, the most striking observation was that five annotated genes encoding for enzymes of the C4 cycle were found in this group: two genes encoding carbonic anhydrases (CAs) and one gene each for phosphoenolpyruvate carboxylase (PEPC), malic enzyme (ME), and pyruvate-inorganic phosphate-dikinase. In addition, a gene encoding phosphoenolpyruvate carboxykinase was on the list of R-SUP-associated genes. This gene was assigned to the MapMan bin gluconeogenesis but has been shown to participate in C4 metabolism in maize (Pick et al., 2011). Thus, in total, six genes for C4 enzymes contained R-SUPs.
We verified this observation for five of the six genes by comparing ChIP-Seq data from this study with previously published chromatin immunoprecipitation-quantitative polymerase chain reaction (ChIP-qPCR) data from our laboratory (Offermann et al., 2008; Heimann et al., 2013). An exemplary profile for ME is shown in Figure 6A, and data for four other genes are shown in Supplemental Figure S2. Red and green dots in Figure 6A represent ChIP-Seq reads, whereas the black line in Figure 6A represents the ChIP-qPCR data. In the blade, beside the TIS region peak, an R-SUP was evident that peaked at around −1,500 relative to the TIS in both ChIP-Seq and ChIP-qPCR data. In this zone, ME transcript levels were highest. At the leaf base, where ME was weakly transcribed, the TIS region peak only slightly changed, whereas the R-SUP completely disappeared. To quantify this effect, we determined H3K9ac fold changes between leaf base and blade for R-SUP and TIS peaks on these C4 genes (Fig. 6B; “Materials and Methods” gives a definition of the R-SUP on one of the CA genes). Whereas fold changes for R-SUPs varied between 9- and 200-fold dependent on the gene, fold changes for TIS region peaks were only between 2- and 10-fold. For each individual C4 gene, regulation of the R-SUP was at least 6-fold stronger than regulation of the TIS region peak. This indicated a strong association of R-SUP regulation and transcriptional regulation for these C4 genes. Another link between R-SUPs and C4 metabolism was provided by our finding that EREs were enriched within the DNA sequences associated with R-SUPs (Table II). Pick et al. (2011) identified transcription factors that were coregulated with prominent C4 genes, and 9 of 18 functionally annotated transcription factors were ERF proteins.
Figure 6.

Regulation of R-SUPs and TIS region peaks on genes encoding C4 enzymes. A, H3K9ac profile on the ME gene at the leaf base and blade. Positions are relative to the TIS in base pairs. Red and green dots represent mapped forward and reverse ChIP-Seq reads. The black line represents previously obtained ChIP-qPCR data from the mature leaf (Heimann et al., 2013). Arrows represent high and low transcript levels. Tx, Transcription. B, H3K9ac fold changes between leaf base and blade for R-SUPs and TIS region peaks on genes encoding C4 enzymes. PEPCK, Phosphenolpyruvate carboxykinase; PPDK, pyruvate-inorganic phosphate-dikinase.
Meta-Analysis of the Connection between Up-Regulated R-SUP Genes and Potential C4 Genes
We hypothesized that R-SUPs were common elements of genes related to C4 photosynthesis and might be involved in coregulation of these genes on the leaf gradient. However, a manual inspection revealed that other C4 genes, such as malate dehydrogenase and small subunit of Rubisco, did not show R-SUPs. We, therefore, screened previously generated lists of candidate genes associated with C4 metabolism based on their expression profile on the leaf developmental gradient or their specificity for M or BS cells. The analysis by Aubry et al. (2014) identified genes that were M or BS specific in both dicotyledoneous (Gynandropsis spp.) and monocotyledoneous (maize) C4 plants. Within this group of 294 genes, 68 genes were transcriptionally up-regulated in the leaf blade and also had an up-regulated H3K9ac peak; 17 of these genes had an up-regulated R-SUP.
This is a significant enrichment of R-SUP genes within the list of potential C4 genes (data not shown; P [χ2] = 0.01). Four of the six R-SUP genes encoding C4 enzymes (Fig. 6) were also found in this comparison. This included PEPC and pyruvate-inorganic phosphate-dikinase but also, both CA genes described above. One of the two CA genes was already described as C4 specific (GRMZM2G121878; Studer et al., 2014). The second CA gene (GRMZM2G414528) was mainly expressed in leaves (Sekhon et al., 2013), and transcripts were highly enriched in M cells compared with BS cells in both maize and Gynandropsis spp. (Aubry et al., 2014). Thus, this gene might contribute to C4-CA activity. This is in line with the minor growth reduction of C4-CA double mutants (Studer et al., 2014), suggesting that some remaining CA activity was present in these double mutants for the formation of C4 cycle substrates.
Another R-SUP-associated gene in the list by Aubry et al. (2014) was a sulfate transporter (GRMZM2G395114), which was specifically expressed in BS cells of maize and Gynandropsis spp. Transcription of this gene was 10-fold higher in the blade compared with the base of the leaf, but H3K9ac at the TIS was unaffected by the transcriptional up-regulation. Instead, we observed a 7-fold up-regulation of the R-SUP that was positioned at −1,600 bp relative to the TIS. This gene might also play a role in C4 photosynthesis, consistent with the confinement of sulfate metabolism to BS cells in C4 plants (Kopriva and Koprivova, 2005).
A BS-specific DNA binding with one finger (DOF) transcription factor (ZmDOF30; AC233935.1_FG005) that had previously been suggested as a candidate C4 regulator also had an up-regulated R-SUP. DOF transcription factors have been shown to control expression of genes involved in carbon metabolism (Yanagisawa, 2000) and specifically, the C4-PEPC gene (Yanagisawa and Sheen, 1998). Both activating and repressive functions have been suggested (Yanagisawa and Sheen, 1998). ZmDOF30 might, therefore, act as an activator of BS-specific gene expression and/or a repressor of M-specific genes in BS cells.
In total, we found 25 transcription factors from the GRASSIUS Database (Yilmaz et al., 2009) that were associated with an up-regulated R-SUP. We, therefore, also compared these transcription factors with a list of transcription factors that were preferentially expressed in M or BS cells in maize (Li et al., 2010). Nine transcription factor genes were found on both lists. Again, an ERF gene was among these candidates (maize ethylene response element binding protein17; GRMZM2G029323). Together with the multiple ERFs identified as candidate C4 regulators by Pick et al. (2011) and the ERE sequences enriched in R-SUP regions (see above), this underlines the potential role of EREs as C4 regulatory elements.
The ZmDOF30 (AC233935.1_FG005) transcription factor was again in the overlap. We furthermore identified two R-SUP-associated genes encoding BS-specific basic leucine zipper (bZIP) transcription factors (GRMZM2G092137 and AC203957.3_FG004). Transcription factors from the bZIP family have been shown to interact with DOF transcription factors (Zhang et al., 1995) and be involved in the regulation of histone acetylation and chromatin structure on maize genes (Casati and Walbot, 2008). Thus, complexes of ZmDOF30 and bZIP factors might be involved in the control of C4 gene expression.
CONCLUSION
During evolution of C4 photosynthesis, C4 genes evolved from C3 ancestors (Brown et al., 2010; Christin et al., 2012) but had to adopt new promoter elements to control higher and more regulated gene expression (Ku et al., 1996; Hibberd and Covshoff, 2010). The comparative analysis of regulated histone modifications in different developmental zones of the maize leaf in this study allowed for the identification of R-SUPs that are distinct from the often-described acetylation peaks in TIS regions. R-SUPs are often associated with EREs and might, therefore, constitute sites for binding of regulatory transcription factors. This binding would be facilitated by the strong acetylation of histones that increases accessibility of DNA in the chromatin context (Dion et al., 2005). Moreover, R-SUPs were identified on many photosynthetic and C4-related genes, suggesting that they might contribute to regulation of these genes. Our data suggest an evolutionary scenario, where parallel recruitment of upstream promoter elements from photosynthetic genes contributed to reprogramming of gene expression during establishment of C4 photosynthesis in maize. Comparative analyses on the chromatin structure of orthologs of C4 promoters in C3 grasses and transgenic studies with promoter deletion constructs will help to test this hypothesis.
MATERIALS AND METHODS
Plant Material and Growth Conditions
Maize (Zea mays) ‘B73’ was grown in growth chambers with a photoperiod of 16 h of light at 25°C and 8 h of dark at 20°C. The photon flux intensity was adjusted to 130 µmol m−2 s−1. Water-soaked seeds were transferred to soil, and plants were harvested when the third leaf was fully expanded (13–14 d after sowing); 4 cm from the middle of the illuminated area of the third leaf were harvested for analysis of the leaf blade. Base samples were isolated from the same leaves. Plants were cut 0.5 cm above soil, and the lowest 1 to 2 cm of the leaf area without light access was harvested.
RNA Isolation and RNA Sequencing
Harvested leaf sections from six plants per biological replicate were pooled and ground in liquid nitrogen. About 50 to 60 mg of ground plant material was dissolved in 1 mL of Trizol and agitated for 15 min; 0.2 volumes of chloroform was added. Samples were agitated for 10 min, and phases were separated by centrifugation (16,100g at 4°C for 15 min); 400 µL of the aqueous phase was transferred to a new reaction tube and washed with 1 volume of chloroform. The RNA was precipitated by addition of 2 volumes of 96% (v/v) ice-cold ethanol, incubation at −20°C for 30 min, and centrifugation (16,100g at 4°C for 15 min). The precipitate was washed with 70% (v/v) ice-cold ethanol and dissolved in 30 µL of RNase-free water. The isolated RNA was treated with the RNase Free DNase Set (79254; Qiagen), purified with the RNeasy MiniElute Cleanup Kit (74204; Qiagen), and eluted two times in 14 µL of RNase-free water. Kits were used according to the manufacturer’s instructions. The integrity of the RNA was analyzed by agarose gel electrophoresis, and the concentration was measured photometrically; 2 µg of RNA per biological replicate was submitted to sequencing on an Illumina HiSeq2000 Instrument with 50-bp single-end reads (GATC Biotech). The service included quality assessment on a 2100 Bioanalyzer, complementary DNA synthesis from fragmented poly(A)+ RNA, and library preparation.
ChIP and ChIP Sequencing
We generated two biological replicates for the analysis of the histone modifications, which is in accordance with the guidelines for ChIP-Seq experiments from the Encyclopedia of DNA Elements and Model Organism Encyclopedia of DNA Elements Consortia (Landt et al., 2012). Harvested leaf sections from 10 plants per biological replicate were pooled, and chromatin was cross linked as described (Horst et al., 2009). Chromatin isolation and immunoprecipitation were performed as described (Heimann et al., 2013), with the exception that protein A agarose was not blocked with salmon sperm DNA. Modifications were detected with 5 µL of antiacetyl H3K9 (07-352; Merck Millipore) or 2.5 µL of anti-H3K4me3 (04-745; Merck Millipore). After washing and reversion of the cross link, samples were treated for 2 h with RNase A (R4642; Sigma-Aldrich) according to the manufacturer’s instructions. The precipitated DNA was purified with the MiniElute PCR Purification Kit (428004; Qiagen). For each biological replicate, eight ChIP reactions were pooled; 10 ng of precipitated DNA was submitted to sequencing on an Illumina HiSeq2000 (for H3K9ac) or Illumina HiSeq2500 (for H3K4me3) Instrument with 50-bp single-end reads (GATC Biotech). The service included quality assessment on a 2100 Bioanalyzer and library preparation.
Processing and Analysis of RNA-Seq Data
Quality of raw sequencing reads was checked with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and reads were processed with the Trimmomatic software (Bolger et al., 2014). The adapter clipper, leading trimmer, trailing trimmer, and sliding window trimmer modules were used with their default settings to trim the raw sequences. Processed sequences were imported into the CLC Genomics Workbench 7.5.1 (http://www.clcbio.com/products/clc-genomics-workbench/; Qiagen). Read mapping was carried out with the RNA-Seq Analysis Tool. Reads were mapped with default settings only to regions that were covered by gene annotations of the maize reference genome (Zea_mays.AGPv3.21.dna.toplevel; ftp://ftp.ensemblgenomes.org/pub/plants/release-21/). Reads that mapped equally to multiple positions were ignored. Raw read counts were chosen as expression values. RPKM values were calculated for each gene to quantify absolute expression (Mortazavi et al., 2008). Statistical evaluation of differentially expressed genes was carried out with the Empirical Analysis of DGE Tool, which implements edgeR analysis (Robinson et al., 2010) and Benjamini-Hochberg correction of P values (Benjamini and Hochberg, 1995). For additional analyses, only genes annotated as protein coding and with an RPKM > 0.1 in at least one of the developmental zones were selected. This threshold was determined based on the coefficient of variation between RPKM values of the biological replicates that strongly increased at RPKM ≤ 0.1.
Processing and Analysis of ChIP-Seq Data
Quality assessment and processing of raw ChIP-Seq sequencing reads were carried out as for RNA-Seq data. Processed reads were imported to the CLC Genomics Workbench. Duplicated reads were removed with the tool Remove Duplicate Reads using default settings. The reads were mapped to the maize reference genome (Zea_mays.AGPv3.21.dna.toplevel; ftp://ftp.ensemblgenomes.org/pub/plants/release-21/) using default parameters with two exceptions: reads were mapped without masking, and reads that mapped equally to multiple positions were ignored. The Peak Shape Tool from the CLC Genomics Workbench was used to call peaks. This tool uses the Hotelling observer to account for variable shapes of peaks (Hotelling, 1931). This approach was successfully used before to call peaks with varying shapes (Kumar et al., 2013). Peaks were initially called with a P value of 0.0001. Afterward, the Advanced Peak Shape Tools were used to optimize peak calling according to the manual. For each sample, a subset of 1,000 called peaks with the highest P values was used as positive regions to define a unique filter with the tool Learn Peak Shape Filter. The number of bins was calculated for each sample as window size per size of bins (35 bp). This optimized filter was used for the definite peak calling with the tool Apply Peak Shape Filter again with a P value of 0.0001.
Differential analysis of regulated peaks between the blade and base of the maize leaf was carried out with the R Bioconductor Package DiffBind following the manual (Stark and Brown, 2011). DiffBind merged overlapping peaks from different samples if peak coordinates overlapped in at least two samples. Reads within the coordinates of merged consensus peaks were counted and normalized for each sample, even if the peak was not called in this sample. Quantitative differences of the normalized reads in the merged peak areas were statistically evaluated using the implemented R Package edgeR (Robinson et al., 2010). Consensus peaks were annotated to genes using the PeakAnalyzer software (Salmon-Divon et al., 2010) with options TSS-Nearest Transcription Start Site and Coding and Non-Coding Genes. If several transcript variants were annotated for one gene, peak distances to the TIS were always calculated for the most 5′-located transcript variant. Only peaks that were located a maximum of 4,000 bp upstream of the TIS or within the transcribed region of the annotated gene were included. As described for RNA-Seq data, the analysis was focused on protein-coding genes with an average RPKM > 0.1. We checked the core C4 genes manually and realized that a strong H3K9ac peak upstream of the CA gene (GRMZM2G121878) was wrongly annotated to a neighboring gene (GRMZM2G122025) that was not expressed (RPKM = 0) and annotated as a low-confidence gene. This peak was reannotated to GRMZM2G121878.
Enrichment Analysis of MapMan Processes
MapMan bins were used to assign genes to processes (Thimm et al., 2004). The χ2 test was used to test for enriched processes. Only processes were taken into account if their expectation in the analyzed group was ≥5. The calculated P values were adjusted with the Benjamini-Hochberg correction to account for multiple testing (Benjamini and Hochberg, 1995).
Motif Identification
De novo identification of motifs was performed with MEME-ChIP (Machanick and Bailey, 2011; http://meme-suite.org). Genomic sequences that were covered by R-SUPs were used as input. MEME-ChIP uses 100 bp from the center of the sequences and performs de novo motif discovery using the algorithms MEME and DREME. Reported motifs were automatically compared with known motifs from the JASPAR CORE Plantae Database (Mathelier et al., 2014) with TOMTOM. MEME-ChIP was used with its default settings, except for limitation of maximum width to 10 nucleotides in MEME.
The original data sets from this article can be found in the National Institutes of Health Gene Expression Omnibus database under the accession number GSE67551.
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. Read mapping statistics of ChIP-Seq and RNA-Seq libraries and comparison of RNA-Seq data from this study with data from Li et al., 2010.
Supplemental Figure S2. H3K9ac profiles on selected C4 genes at the leaf base and blade.
Supplemental Table S1. List of genes with R-SUPs.
Supplemental Table S2. List of R-SUP-associated genes with induction of both transcription and R-SUP acetylation in the leaf blade compared with the base and their functional annotations; additionally, overlaps between these genes and gene lists from two C4-related studies and induced R-SUP genes assigned to the photosynthesis bin are shown.
Supplementary Material
Glossary
- BS
bundle sheath
- CA
carbonic anhydrase
- H3K4me3
trimethylated Lys 4 on histone H3
- H3K9ac
acetylation of H3K9
- M
mesophyll
- ME
malic enzyme
- PEPC
phosphoenolpyruvate carboxylase
- R-SUP
regulated secondary upstream peak
- TIS
transcription initiation site
- ERE
ethylene-responsive element
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (grant no. PE819/5–1) and the European Union FP7 Program (Project 3to4; contract no. 289582).
Articles can be viewed without a subscription.
References
- Aubry S, Kelly S, Kümpers BM, Smith-Unna RD, Hibberd JM (2014) Deep evolutionary comparison of gene expression identifies parallel recruitment of trans-factors in two independent origins of C4 photosynthesis. PLoS Genet 10: e1004365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL. (2011) DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics 27: 1653–1659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TM, Elkan C (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 2: 28–36 [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol 57: 289–300 [Google Scholar]
- Berger SL. (2007) The complex language of chromatin regulation during transcription. Nature 447: 407–412 [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown NJ, Palmer BG, Stanley S, Hajaji H, Janacek SH, Astley HM, Parsley K, Kajala K, Quick WP, Trenkamp S, et al. (2010) C acid decarboxylases required for C photosynthesis are active in the mid-vein of the C species Arabidopsis thaliana, and are important in sugar and amino acid metabolism. Plant J 61: 122–133 [DOI] [PubMed] [Google Scholar]
- Brusslan JA, Bonora G, Rus-Canterbury AM, Tariq F, Jaroszewicz A, Pellegrini M (March 23, 2015) A genome-wide chronological study of gene expression and two histone modifications, H3K4me3 and H3K9ac, during developmental leaf senescence. Plant Physiol 168: 1246–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casati P, Walbot V (2008) Maize lines expressing RNAi to chromatin remodeling factors are similarly hypersensitive to UV-B radiation but exhibit distinct transcriptome responses. Epigenetics 3: 216–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christin PA, Edwards EJ, Besnard G, Boxall SF, Gregory R, Kellogg EA, Hartwell J, Osborne CP (2012) Adaptive evolution of C(4) photosynthesis through recurrent lateral gene transfer. Curr Biol 22: 445–449 [DOI] [PubMed] [Google Scholar]
- Danker T, Dreesen B, Offermann S, Horst I, Peterhänsel C (2008) Developmental information but not promoter activity controls the methylation state of histone H3 lysine 4 on two photosynthetic genes in maize. Plant J 53: 465–474 [DOI] [PubMed] [Google Scholar]
- Dion MF, Altschuler SJ, Wu LF, Rando OJ (2005) Genomic characterization reveals a simple histone H4 acetylation code. Proc Natl Acad Sci USA 102: 5501–5506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Li H, Wei Q, Zhao X, Wang C, Zhu Q, Yi X, Xu W, Liu XS, Jin W, et al. (2013) Genome-wide analysis of histone modifications: H3K4me2, H3K4me3, H3K9ac, and H3K27ac in Oryza sativa L. Japonica. Mol Plant 6: 1463–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutterson N, Reuber TL (2004) Regulation of disease resistance pathways by AP2/ERF transcription factors. Curr Opin Plant Biol 7: 465–471 [DOI] [PubMed] [Google Scholar]
- Hart CM, Nagy F, Meins F Jr (1993) A 61 bp enhancer element of the tobacco β-1,3-glucanase B gene interacts with one or more regulated nuclear proteins. Plant Mol Biol 21: 121–131 [DOI] [PubMed] [Google Scholar]
- Hassan AH, Awad S, Al-Natour Z, Othman S, Mustafa F, Rizvi TA (2007) Selective recognition of acetylated histones by bromodomains in transcriptional co-activators. Biochem J 402: 125–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G, Chen B, Wang X, Li X, Li J, He H, Yang M, Lu L, Qi Y, Wang X, et al. (2013) Conservation and divergence of transcriptomic and epigenomic variation in maize hybrids. Genome Biol 14: R57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimann L, Horst I, Perduns R, Dreesen B, Offermann S, Peterhansel C (2013) A Common histone modification code on C4 genes in maize and its conservation in Sorghum and Setaria italica. Plant Physiol 162: 456–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibberd JM, Covshoff S (2010) The regulation of gene expression required for C4 photosynthesis. Annu Rev Plant Biol 61: 181–207 [DOI] [PubMed] [Google Scholar]
- Horst I, Heimann L, Peterhansel C (2013) Signal integration on plant promoters: a case study in maize. Plant Signal Behav 8: e25389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horst I, Offermann S, Dreesen B, Niessen M, Peterhansel C (2009) Core promoter acetylation is not required for high transcription from the phosphoenolpyruvate carboxylase promoter in maize. Epigenetics Chromatin 2: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotelling H. (1931) The generalization of student’s ratio. Ann Math Stat 2: 360–378 [Google Scholar]
- Hussey SG, Mizrachi E, Groover A, Berger DK, Myburg AA (2015) Genome-wide mapping of histone H3 lysine 4 trimethylation in Eucalyptus grandis developing xylem. BMC Plant Biol 15: 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopriva S, Koprivova A (2005) Sulfate assimilation and glutathione synthesis in C4 plants. Photosynth Res 86: 363–372 [DOI] [PubMed] [Google Scholar]
- Kouzarides T. (2007) Chromatin modifications and their function. Cell 128: 693–705 [DOI] [PubMed] [Google Scholar]
- Ku MS, Kano-Murakami Y, Matsuoka M (1996) Evolution and expression of C4 photosynthesis genes. Plant Physiol 111: 949–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Külahoglu C, Denton AK, Sommer M, Maß J, Schliesky S, Wrobel TJ, Berckmans B, Gongora-Castillo E, Buell CR, Simon R, et al. (2014) Comparative transcriptome atlases reveal altered gene expression modules between two Cleomaceae C3 and C4 plant species. Plant Cell 26: 3243–3260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Muratani M, Rayan NA, Kraus P, Lufkin T, Ng HH, Prabhakar S (2013) Uniform, optimal signal processing of mapped deep-sequencing data. Nat Biotechnol 31: 615–622 [DOI] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al. (2012) ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 22: 1813–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langdale JA. (1998) Cellular differentiation in the leaf. Curr Opin Cell Biol 10: 734–738 [DOI] [PubMed] [Google Scholar]
- Lauberth SM, Nakayama T, Wu X, Ferris AL, Tang Z, Hughes SH, Roeder RG (2013) H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell 152: 1021–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauria M, Rossi V (2011) Epigenetic control of gene regulation in plants. Biochim Biophys Acta 1809: 369–378 [DOI] [PubMed] [Google Scholar]
- Li P, Ponnala L, Gandotra N, Wang L, Si Y, Tausta SL, Kebrom TH, Provart N, Patel R, Myers CR, et al. (2010) The developmental dynamics of the maize leaf transcriptome. Nat Genet 42: 1060–1067 [DOI] [PubMed] [Google Scholar]
- Machanick P, Bailey TL (2011) MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27: 1696–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeran W, Cai Y, Sun Q, van Wijk KJ (2005) Functional differentiation of bundle sheath and mesophyll maize chloroplasts determined by comparative proteomics. Plant Cell 17: 3111–3140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeran W, Friso G, Ponnala L, Connolly B, Huang M, Reidel E, Zhang C, Asakura Y, Bhuiyan NH, Sun Q, et al. (2010) Structural and metabolic transitions of C4 leaf development and differentiation defined by microscopy and quantitative proteomics in maize. Plant Cell 22: 3509–3542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley-Hunt R, Arenillas DJ, Buchman S, Chen CY, Chou A, Ienasescu H, et al. (2014) JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res 42: D142–D147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621–628 [DOI] [PubMed] [Google Scholar]
- Nelissen H, Rymen B, Jikumaru Y, Demuynck K, Van Lijsebettens M, Kamiya Y, Inzé D, Beemster GT (2012) A local maximum in gibberellin levels regulates maize leaf growth by spatial control of cell division. Curr Biol 22: 1183–1187 [DOI] [PubMed] [Google Scholar]
- Offermann S, Danker T, Dreymüller D, Kalamajka R, Töpsch S, Weyand K, Peterhänsel C (2006) Illumination is necessary and sufficient to induce histone acetylation independent of transcriptional activity at the C4-specific phosphoenolpyruvate carboxylase promoter in maize. Plant Physiol 141: 1078–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offermann S, Dreesen B, Horst I, Danker T, Jaskiewicz M, Peterhansel C (2008) Developmental and environmental signals induce distinct histone acetylation profiles on distal and proximal promoter elements of the C4-Pepc gene in maize. Genetics 179: 1891–1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papait R, Cattaneo P, Kunderfranco P, Greco C, Carullo P, Guffanti A, Vigano V, Stirparo GG, Latronico MV, Hasenfuss G, et al. (2013) Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc Natl Acad Sci USA 110: 20164–20169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pick TR, Bräutigam A, Schlüter U, Denton AK, Colmsee C, Scholz U, Fahnenstich H, Pieruschka R, Rascher U, Sonnewald U, et al. (2011) Systems analysis of a maize leaf developmental gradient redefines the current C4 model and provides candidates for regulation. Plant Cell 23: 4208–4220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. (2012) Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481: 389–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roudier F, Ahmed I, Bérard C, Sarazin A, Mary-Huard T, Cortijo S, Bouyer D, Caillieux E, Duvernois-Berthet E, Al-Shikhley L, et al. (2011) Integrative epigenomic mapping defines four main chromatin states in Arabidopsis. EMBO J 30: 1928–1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon-Divon M, Dvinge H, Tammoja K, Bertone P (2010) PeakAnalyzer: genome-wide annotation of chromatin binding and modification loci. BMC Bioinformatics 11: 415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekhon RS, Briskine R, Hirsch CN, Myers CL, Springer NM, Buell CR, de Leon N, Kaeppler SM (2013) Maize gene atlas developed by RNA sequencing and comparative evaluation of transcriptomes based on RNA sequencing and microarrays. PLoS One 8: e61005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Angel A, Howard M, Dean C (2012) Vernalization - a cold-induced epigenetic switch. J Cell Sci 125: 3723–3731 [DOI] [PubMed] [Google Scholar]
- Stark R, Brown G (2011) DiffBind: differential binding analysis of ChIP-Seq peak data. http://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf (March 2, 2015)
- Studer AJ, Gandin A, Kolbe AR, Wang L, Cousins AB, Brutnell TP (2014) A limited role for carbonic anhydrase in C4 photosynthesis as revealed by a ca1ca2 double mutant in maize. Plant Physiol 165: 608–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tausta SL, Li P, Si Y, Gandotra N, Liu P, Sun Q, Brutnell TP, Nelson T (2014) Developmental dynamics of Kranz cell transcriptional specificity in maize leaf reveals early onset of C4-related processes. J Exp Bot 65: 3543–3555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, Selbig J, Müller LA, Rhee SY, Stitt M (2004) MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J 37: 914–939 [DOI] [PubMed] [Google Scholar]
- To TK, Kim JM (2014) Epigenetic regulation of gene responsiveness in Arabidopsis. Front Plant Sci 4: 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner BM. (2002) Cellular memory and the histone code. Cell 111: 285–291 [DOI] [PubMed] [Google Scholar]
- Vermeulen M, Mulder KW, Denissov S, Pijnappel WW, van Schaik FM, Varier RA, Baltissen MP, Stunnenberg HG, Mann M, Timmers HT (2007) Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 131: 58–69 [DOI] [PubMed] [Google Scholar]
- Wang L, Czedik-Eysenberg A, Mertz RA, Si Y, Tohge T, Nunes-Nesi A, Arrivault S, Dedow LK, Bryant DW, Zhou W, et al. (2014) Comparative analyses of C₄ and C₃ photosynthesis in developing leaves of maize and rice. Nat Biotechnol 32: 1158–1165 [DOI] [PubMed] [Google Scholar]
- Wang X, Elling AA, Li X, Li N, Peng Z, He G, Sun H, Qi Y, Liu XS, Deng XW (2009) Genome-wide and organ-specific landscapes of epigenetic modifications and their relationships to mRNA and small RNA transcriptomes in maize. Plant Cell 21: 1053–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa S. (2000) Dof1 and Dof2 transcription factors are associated with expression of multiple genes involved in carbon metabolism in maize. Plant J 21: 281–288 [DOI] [PubMed] [Google Scholar]
- Yanagisawa S, Sheen J (1998) Involvement of maize Dof zinc finger proteins in tissue-specific and light-regulated gene expression. Plant Cell 10: 75–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz A, Nishiyama MY Jr, Fuentes BG, Souza GM, Janies D, Gray J, Grotewold E (2009) GRASSIUS: a platform for comparative regulatory genomics across the grasses. Plant Physiol 149: 171–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Chen W, Foley RC, Büttner M, Singh KB (1995) Interactions between distinct types of DNA binding proteins enhance binding to ocs element promoter sequences. Plant Cell 7: 2241–2252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. (2008) The epigenetic landscape of plants. Science 320: 489–492 [DOI] [PubMed] [Google Scholar]
- Zhou J, Tang X, Martin GB (1997) The Pto kinase conferring resistance to tomato bacterial speck disease interacts with proteins that bind a cis-element of pathogenesis-related genes. EMBO J 16: 3207–3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.