

A method is presented to detect peptide bonds that need either a trans–cis flip or a peptide-plane flip.
Keywords: peptide conformation, cis peptide bond, structure validation, structure correction
Abstract
A coordinate-based method is presented to detect peptide bonds that need correction either by a peptide-plane flip or by a trans–cis inversion of the peptide bond. When applied to the whole Protein Data Bank, the method predicts 4617 trans–cis flips and many thousands of hitherto unknown peptide-plane flips. A few examples are highlighted for which a correction of the peptide-plane geometry leads to a correction of the understanding of the structure–function relation. All data, including 1088 manually validated cases, are freely available and the method is available from a web server, a web-service interface and through WHAT_CHECK.
1. Introduction
Peptide bonds connect adjacent amino acids in proteins. The partial double-bond character of the peptide bond restricts its torsion. The dihedral angle ω (Cα i−1—Ci−1—Ni—Cα i) typically has values around 180° (trans) or 0° (cis), although exceptions are possible (Berkholz et al., 2012 ▸). The Cα i–1—Cα i distance is around 3.81 Å in the trans conformation and around 2.94 Å in the cis conformation. The trans conformation is energetically preferred over the cis conformation owing to unfavourable nonbonded interactions between the two Cα—Hα moieties flanking the peptide bond (Zimmerman & Scheraga, 1976 ▸; Stewart et al., 1990 ▸ and references therein; Jabs et al., 1999 ▸ and references therein). The cis imide bond X—Pro is more frequently observed than the cis amide bond X—Xnp (where Xnp is any residue except Pro) because the Cα i−1 and Oi−1 atoms have similar third neighbours in either X—Pro conformation (Ramachandran & Sasisekharan, 1968 ▸).
In the early days of protein crystallography cis peptides were almost completely absent in the available protein crystal structures (Ramachandran & Mitra, 1976 ▸). Therefore, characterization of cis-peptide geometry in protein crystal structures was very difficult. In the early 1990s the number of structures deposited in the PDB allowed the first studies of the local protein environment of cis peptides. Stewart et al. (1990 ▸) characterized 17 cis X—Xnp and 99 cis X—Pro peptides and MacArthur & Thornton (1991 ▸) analysed 58 cis X—Pro residues in a set of nonhomologous structures. Many surveys showed that the computationally derived energy differences between the cis and trans isomers are only partially reflected in their frequency of occurrence in the PDB. Jabs and coworkers reviewed the arguments for the rarer than expected occurrence of cis peptide bonds in protein structures (Jabs et al., 1999 ▸). These arguments were mainly related to protein energetics and to protein function, as several studies had observed cis peptides at functionally important locations such as active sites or protein binding interfaces (Stoddard & Pietrokovski, 1998 ▸ and references therein; Weiss et al., 1998 ▸ and references therein). Several studies showed a correlation between the resolution of the crystal structure and the number of cis peptides (Stewart et al., 1990 ▸; Weiss et al., 1998 ▸; Pal & Chakrabarti, 1999 ▸). Several authors, including Jabs and coworkers, have noted that the underrepresentation of cis peptides is partly the result of the a priori assumption often made upon determining X-ray crystal structures that all peptides have a trans conformation (Huber & Steigemann, 1974 ▸; Stewart et al., 1990 ▸; Weiss et al., 1998 ▸; Jabs et al., 1999 ▸). In the 1970s it had already been noted that reinterpretation of electron-density maps might reveal many previously unnoticed cis peptides in protein structures (Huber & Steigemann, 1974 ▸; Ramachandran & Mitra, 1976 ▸).
An incorrectly assigned peptide conformation can be in need of any of a number of possible corrections. The first and most common is a rotation of the entire peptide plane by 180°, referred to as an amide flip (McCammon et al., 1977 ▸), peptide-plane flip (Hayward, 2001 ▸) or peptide flip (Joosten et al., 2011 ▸). In the following these peptide-plane flips are called tt+ [Fig. 1 ▸; the first two symbols are either c for cis or t for trans, reflecting the situation before and after the flip, respectively; the third symbol is a plus (+) sign if the conformational change includes a flip of the backbone carbonyl C=O, otherwise it is a minus (−) sign]. The other two corrections are trans↔cis flips that can either constitute a flip of the backbone C=O (tc+ or ct+) or a flip of the backbone amide N—H (tc− or ct−). A correct peptide is either tt− or cc−. Every type of flip was observed in crystal structures deposited in the Protein Data Bank (PDB; Berman et al., 2007 ▸) except cc+ (cis-peptide flips that include a flip of the C=O). Fig. 1 ▸ shows representative examples for the five possible codes that include a flip and that have actually been observed in the PDB. More examples can be found on the associated website (http://swift.cmbi.ru.nl/gv/flips/).
Figure 1.

Representative peptide flips. The left figures show the peptide conformation found in the PDB (pink structures) and electron-density maps obtained from the Uppsala Electron Density Server (Kleywegt et al., 2004 ▸). The figures on the right show the conformation and electron density of the corresponding re-refined structure models (green) from the PDB_REDO databank (Joosten et al., 2011 ▸). The flip type is indicated (tt− and cc− are not flips and thus are not illustrated). A cc+ example could not be found in the PDB. (a) Gly90, chain A, PDB entry 3hr7 (Cheng et al., 2012 ▸), 1.8 Å resolution. (b) Glu175, chain C, PDB entry 3k2g, 1.8 Å resolution. (c) Gln541, chain A, PDB entry 1w0o (Moustafa et al., 2004 ▸), 1.9 Å resolution. (d) Ala82, chain A, PDB entry 1v6i (Kundhavai Natchiar et al., 2004 ▸), 2.15 Å resolution. (e) Pro55, chain B, PDB entry 1j1j (Sugiura et al., 2004 ▸), 2.2 Å resolution. The PDB_REDO program pepflip (Joosten et al., 2011 ▸) performs tt+ peptide-plane flips, which are a combination of both a C=O flip and an N—H flip. Re-refinement alone can lead to N—H flips (tc− and ct− flips) when the signal in the X-ray data is strong enough. Therefore, the net result of a tt+ flip and subsequent refinement may become a tc+ flip. Consequently, PDB_REDO is able to correct many (but certainly not all) peptides in need of a trans–cis flip. The 2mFo − DF c (grey mesh) and mFo − DF c maps (+, green mesh; −, red mesh) have been contoured at 1.5σ and ±3σ, respectively, and have been rendered with a grid size of 0.2 Å for visualization purposes. The figures were prepared with CCP4mg (McNicholas et al., 2011 ▸).
Frömmel & Preissner (1990 ▸) were the first to predict cis X—Pro peptides based on just the amino-acid sequence. Later, the prediction algorithms were expanded to take into account X—Xnp peptides (Pahlke et al., 2005 ▸; Exarchos et al., 2009 ▸). Machine-learning approaches have been applied that included multiple sequence alignments, predicted secondary structure and predicted solvent accessibility (Wang et al., 2004 ▸; Song et al., 2006 ▸; Exarchos et al., 2009 ▸). These sequence-based algorithms correctly predict the conformation of about three quarters of the tested peptides. A detailed comparison of these algorithms is impossible since the only software still available today is that of Song et al. (2006 ▸). Sequence-based algorithms have probably become obsolete because much higher prediction accuracy is required in the everyday practice of crystallographers.
Jabs and coworkers were the first to discuss geometric aspects that could be used to detect incorrectly modelled X—Xnp tc− flips (Jabs et al., 1999 ▸). Their algorithm was the first coordinate-based flip-prediction method that took into account the locally distorted geometrical environment of a misassigned peptide bond. Initially, the algorithm was based on just four cis peptides in coagulation factor XIII (Jabs et al., 1999 ▸). This method was implemented in WHAT IF but suffered from many false positives, i.e. incorrectly predicted trans-to-cis flips. Weiss and Hilgenfeld (WH) later refined their algorithm using a set of 17 incorrectly assigned trans peptides (Weiss & Hilgenfeld, 1999 ▸). This method was implemented for comparison purposes, but it was found to give many false-negative predictions, i.e. it overlooked necessary tc− flips (see Table 5). The WH method was designed for X—Xnp peptides only. A structure-based algorithm predicting trans-to-cis flips in X—Pro peptides does not yet exist. Cis-to-trans flips are much rarer than trans-to-cis flips, mostly as a consequence of the a priori assumption that peptides are in the trans isomer. Cis-to-trans flips have not been predicted by any algorithm known to date.
With the introduction of PDB_REDO (Joosten & Vriend, 2007 ▸; Joosten et al., 2009 ▸), it became possible to reinterpret experimental X-ray data in an automated way, and when Joosten and coworkers developed a method called pepflip, peptide-plane tt+ flips could be detected and corrected based on the fit to the local electron density (Joosten et al., 2011 ▸). It should be noted, however, that pepflip does not perform trans↔cis flips, and occasionally performs a tt+ flip when actually a trans–cis flip is needed.
All crystallographic PDB structures were compared with their PDB_REDO counterparts and many examples were observed of the different flip types in Fig. 1 ▸. With the large set of peptide flips in hand, we asked whether these flips could be used to obtain a large training set for a Random Forest (RF; Breiman, 2001 ▸) machine-learning approach for the structure-based prediction of peptide-plane inversions. The method predicts 70 461 peptide-plane flips and 4617 trans–cis flips in the PDB.
2. Methods
2.1. Data selection
Pairs of X-ray structures were obtained from the PDB and PDB_REDO releases of 20 October 2014 and were used only if they met the selection criteria listed in Table 1 ▸.
Table 1. Selection criteria for PDB entries.
| Selection parameter | Criterion |
|---|---|
| Experimental method | X-ray |
| Resolution | 3.5 or better |
| PDB_REDO entry | Must exist |
| DSSP entry | Must be determinable |
| BDB entry | Must exist |
| Composition | At least one chain with 25 amino acids |
| Flip (tt+, tc, tc+, cc+, ct or ct+) | At least one detected by WHAT IF |
From these PDB files, stretches of four residues were selected if they met the selection criteria listed in Table 2 ▸.
Table 2. Selection criteria for tetrapeptides.
| Selection parameter for residues | Criterion |
|---|---|
| Position in structure | Not a C-terminus or an N-terminus; not adjacent to a chain break |
| Amino-acid type | Must be canonical |
| Angles, dihedrals, improper dihedrals | Must be determinable |
| Atoms | All must be present; all B factors > 02; all occupancies = 1.0 |
| Covalently bound atoms | Only canonical bonds and no other bonds, not even disulfide bonds |
| Anything outside own molecule | Not within 2.5 of O atom in central peptide plane |
The tetrapeptides in the data set were divided into X-X-Pro-X, X-X-Gly-X and X-X-Xnpg-X (where Xnpg is any residue except for Pro or Gly), which, for brevity, are called X-Pro, X-Gly and X-Xnpg, respectively.
A large number of tetrapeptides were manually validated. These tetrapeptides represented both correct and incorrect conformations. 173 peptides in 81 PDB_REDO files were rebuilt and re-refined because visual inspection suggested that the PDB_REDO conformation was not plausible (see, for example, Figs. 2 and 4). This resulted in a validation data set consisting of 1088 tetrapeptides (see Table 3 ▸) in 438 PDB structures, 192 of which contained at least one genuine flip. Many more peptides were inspected, but were not included because the quality of the electron density was not good enough. The validation data were gathered over the course of this study. Many tetrapeptides were expected to be difficult for an automated method to predict correctly. These difficult cases were deliberately added to the validation set and included not only incorrect tetrapeptides but also correct ones. About 400 cases were included that had been incorrectly classified by earlier versions of the classification algorithm developed here or by the first Jabs, Weiss and Hilgenfeld algorithm.
Table 3. Peptide-conformation data.
For each peptide class three rows are given. The first row shows the counting statistics for peptide-conformation differences between PDB_REDO and PDB in the 16688 structure pairs that share at least one trans cis difference or a peptide-plane flip. The flip types are illustrated in Fig. 1 ▸. The second row shows the subset of these cases that has been used to train Random Forest classifiers. The tt cases were needed to teach the method what correct (trans) peptide planes look like. The third row shows the independent cases used to test the method. The 1088 cases in the test set of 438 structures have been validated manually and were corrected and re-refined when necessary. The test cases have been derived from PDB_REDOPDB comparison (the test cases are not included in the first row) or were otherwise detected over the course of this study. Entries in bold indicate that WHAT_CHECK can now validate cases that fall into this category; for the other classes insufficient data are available for proper training and testing. The tc cases for X-Pro were solved with a very simple, manually designed decision tree, as explained in the text.
| Peptide class | tt | tt+ | tc | tc+ | cc | cc+ | ct | ct+ | |
|---|---|---|---|---|---|---|---|---|---|
| X-Xnpg | Found | 13875524 | 24742 | 176 | 0 | 8001 | 0 | 0 | 0 |
| Train | 4307 | 4131 † | 176 | 0 | 0 | 0 | 0 | 0 | |
| Test | 435 | 65 | 122 | 12 | 6 | 0 | 21 | 3 | |
| X-Pro | Found | 696375 | 0 | dt | 88 | 33236 | 0 | 0 | 0 |
| Train | 88 | 0 | dt | 88 | 0 | 0 | 0 | 0 | |
| Test | 90 | 1 | dt | 69 | 74 | 0 | 3 | 13‡ | |
| X-Gly | Found | 1141604 | 11869 | 0 | 0 | 2329 | 0 | 0 | 0 |
| Train | 1049 | 1049 § | 0 | 0 | 0 | 0 | 0 | 0 | |
| Test | 77 | 31 | 7¶ | 0 | 0 | 0 | 4 | 0 |
Training and testing examples were only taken from structures solved at 2.2 resolution or better.
Training and testing examples were only taken from structures solved at better than 2.0 resolution.
Six occurrences in different chains of PDB entry 2ef5.
A special menu was added to WHAT IF (Vriend, 1990 ▸) that compares PDB entries with their PDB_REDO mates. Options in this menu allow the detection of many differences in coordinates, angles, torsion angles, B factors etc. between PDB and PDB_REDO pairs. The WHAT IF procedure that compares the peptide conformations in the ∼71 000 PDB–PDB_REDO pairs of protein structures and that assigns the flip types was based on three variables describing the difference between the central peptide planes in the corresponding tetrapeptides: the (C=O, C=O) angle, the (N—H, N—H) angle and the ω torsion-angle difference. The training examples were taken from structures solved at 3.5 Å resolution or better, except for the examples for peptide-plane flips because validation of flip assignment and prediction was found to be more accurate when only structures solved at 2.2 Å resolution or better were included. In total, at least one clearly flipped peptide was observed in 16 688 PDB_REDO entries.
Structures were rebuilt manually with Coot (Emsley et al., 2010 ▸) and re-refined with REFMAC (Murshudov et al., 2011 ▸). The refinement strategy and parameters were obtained from the PDB_REDO protocol (Joosten et al., 2012 ▸). The CCP4 (Winn et al., 2011 ▸) program EDSTATS (Tickle, 2012 ▸) was used to calculate real-space correlation coefficients.
2.2. Prediction
For each tetrapeptide a large number of features was calculated using WHAT IF, including Cα—Cα distances, Cβ—Cβ distances, O—O distances, backbone torsion angles, backbone bond lengths, backbone bond angles up to Cβ atoms, chiral volumes, C—O—C—O angles, the Oi−1 bump score and the carbonyl alignment with an α-helix nearby in the sequence, three-state secondary structure as derived from DSSP (Kabsch & Sander, 1983 ▸; Touw et al., 2015 ▸) and B factors from BDB entries (Touw & Vriend, 2014 ▸), which consistently have full isotropic B factors, unlike PDB entries that can have residual B factors from TLS (Schomaker & Trueblood, 1968 ▸) refinement. The WH method was implemented in WHAT IF as described by Weiss & Hilgenfeld (1999 ▸). The WH ‘penalty-function score’ (D tot) is also one of its features. Random Forest (Breiman, 2001 ▸) classifiers were constructed using the R (R Core Team, 2015 ▸) package randomForest (Liaw & Wiener, 2002 ▸) and tuned using repeated fivefold cross-validation. The classifier objects were automatically converted into Fortran code for inclusion in WHAT IF and WHAT_CHECK (Hooft et al., 1996 ▸).
3. Results
3.1. Peptide-plane inversion examples
Thousands of peptide-plane inversions were observed by comparing PDB structures with their PDB_REDO counterparts (Table 3 ▸).
Visual inspection of many peptide-plane inversions indicated that about 90% of the flips introduced by PDB_REDO are correct. Sometimes the PDB_REDO peptide conformation is suboptimal. In some of the tc− cases, for example, the ω angle can end up at around 90° in the PDB_REDO output model (Fig. 2 ▸). These conformations, which are essentially halfway between the wrong and the right conformation, are the result of trans-peptide restraints outweighing the crystallographic data during refinement. The problems can be resolved by additional refinement with cis-peptide restraints.
Figure 2.
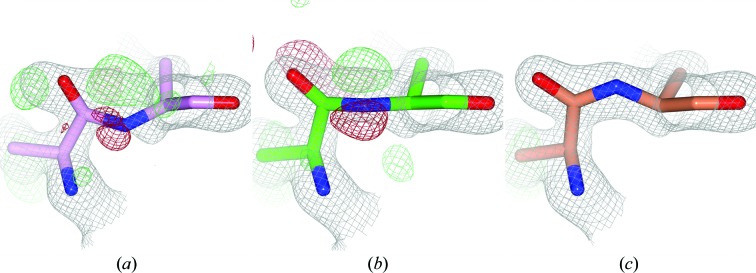
Stepwise improvements from trans to cis. (a) The peptide bond between Arg71 and Phe72 (ω = 132°) in PDB entry 2bmd (Huber & Scheidig, 2005 ▸) needs a tc− flip. (b) The peptide has been flipped only ‘halfway’ in PDB_REDO (ω = 81°). (c) The fully flipped and refined cis peptide (ω = 9°). The side chains have been omitted beyond Cβ for clarity. Maps are as in Fig. 1 ▸.
3.2. Prediction of peptide flips
The studies by Weiss & Hilgenfeld (1999 ▸), and our own visual inspection of hundreds of peptide planes that needed a flip to better agree with the X-ray data, revealed that a number of geometric variables tend to deviate from their common values when a peptide plane has been built in the wrong conformation. For instance, the angle Cα i−1—Ci−1—Ni tends to be smaller than normal for X-Xnpg tc− peptides, and the B factor of the O atom in the plane tends to be high if the peptide plane needs a tt+ flip. Therefore, all features were collected that could possibly characterize the local distortion of an incorrectly modelled peptide plane. These features were not limited to geometric variables and B factors, but also included secondary structure and a description of the environment of the O atom in the peptide plane. Other variables such as the hydrogen-bonding status and rotamericity of the side chains can be added, but they are computationally intensive and, for reasons that we do not yet fully understand, do not influence the prediction accuracy of the method very much. A comprehensive list of variables is available on the project’s website. For each flip type a classifier was trained to determine variable combinations that can separate peptides in need of a flip from correct peptides. The flip-type specific classifiers were combined into one classifier per residue class (X-Xnpg, X-Pro and X-Gly). All classifiers were validated using an independent test set. Table 3 ▸ shows the number of peptides in the training test sets. Classifiers for cis-to-trans flips and other small categories were not constructed because classifiers fitted to too few training examples will not be generally applicable. The full details of the design, implementation and use of the classifiers for the four situations for which adequate data was available and the manual decision tree for X-Pro tc− are given on the project’s website.
Table 4 ▸ lists the results for the four residue and flip-type specific RF classifiers. The combined classifiers predict X-Xnpg flip types (tt−, tt+, tc−, tc+) with an accuracy of 93%. This includes all 12 tc+ cases that were found in the PDB and that were not in the training set. The accuracy is 95% without these X-Xnpg tc+ cases. X-Pro tt−, tt+, tc− and tc+ flips in the test set can be classified with an overall accuracy of 93%.
Table 4. Test-set performance.
The performance on the test set is shown for the four RF classifiers. The performances of the WH method with the original threshold and with the threshold determined in the present study, respectively, are shown in parentheses. Note that the classification accuracy is sensitive to class imbalance, while the area under the receiver operating characteristic curve (AUC) and the Matthews correlation coefficient (MCC; Matthews, 1975 ▸) are not. The values for several other performance metrics and the confusion tables for the combined predictions can be found on the project’s website.
| X-Xnpg | X-Pro | X-Gly | ||
|---|---|---|---|---|
| tt+ | tc | tc+ | tt+ | |
| AUC | 0.99 | 0.98 (0.97/0.97) | 0.94 | 0.98 |
| MCC | 0.91 | 0.89 (0.31/0.82) | 0.85 | 0.93 |
| Accuracy | 0.98 | 0.96 (0.80/0.94) | 0.92 | 0.97 |
3.2.1. X-Xnpg tc−
Table 5 ▸ shows the prediction outcome in terms of true positives (TP), true negatives (TN), false positives (FP) and false negatives (FN) for the X-Xnpg tc− and tt− test peptides. The confusion tables for the results obtained with the WH method are also shown. Weiss & Hilgenfeld (1999 ▸) mentioned that their cutoff could not be validated, as the experimental data for most of the structures in their data set were not available. The optimal cutoff for the WH D tot score could be determined using the 539 cases that were manually validated using electron density. With the new threshold both the TP and FN rates improved more than 75% compared with the original WH threshold (Table 5 ▸). The RF-based method further increased this performance by decreasing the FP rate at the cost of a small increase in the FN rate. Note that a low FP rate is more important for protein structure-validation purposes than for prediction-assisted rebuilding and re-refinement.
Table 5. X-Xnpg test-set predictions.
The rows give the true class and the columns give the predicted class. RF, the method developed in this study (MCC = 0.89). WH, the method developed by Weiss Hilgenfeld (1999 ▸) with a D tot score threshold of 143.10 (MCC = 0.31). WH, WH with a redetermined D tot cutoff of 82.256 (MCC = 0.82).
| RF | WH | WH | ||||
|---|---|---|---|---|---|---|
| tc | tt | tc | tt | tc | tt | |
| tc | 107 | 14 | 15 | 106 | 110 | 11 |
| tt | 6 | 412 | 0 | 418 | 24 | 394 |
The variables that were most important for separating X-Xnpg tc− from tt− were also used in the WH algorithm: ϕi, the backbone angles Oi−1—Ci−1—Ni, Cα i−1—Ci−1—Ni−1, Cα i−1—Ci−1—Oi−1 and Ci−1—Ni—Cα i, and the Cα i−1—Cα i distance. In addition, the Cα i—Ci—Ni angle, the Cα i chiral volume and the Cβ i−1—Cβ i distance were found to be important for the RF method. Other WH bond lengths and angles were found to be less important. The full list of variables and their importance can be found on the associated website. In general, and not unexpectedly, the variables extracted from the inner two residues in tetrapeptides contributed most to the prediction accuracy of all flip types.
Application of the X-Xnpg tc− method to X-Gly cases did not reveal any new X-Gly tc− flips. This result suggests that the method might not be able to detect X-Gly tc− flips; after all, it was trained only on X-Xnpg tetrapeptides. Another explanation is that X-Gly tc− flips are simply very rare. This explanation is supported by the observation that a cis peptide modelled in the trans conformation is more easily corrected to the cis conformation automatically during refinement when the residue type is Gly rather than any other type.
3.2.2. X-Pro tc+
X-Pro tc+ cases in the test set could be classified without any FP. Important variables are the angle between the carbonyl of the central peptide bond and the carbonyl before that, the Ci−1—Ni—Cα i and Ni−1—Cα i−1—Ci−1 angles, ψi−1, the Cα i−1—Cα i distance and the Oi−1 bump score. Engh & Huber (2001 ▸) observed that the bimodal distribution of Ci−1—Ni—Cα i in high-resolution peptide fragments was caused by differences between the cis and trans forms. The median Ci−1—Ni—Cα i angle for tc+ cases in the test set (116.0°) was smaller than the median for tt− cases in the PDB (121.0°) and the value Engh & Huber (2001 ▸) reported for trans proline (119.3 ± 1.5°), but after re-refinement the median (129°) was just above the value that Engh and Huber reported for cis proline (127.0 ± 2.4°). This is illustrated in Fig. 3 ▸. The figures on the website show the change in all variables before and after correction and re-refinement of the tetrapeptides in the test set.
Figure 3.

The Ci−1—Ni—Cα i angle before and after correction of X-Pro tc+ cases. The curved lines show Gaussian kernel density estimates for the Ci−1—Ni—Cα i backbone angle for 25 X-Pro tc+ cases in the test set before (solid line) and after (dashed line) correction and re-refinement. The vertical lines show the values for trans-Pro (119.3 ± 1.5°) and cis-Pro (127.0 ± 2.4°) reported by Engh & Huber (2001 ▸).
3.2.3. X-Xnpg tt+
The 480 X-Xnpg tt+ cases in the test set could be classified with seven FP and three FN. One FN and one FP are next to a trans–cis flip. The eight X-Xnpg cases in the test set that PDB_REDO failed to flip were correctly predicted by the RF method. The most important variables for predicting X-Xnpg tt+ cases are the B factors of the O and C atoms in the central peptide plane, ϕ, ψ, the secondary structure and the Cβ i−1—Cβ i distance.
3.2.4. X-Gly tt+
The B factor of the central O atom is also very important for X-Gly classification, as are the Ci−1 B factor, the Ci−1—Ni—Cα i and Ni−1—Cα i−1—Ci−1 angles and the secondary structure of residue i − 1. Gunasekaran et al. (1998 ▸) studied in vivo conversion between type I and type II β-turns and between type I′ and type II′ β-turns. They reported the importance of the B-factor distribution of the central O atom in flippable β-turns (Gunasekaran et al., 1998 ▸). The best classification results were obtained for X-Gly (two FP and one FN) when the tt+ classifier was trained with data from structures solved at a resolution better than 2 Å, probably because the backbone is generally less well defined in low-resolution structures, resulting in higher B factors caused by the low resolution rather than by a peptide in need of a flip. The inherent mobility of Gly may also explain the fact that many surface-located X-Gly were found that could not be interpreted well because of electron density that was too poor.
3.2.5. X-Pro tc−
All 40 X-Pro tc− in the test set derived from the PDB–PDB_REDO comparison had a positive ϕi, while the ϕi for tt− and tc+ cases is always around −60°. Remarkably, in the entire set of crystal structures with deposited structure factors 904 X-Pro cases were found with a positive ϕi, 86 of which were in structures solved at a resolution of between 1.2 and 2.0 Å. These 904 cases all were either X-Pro tc− flips (Fig. 4 ▸ a) or trans X-Pro with an otherwise incorrect nitrogen chirality (‘NCh’).
Figure 4.
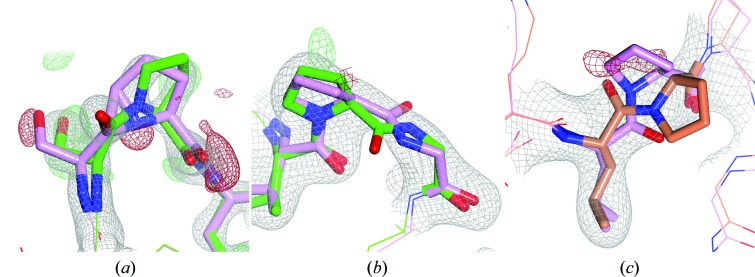
X-Pro problems characterized by positive ϕi angles. (a) X-Pro tc− flip; Ser339-Pro340, chain A, PDB entry 1se6 (Zhao et al., 2005 ▸), 1.75 Å resolution. (b) Pro-Gly tt+ flip; Val53-Pro54-Gly55, chain A, PDB entry 1hxd (Weaver et al., 2001 ▸), 2.40 Å resolution. (c) X-Pro tt+ flip; Leu203-Pro204, chain B, PDB entry 1cdd (Almassy et al., 1992 ▸), 2.80 Å resolution. Electron-density maps were calculated using the PDB structure and are rendered as in Fig. 1 ▸. The PDB structures are shown in pink and the PDB_REDO structures in green; the manually corrected and re-refined structure is shown in orange.
The ‘NCh’ class includes tetrapeptides where residue i + 1 needs a tc+ flip (e.g. His173-Leu174-Pro175-Pro176 in PDB entries 1bug and 1bt2; Klabunde et al., 1998 ▸) or a tt+ flip [e.g. Thr52-Val53-Pro54-Gly55 (see Fig. 4 ▸ b) in chain A of PDB entry 1hxd (Weaver et al., 2001 ▸) and Gly152-Ala153-Pro154-Gly155 in chain B of PDB entry 4le4 (T. Jiang, H.-C. Chan, C.-H. Huang, T.-P. Ko, T.-Y. Huang, J.-R. Liu & R.-T. Guo, unpublished work)]. Surprisingly, one of the examples was even an X-Pro tt+ flip (Pro204 in chain B of PDB entry 1cdd; Almassy et al., 1992 ▸; Fig. 4 ▸ c). The ‘wrinkled’ tc− prolines with positive ϕi often have an almost straight Ci−1—Ni—Cα i angle (Fig. 4 ▸ a), which is probably the result of very tight ω restraints, and are reminiscent of the intermediate structure of the trans-to-cis transition of Gly78-Ile79 observed during the refinement of rubrerythrin (Stenkamp, 2005 ▸). From the 904 cases, 59 tc− flips and 22 ‘NCh’ X-Pro cases were visually inspected. If the angle τ (Ni—Cα i—Ci) is larger than 112.5° and the bump score of the O atom in the peptide plane is larger than 0.26 WHAT IF bump score units, then the X-Pro with a positive ϕi is not a tc− peptide but an ‘NCh’ X-Pro. This rule predicts 404 X-Pro tc− flips and 500 ‘NCh’ X-Pro.
3.3. Cis→trans flips
44 clear cis-to-trans flips have been found in this study. For trans X-Xnpg tetrapeptides modelled as cis tetrapeptides the median Cα i−1—Cα i distance (3.34 Å) tends to be larger than the median Cα i−1—Cα i distance for correct cis X-Xnpg tetrapeptides (2.95 Å). Similarly, the median Ci−1—Ni—Cα i angle (131°) tends to be larger than normal (125°). The median Cα i−1—Cα i distance (3.55 Å) and Ci−1—Ni—Cα i angle (159°) for ct− and ct+ X-Pro tetrapeptides also tend to be larger than normal (2.95 Å and 127°, respectively).
3.4. Molecular replacement
Molecular replacement (MR) using a trans peptide is a very common reason for failing to model a cis peptide correctly. This section describes a few examples of this problem.
In the Escherichia coli family 31 α-glycosidase Yicl, Cys316 and Val477 adopt a cis conformation in both the free form (PDB entries 1xsi and 1xsj; Lovering et al., 2005 ▸) and when bound to the sugar adduct eq-5-fluoroxylosyl (PDB entry 1xsk; Lovering et al., 2005 ▸). The authors listed both residues as part of the active site of the α-glycosidase and mentioned that cis-Cys316 orients the side chain of Trp315 to direct Cys307 toward the sugar-binding site (Lovering et al., 2005 ▸). Notably, PDB entry 1xsi was the MR search model for PDB entries 1xsj and 1xsk, but in this process the one Cys316 in chain A that was correctly in the cis conformation became trans in the latter two structures. Val477 was in the incorrect trans conformation in all six chains related by noncrystallographic symmetry (NCS) in each of the three PDB structures.
The PDB_REDO structure of PDB entry 1uyq (P. Isorna, J. Polaina & J. Sanz-Aparicio, unpublished work) clearly showed that a tc− flip should be performed for Ser399. The same flip was predicted in all β-glucosidase A molecules listed in the PDB file as related structures [PDB entries 1bga and 1bgg (Sanz-Aparicio, Hermoso, Martínez-Ripoll, Lequerica et al., 1998 ▸) and 1tr1 and 1e4i (Sanz-Aparicio, Hermoso, Martínez-Ripoll, González et al., 1998 ▸)]. However, the structure factors are not available for any of these structures. Although there is no paper to support it, it seems very likely that one of the related structures was used as a search model to solve the structure 1uyq and Ser399 should be cis in all related structures.
Even though it is more likely that a cis peptide will be accidentally refined as a trans peptide, cis-to-trans flips were observed in four different chains of PDB entry 1v6i (Kundhavai Natchiar et al., 2004 ▸) at residues Lys77-Asp78 and one additional ct− flip only in chain A at Pro81-Ala82. The structure was solved at 2.15 Å resolution using MR with PDB entry 2pel (Banerjee et al., 1996 ▸) as a starting structure. However, the corresponding residues in PDB entry 2pel all have the correct trans conformation. It is therefore unclear to us how the cis peptides have been introduced into the 1v6i model.
3.5. Crystallographic improvement
The real- and reciprocal-space correlation of several representative corrected and re-refined PDB structure models was analysed to investigate the effect on crystallographic quality metrics of flip correction and re-refinement. As an example, the local improvement after a flip in terms of fit to the electron density is shown for PDB entry 2z81 (Jin et al., 2007 ▸) in Fig. 5 ▸. Similar figures for the other re-refinement examples can be found on the website.
Figure 5.

Improvement of the real-space correlation coefficient (RSCC) in PDB entry 2z81 (Jin et al., 2007 ▸; 1.80 Å resolution) after rebuilding and re-refining incorrect peptide bonds. The panels show for six peptide bonds in a region of 20 surrounding residues the increase in average backbone-atom RSCC (including Cβ) when the peptides are corrected and the structure is re-refined, compared with re-refinement of the structure only. The residue after the central peptide bond is indicated in the top bar. Pro540 was corrected by a tc+ flip and all other peptides by tt+ flips. Re-refinement alone already resulted in correction of the conformation of Arg541. Re-refinement details and figures showing the local backbone of the peptide bonds can be found on the associated website.
The improvement in R work/R free as a result of flipping and re-refining was 0.14/0.41% with respect to re-refining only. The work/free reciprocal-space correlation improvement was 0.08/0.22%. The largest improvement in R work/R free (0.23/0.43%) and work/free reciprocal-space correlation (0.18/0.28%) was found for PDB entry 1hi8 (Butcher et al., 2001 ▸), in which 16 flips were necessary. Although the global refinement metrics improve only marginally, the local metrics show a clear improvement upon flipping and, more importantly, sometimes a flip alters our understanding of the relationship between structure and function of a protein.
3.6. Biological implications of newly detected flips
3.6.1. Rab4a
A tc− flip was predicted for Phe72 in the GDP-bound state of human Rab4a (PDB entry 2bmd). This residue is referred to as Phe70 in the associated paper (Huber & Scheidig, 2005 ▸). Phe72 is located at the start of α-helix H2 in the switch 2 region of the small GTPase (Fig. 6 ▸). Fig. 2 ▸ shows Phe72 in the GDP-bound Rab4a before and after correction and re-refinement.
Figure 6.

Rab4a. (a) The active state (PDB entry 2bme; Huber & Scheidig, 2005 ▸) with trans-Phe72 (inset). The grey sphere is magnesium. (b) The inactive state (PDB entry 2bmd; Huber & Scheidig, 2005 ▸) with the corrected and re-refined cis-Phe72 in orange (inset). The incorrect trans-Phe72 backbone is shown in pink.
In the GppNHp-bound Rab4a (PDB entry 2bme; Huber & Scheidig, 2005 ▸) Phe72 has the trans conformation (Fig. 6 ▸ a). Upon GTP hydrolysis, the hydrogen bond between the γ-phosphate and Gly68 is lost and conformational rearrangements take place in the switch 2 region (Huber & Scheidig, 2005 ▸). We cannot exclude that the cis-form was selected during crystallization, but our findings could also suggest that Arg71-Phe72 trans–cis isomerization might be part of this rearrangement (Figs. 2 ▸ and 6 ▸ b), which would indicate that the rearrangement process might be more complicated than previously thought. Phe72 is 97% conserved in the HSSP alignment (Touw et al., 2015 ▸) and is part of the homologous effector-binding epitope of Rab5a (Huber & Scheidig, 2005 ▸), suggesting a role in discrimination between different effector proteins.
3.6.2. Inosine 5′-monophosphate dehydrogenase
A tc− flip is predicted for Asn291 (Fig. 7 ▸ a) in all four copies of Tritrichomonas foetus inosine 5′-monophosphate dehydrogenase (IMPDH; PDB entry 1lrt; Gan et al., 2002 ▸). Structure factors were not deposited for PDB entry 1lrt. We nevertheless believe that Asn291 adopts the cis conformation because the corresponding peptide plane in the MR search model (PDB entry 1ak5; Whitby et al., 1997 ▸) should also be cis. Further, the homologous Asn in human type II IMPDH (PDB entry 1b3o; Colby et al., 1999 ▸) should be cis as well (Fig. 7 ▸ b).
Figure 7.

Part of the active site of inosine 5′-monophosphate (IMP) dehydrogenase (IMPDH). (a) T. foetus IMPDH (PDB entry 1lrt; Gan et al., 2002 ▸). (b) Human IMPDH (PDB entry 1b3o; Colby et al., 1999 ▸). The correct cis-Asn303 from PDB_REDO is shown in green. The NAD+ analogue is β-methylene thiazole-4-carboxamide adenine dinucleotide (β-Me-TAD) in PDB entry 1lrt and selenazole-4-carboxamide adenine dinucleotide (SAD) in PDB entry 1b3o. The IMP analogue 6-chloropurine riboside 5′-monophosphate (6-Cl-IMP) is covalently bound to Cys331 in PDB entry 1b3o. The wires trace the Cα atoms. Water molecules have been omitted for clarity.
Asn91 is part of the β-Me-TAD binding site. The Asn291 side chain hydrogen-bonds to the conserved Gly312 carbonyl in the so-called active-site loop and is located less than 4 Å away from the carboxamide of β-Me-TAD (Fig. 7 ▸ a). The authors write that the interactions between the active-site loop and this carboxamide are the ‘most striking feature of the ternary complex’ (Gan et al., 2002 ▸). They also write that the homologous Asn in PDB entry 1b3o directly hydrogen-bonds to the carboxamide (Fig. 7 ▸ b). The authors extensively allude to the importance of Asn291 in the binding differences of the two ligands. They however fail to notice the peptide-plane flip between PDB entries 1b3o and 1lrt and that in both structures Asn291 is most likely to be a cis peptide, a biological feature that in an active site surely is of importance.
4. Discussion
The present study shows that there is a great need for algorithms that can point out peptide bonds that might need flipping and require a crystallographer’s attention. The usefulness of such algorithms is not limited to lower resolution, as flips are sometimes needed at atomic resolution as well (Fig. 8 ▸).
Figure 8.

X-Pro C=O flip at atomic resolution. The central peptide Val129-Pro130 is shown in (a) the 1.2 Å resolution PDB entry 4gqr (Williams et al., 2012 ▸) and (b) the corresponding PDB_REDO structure. Note that in the PDB_REDO conformation not only the local backbone, but also the Val (Cγ1 pointing towards the reader) and Pro side chains fit the density much better. Colours and maps are as in Fig. 1 ▸.
The validation set is not free of selection bias. The residue composition in the validation set has not been matched to the PDB-wide average nor to the training-set average. As mentioned before, the validation set contains relatively difficult cases. Therefore, the ‘true’ performance of the method is presumably even better than the performance reported in Table 4 ▸.
The PDB_REDO rebuilding stage explicitly checks whether the real-space correlation of a peptide plane is better before or after a peptide-plane flip (Joosten et al., 2011 ▸). In cases where a tc+ flip is actually needed, a tt+ flip often still fits the density better than no flip at all (see Fig. 1 ▸). Further refinement can often lead to the additionally required N—H flip. This explains why PDB_REDO solves many trans–cis flip problems using only a peptide-plane flip search algorithm. The RF method was trained using peptides that were flipped by PDB_REDO and therefore had never seen any cases that PDB_REDO failed to correct. One might therefore expect that the classifier might have been biased towards the training set and might have learned to only recognize incorrect peptide conformations that are correctable by PDB_REDO. The independent test set contained 69 manually validated X-Xnpg cases that needed a tc− flip in both the PDB structure and the PDB_REDO structure. 63 of these cases were classified correctly, which suggests that the method generalizes sufficiently to augment the PDB_REDO process. As new crystal structures are continuously being solved and re-refined iteratively by PDB_REDO, the method can easily be iteratively improved as well.
12 of the 14 X-Xnpg tc− FN corresponded to cases for which a flip was observed or predicted in an NCS-related chain or in the MR search model [Asn267 in chain A and Glu435 in chain D of PDB entry 1fwx (Brown et al., 2000 ▸), Ser412 in PDB entries 1q7z and 1q85 (Evans et al., 2004 ▸), Ala458 in PDB entries 1w9b and 1w9d (Bourderioux et al., 2005 ▸) and Asp273 in PDB entries 3fx6 (Wang et al., 2009 ▸) and 3fvl (Wang et al., 2010 ▸)]. These FN will therefore in practice not be a large problem. For example, if WHAT_CHECK suggests a flip for the same NCS-related residue in chains B, C and D, the crystallographer will of course also check the residue in chain A (e.g. Asn267 in PDB entry 1fwx). Conversely, a predicted flip might turn out to be an FP when homologous chains are inspected. The FP Glu277 in arginase 1 (PDB entry 3lp4; Di Costanzo et al., 2010 ▸), for example, is predicted to be tc− in chain A but not in chain B. The FP His188 in the Y364F mutant of 12-oxophytodienoate reductase 3 (PDB entry 2hs8; Breithaupt et al., 2006 ▸) is not predicted to be a tc− flip in another mutant, the wild type or the MR search model. Similarly, Asn137 in the light chain of the antibody structure of PDB entry 2fbj is tt− in other immunoglobulin light chains and Asp383 in the caspase 8 chain of PDB entry 3h11 (Yu et al., 2009 ▸) is tt− in the MR search model (PDB entry 1i4e; Xu et al., 2001 ▸). WHAT_CHECK additionally shows plots in which the backbone torsion angles are compared between NCS-related chains. A peptide flip in just one of the two chains leads to a massive peak in this plot.
As mentioned before, trans peptides are energetically favoured over cis peptides and model-building software by default attempts to build trans peptides first. One could also argue that experimentalists pay more attention to cis peptides, if these are recognized as such, than to trans peptides. The combination of these arguments results in a very small chance of observing incorrectly modelled cis peptides. Indeed, only very few clear cis-to-trans flips were observed in the present study. The fact that automated re-refinement without rebuilding results in few cis-to-trans corrections suggests that the crystallographic data seldom indicate a strong preference for the trans conformation in these cases. Visual inspection also seems to suggest that true cis peptides typically occur in well resolved locations. Croll (2015 ▸) recently reported the high rate of X-Xnpg cis peptides in the PDB. His study suggests that an exhaustive manual search is likely to identify more cis-to-trans flips. For example, some structure models in the PDB have an unexpected large number of cis peptides in regions with poor density. These are likely to be incorrect, but could not be used to train or validate the RF classifiers. An exhaustive manual search for additional cis-to-trans flips is beyond the scope of this project, but simply rebuilding all cis peptides in the PDB_REDO pipeline is computationally feasible. Many cis–trans flips will be available for training RF classifiers when all PDB structures have been re-refined using the new PDB_REDO pipeline.
The number of observed flips was very large in some classes, while some flip classes were almost completely absent. To increase the number of observations in the smaller classes, observations were fabricated by automatically performing unnecessary flips and subsequent extensive re-refinement. The simulated peptides resulting from this very time-consuming process unfortunately could not be used to successfully predict peptides of the same class observed in the PDB. The small classes will be monitored and in due course the analysis will be repeated when sufficient examples have become available.
The recommendations given recently by Croll (2015 ▸) will help crystallographers to identify spurious cis peptides. The method presented here may help to detect trans peptides in need of a flip. We believe that these results can help everyday crystallographic practice if they are used. However, the true solution to the problem of incorrect peptide conformations is the training and good supervision of inexperienced crystallographers.
5. Availability
The peptide-validation method has been implemented in WHAT_CHECK (http://swift.cmbi.ru.nl/gv/whatcheck/) and is available as a web server (http://swift.cmbi.ru.nl/servers/html/flpchk.html) and as a web service (http://wiws.cmbi.ru.nl/wsdl/). The functionality to perform flips of the tc− and tc+ type will be added to PDB_REDO. The Coot visualization scripts for PDB_REDO entries (Joosten et al., 2014 ▸) show peptide-plane flips and trans↔cis flips since PDB_REDO version 5.43.
The details of classifier training, the resulting classifiers, all tetrapeptide data used for training and validation, the details and pseudo-code for comparing peptide conformations and all re-refinement example data are available from the associated website at http://swift.cmbi.ru.nl/gv/flips/. WHAT IF, including the PDB–PDB_REDO comparison menu and peptide-validation method, is freely available from http://swift.cmbi.ru.nl/gv/facilities/.
6. Conclusion
When applied to the 46 418 233 peptide planes in the PDB, the method predicts 1527 X-Xnpg tc− flips, 53 974 X-Xnpg tt+ flips, 517 X-Pro tc− flips, 2573 X-Pro tc+ flips and 16 487 X-Gly tt+ flips. PDB_REDO has already corrected ∼14% of the peptide-plane flips and ∼8% of the trans-to-cis flips. Peptide-conformation correction leads to a small improvement in the R factors, but more importantly surprisingly often provides a better insight into the structure–function relationship.
Acknowledgments
We thank the RIKEN Structural Genomics/Proteomics Initiative for depositing PDB entry 2ef5, the New York Structural Genomics Research Consortium for depositing PDB entry 3k2g and the depositors of PDB entries 4le4, 1uyq and 2fbj. GV acknowledges financial support from NewProt, which is funded by the European Commission within its FP7 Programme under the thematic area KBBE-2011-5 with contract No. 289350, and from the research programme 11319, which is financed by STW. RPJ is supported by Vidi 723.013.003 from Netherlands Organization for Scientific Research (NWO)
References
- Almassy, R. J., Janson, C. A., Kan, C. C. & Hostomska, Z. (1992). Proc. Natl Acad. Sci. USA, 89, 6114–6118. [DOI] [PMC free article] [PubMed]
- Banerjee, R., Das, K., Ravishankar, R., Suguna, K., Surolia, A. & Vijayan, M. (1996). J. Mol. Biol. 259, 281–296. [DOI] [PubMed]
- Berkholz, D. S., Driggers, C. M., Shapovalov, M. V., Dunbrack, R. L. & Karplus, P. A. (2012). Proc. Natl Acad. Sci. USA, 109, 449–453. [DOI] [PMC free article] [PubMed]
- Berman, H., Henrick, K., Nakamura, H. & Markley, J. L. (2007). Nucleic Acids Res. 35, D301–D303. [DOI] [PMC free article] [PubMed]
- Bourderioux, A., Lefoix, M., Gueyrard, D., Tatibouët, A., Cottaz, S., Arzt, S., Burmeister, W. P. & Rollin, P. (2005). Org. Biomol. Chem. 3, 1872–1879. [DOI] [PubMed]
- Breiman, L. (2001). Mach. Learn. 45, 5–32.
- Breithaupt, C., Kurzbauer, R., Lilie, H., Schaller, A., Strassner, J., Huber, R., Macheroux, P. & Clausen, T. (2006). Proc. Natl Acad. Sci. USA, 103, 14337–14342. [DOI] [PMC free article] [PubMed]
- Brown, K., Djinovic-Carugo, K., Haltia, T., Cabrito, I., Saraste, M., Moura, J. J. G., Moura, I., Tegoni, M. & Cambillau, C. (2000). J. Biol. Chem. 275, 41133–41136. [DOI] [PubMed]
- Butcher, S. J., Grimes, J. M., Makeyev, E. V., Bamford, D. H. & Stuart, D. I. (2001). Nature (London), 410, 235–240. [DOI] [PubMed]
- Cheng, W.-C., Chen, Y.-F., Wang, H.-J., Hsu, K.-C., Lin, S.-C., Chen, T.-J., Yang, J.-M. & Wang, W.-C. (2012). PLoS One, 7, e33481. [DOI] [PMC free article] [PubMed]
- Cheon, Y.-H., Kim, H.-S., Han, K.-H., Abendroth, J., Niefind, K., Schomburg, D., Wang, J. & Kim, Y. (2002). Biochemistry, 41, 9410–9417. [DOI] [PubMed]
- Colby, T. D., Vanderveen, K., Strickler, M. D., Markham, G. D. & Goldstein, B. M. (1999). Proc. Natl Acad. Sci. USA, 96, 3531–3536. [DOI] [PMC free article] [PubMed]
- Croll, T. I. (2015). Acta Cryst. D71, 706–709. [DOI] [PubMed]
- Di Costanzo, L., Ilies, M., Thorn, K. J. & Christianson, D. W. (2010). Arch. Biochem. Biophys. 496, 101–108. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Engh, R. A. & Huber, R. (2001). International Tables for Crystallography, Vol. F, edited by M. G. Rossmann & E. Arnold, pp. 382–392. Dordrecht: Kluwer Academic Publishers.
- Evans, J. C., Huddler, D. P., Hilgers, M. T., Romanchuk, G., Matthews, R. G. & Ludwig, M. L. (2004). Proc. Natl Acad. Sci. USA, 101, 3729–3736. [DOI] [PMC free article] [PubMed]
- Exarchos, K. P., Papaloukas, C., Exarchos, T. P., Troganis, A. N. & Fotiadis, D. I. (2009). J. Biomed. Inform. 42, 140–149. [DOI] [PubMed]
- Frömmel, C. & Preissner, R. (1990). FEBS Lett. 277, 159–163. [DOI] [PubMed]
- Gan, L., Petsko, G. A. & Hedstrom, L. (2002). Biochemistry, 41, 13309–13317. [DOI] [PubMed]
- Gunasekaran, K., Gomathi, L., Ramakrishnan, C., Chandrasekhar, J. & Balaram, P. (1998). J. Mol. Biol. 284, 1505–1516. [DOI] [PubMed]
- Hayward, S. (2001). Protein Sci. 10, 2219–2227. [DOI] [PMC free article] [PubMed]
- Hooft, R. W. W., Vriend, G., Sander, C. & Abola, E. E. (1996). Nature (London), 381, 272. [DOI] [PubMed]
- Huber, S. K. & Scheidig, A. J. (2005). FEBS Lett. 579, 2821–2829. [DOI] [PubMed]
- Huber, R. & Steigemann, W. (1974). FEBS Lett. 48, 2–4. [DOI] [PubMed]
- Jabs, A., Weiss, M. S. & Hilgenfeld, R. (1999). J. Mol. Biol. 286, 291–304. [DOI] [PubMed]
- Jin, M. S., Kim, S. E., Heo, J. Y., Lee, M. E., Kim, H. M., Paik, S.-G., Lee, H. & Lee, J.-O. (2007). Cell, 130, 1071–1082. [DOI] [PubMed]
- Joosten, R. P., Joosten, K., Cohen, S. X., Vriend, G. & Perrakis, A. (2011). Bioinformatics, 27, 3392–3398. [DOI] [PMC free article] [PubMed]
- Joosten, R. P., Joosten, K., Murshudov, G. N. & Perrakis, A. (2012). Acta Cryst. D68, 484–496. [DOI] [PMC free article] [PubMed]
- Joosten, R. P. et al. (2009). J. Appl. Cryst. 42, 376–384. [DOI] [PMC free article] [PubMed]
- Joosten, R. P. & Vriend, G. (2007). Science, 317, 195–196. [DOI] [PubMed]
- Kabsch, W. & Sander, C. (1983). Biopolymers, 22, 2577–2637. [DOI] [PubMed]
- Klabunde, T., Eicken, C., Sacchettini, J. C. & Krebs, B. (1998). Nature Struct. Mol. Biol. 5, 1084–1090. [DOI] [PubMed]
- Kleywegt, G. J., Harris, M. R., Zou, J., Taylor, T. C., Wählby, A. & Jones, T. A. (2004). Acta Cryst. D60, 2240–2249. [DOI] [PubMed]
- Kundhavai Natchiar, S., Arockia Jeyaprakash, A., Ramya, T. N. C., Thomas, C. J., Suguna, K., Surolia, A. & Vijayan, M. (2004). Acta Cryst. D60, 211–219. [DOI] [PubMed]
- Liaw, A. & Wiener, M. (2002). R. News, 2, 18–22.
- Lovering, A. L., Lee, S.-S., Kim, Y. W., Withers, S. G. & Strynadka, N. C. J. (2005). J. Biol. Chem. 280, 2105–2115. [DOI] [PubMed]
- MacArthur, M. W. & Thornton, J. M. (1991). J. Mol. Biol. 218, 397–412. [DOI] [PubMed]
- Matthews, B. W. (1975). Biochim. Biophys. Acta, 405, 442–451. [DOI] [PubMed]
- McCammon, J. A., Gelin, B. R. & Karplus, M. (1977). Nature (London), 267, 585–590. [DOI] [PubMed]
- McNicholas, S., Potterton, E., Wilson, K. S. & Noble, M. E. M. (2011). Acta Cryst. D67, 386–394. [DOI] [PMC free article] [PubMed]
- Moustafa, I., Connaris, H., Taylor, M., Zaitsev, V., Wilson, J. C., Kiefel, M. J., von Itzstein, M. & Taylor, G. (2004). J. Biol. Chem. 279, 40819–40826. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Pahlke, D., Leitner, D., Wiedemann, U. & Labudde, D. (2005). Bioinformatics, 21, 685–686. [DOI] [PubMed]
- Pal, D. & Chakrabarti, P. (1999). J. Mol. Biol. 294, 271–288. [DOI] [PubMed]
- Ramachandran, G. N. & Mitra, A. K. (1976). J. Mol. Biol. 107, 85–92. [DOI] [PubMed]
- Ramachandran, G. N. & Sasisekharan, V. (1968). Adv. Protein Chem. 23, 283–438. [DOI] [PubMed]
- R Core Team (2014). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.r-project.org/.
- Sanz-Aparicio, J., Hermoso, J. A., Martínez-Ripoll, M., González, B., López-Camacho, C. & Polaina, J. (1998). Proteins, 33, 567–576. [DOI] [PubMed]
- Sanz-Aparicio, J., Hermoso, J. A., Martínez-Ripoll, M., Lequerica, J. L. & Polaina, J. (1998). J. Mol. Biol. 275, 491–502. [DOI] [PubMed]
- Schomaker, V. & Trueblood, K. N. (1968). Acta Cryst. B24, 63–76.
- Song, J., Burrage, K., Yuan, Z. & Huber, T. (2006). BMC Bioinformatics, 7, 124. [DOI] [PMC free article] [PubMed]
- Stenkamp, R. E. (2005). Acta Cryst. D61, 1599–1602. [DOI] [PubMed]
- Stewart, D. E., Sarkar, A. & Wampler, J. E. (1990). J. Mol. Biol. 214, 253–260. [DOI] [PubMed]
- Stoddard, B. L. & Pietrokovski, S. (1998). Nature Struct. Mol. Biol. 5, 3–5. [DOI] [PubMed]
- Sugiura, I., Sasaki, C., Hasegawa, T., Kohno, T., Sugio, S., Moriyama, H., Kasai, M. & Matsuzaki, T. (2004). Acta Cryst. D60, 674–679. [DOI] [PubMed]
- Tickle, I. J. (2012). Acta Cryst. D68, 454–467. [DOI] [PMC free article] [PubMed]
- Touw, W. G., Baakman, C., Black, J., te Beek, T. A. H., Krieger, E., Joosten, R. P. & Vriend, G. (2015). Nucleic Acids Res. 43, D364–D368. [DOI] [PMC free article] [PubMed]
- Touw, W. G. & Vriend, G. (2014). Protein Eng. Des. Sel. 27, 457–462. [DOI] [PubMed]
- Vriend, G. (1990). J. Mol. Graph. 8, 52–56. [DOI] [PubMed]
- Wang, M.-L., Li, W.-J., Wang, M. L. & Xu, W. B. (2004). J. Pept. Res. 63, 23–28. [DOI] [PubMed]
- Wang, S. F., Jin, J.-Y., Zeng, Z. H. & Tian, G. R. (2010). Chin. Chem. Lett. 21, 159–162.
- Wang, S.-F., Tian, G. R., Zhang, W.-Z. & Jin, J.-Y. (2009). Bioorg. Med. Chem. Lett. 19, 5009–5011. [DOI] [PubMed]
- Weaver, L. H., Kwon, K., Beckett, D. & Matthews, B. W. (2001). Proc. Natl Acad. Sci. USA, 98, 6045–6050. [DOI] [PMC free article] [PubMed]
- Weiss, M. S. & Hilgenfeld, R. (1999). Biopolymers, 50, 536–544. [DOI] [PubMed]
- Weiss, M. S., Jabs, A. & Hilgenfeld, R. (1998). Nature Struct. Mol. Biol. 5, 676. [DOI] [PubMed]
- Whitby, F. G., Luecke, H., Kuhn, P., Somoza, J. R., Huete-Perez, J. A., Phillips, J. D., Hill, C. P., Fletterick, R. J. & Wang, C. C. (1997). Biochemistry, 36, 10666–10674. [DOI] [PubMed]
- Williams, L. K., Li, C., Withers, S. G. & Brayer, G. D. (2012). J. Med. Chem. 55, 10177–10186. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Xu, G., Cirilli, M., Huang, Y., Rich, R. L., Myszka, D. G. & Wu, H. (2001). Nature (London), 410, 494–497. [DOI] [PubMed]
- Yu, J. W., Jeffrey, P. D. & Shi, Y. (2009). Proc. Natl Acad. Sci. USA, 106, 8169–8174. [DOI] [PMC free article] [PubMed]
- Zhao, B. et al. (2005). J. Biol. Chem. 280, 11599–11607. [DOI] [PubMed]
- Zimmerman, S. S. & Scheraga, H. A. (1976). Macromolecules, 9, 408–416. [DOI] [PubMed]