

The crystal structure of recombinant prolidase from T. sibiricus was determined by X-ray diffraction to a resolution of 2.6 Å and deposited in the Protein Data Bank.
Keywords: archaeal proteins, prolidase, Thermococcus sibiricus, crystallization, crystallography
Abstract
The crystal structure of recombinant prolidase from Thermococcus sibiricus was determined by X-ray diffraction at a resolution of 2.6 Å and was found to contain a tetramer in the asymmetric unit. A protein crystal grown in microgravity using the counter-diffusion method was used for X-ray studies. The crystal belonged to space group P21221, with unit-cell parameters a = 97.60, b = 123.72, c = 136.52 Å, α = β = γ = 90°. The structure was refined to an R cryst of 22.1% and an R free of 29.6%. The structure revealed flexible folding of the N-terminal domain of the protein as well as high variability in the positions of the bound metal ions. The coordinates of the resulting model were deposited in the Protein Data Bank as entry 4rgz.
1. Introduction
Prolidases are metallopeptidases that hydrolyze dipeptides with proline at the C-terminus and a nonpolar amino acid at the N-terminus (Ghosh et al., 1998 ▸; Lowther & Matthews, 2002 ▸). The enzymes are ubiquitous in nature and have been isolated from eukaryota, bacteria and archaea, where they are thought to participate in catabolic processes and proline recycling (see the review by Kitchener & Grunden, 2012 ▸). Recombinant prolidases have medical, military and industrial value, and are currently being extensively studied in these areas.
Enzymes from extremophilic archaea have attracted great scientific interest owing to the extreme conditions under which many of these microorganisms exist. The first archaeal prolidase to have its crystal structure determined was the enzyme PF1343 from the hyperthermophilic archaeon Pyrococcus furiosus (Ghosh et al., 1998 ▸; Grunden et al., 2001 ▸; Maher et al., 2004 ▸). Structural and functional properties of two thermostable homologues, PH1149 and PH0974, from P. horikoshii were subsequently determined (Jeyakanthan et al., 2009 ▸; Theriot et al., 2010 ▸). Recently, we reported the crystal structure of the prolidase Tsprol from the thermophilic organotrophic archaeon Thermococcus sibiricus (Trofimov et al., 2012 ▸). The crystallized protein contained a 6×His-tag sequence at the N-terminus, and analysis of the solved structure demonstrated the contribution of amino-acid residues from the 6×His tag and cadmium ions from the crystallization solution to the folding of the Tsprol dimers. In order to verify the impact of the 6×His tag on the intermolecular interactions observed in the Tsprol crystal, we obtained a tag-free form of Tsprol. In this paper, we describe the purification and crystallization in microgravity of recombinant tag-free prolidase from T. sibiricus (Tsprol-1), as well as the comparative analysis of its three-dimensional structure determined by X-ray diffraction at a resolution of 2.6 Å.
2. Materials and methods
2.1. Macromolecule production
The Tsib_0821 gene was produced via PCR amplification of the recombinant plasmid pQE80L_TSIB0821 (Trofimov et al., 2012 ▸) with the primers Tsprol-1_For (5′-GCCCATGGATTACAAGAGAAGGATTCATAAG-3′) and Tsprol-1_Rev (5′-TTTGTCGACCTATAGCGTGATCAATTCCCTATC-3′) containing NcoI and SalI restriction sites, respectively. The resulting PCR product encoding full-length Tsprol-1 was digested by these restriction enzymes and cloned into a polylinker of a modified pET-22b vector (Novagen, Darmstadt, Germany). The vector was modified by replacing the pelB signal peptide with a cassette which included an N-terminal 6×His-tag sequence followed by a tobacco etch virus (TEV) protease digestion site (MSYYHHHHHHDYDIPTTENLYFQGA). The open reading frame was fully sequenced to ensure that the construct was correct.
The Escherichia coli expression strain BL21-CodonPlus (DE3)-RIPL (Stratagene, La Jolla, California, USA) was transformed with the plasmid and the cells were cultured on Luria–Bertani (LB) medium containing 100 mg ml−1 ampicillin in a shaker incubator at 310 K. Protein expression was induced when the culture reached an optical density of 0.7 at 600 nm by the addition of isopropyl β-d-1-thiogalactopyranoside to a final concentration of 1 mM. After overnight incubation at 297 K, the cells were collected by centrifugation, flash-frozen and stored at 190 K.
For Ni–NTA affinity purification of 6×HisTEV-Tsprol-1, the cells were thawed on ice and suspended in lysis buffer [50 mM Tris–HCl, 500 mM NaCl, 10 mM imidazole, 5%(v/v) glycerol, 0.2%(v/v) Triton X-100 pH 8.0 supplemented with 1 mM PMSF]. The cells were disrupted by sonication on ice (40%, 5 min) using an UP200S ultrasonication processor (24 kHz, 200 W; Dr Hielscher GmbH, Teltow, Germany). Cell debris was removed from the crude extract by centrifugation at 25 000g for 30 min at 277 K and the supernatant was loaded onto a 5 ml Ni–NTA Superflow column (Quiagen, The Netherlands) equilibrated with lysis buffer. Nonspecifically bound proteins were removed by washing with five column volumes (CVs) of lysis buffer and 5 CVs of wash buffer (50 mM Tris–HCl, 500 mM NaCl, 40 mM imidazole pH 8.0). The 6×His-tagged protein was eluted with 3 CVs of elution buffer [50 mM Tris–HCl, 500 mM NaCl, 300 mM imidazole, 5%(v/v) glycerol pH 8.0] and incubated overnight at 277 K with TEV protease at a molar ratio of about 10:1. After the removal of imidazole by dialysis, the digested protein was again loaded onto a 5 ml Ni–NTA Superflow column and the flowthrough fraction containing tag-free Tsprol-1 was collected, concentrated using centrifugal concentrators (Amicon Ultra 10 kDa cutoff; Millipore, Ireland) and subjected to final polishing and buffer exchange by size-exclusion chromatography (using a GE Superdex G200 10/300 GL column on an ÄKTAexplorer liquid-chromatography system; GE Healthcare Life Sciences, USA) in buffer consisting of 40 mM Tris–HCl, 200 mM NaCl, 5%(v/v) glycerol pH 8.0. This size-exclusion chromatography demonstrated the apparent mobility of Tsprol-1 (molecular weight 39 kDa) as a 70–75 kDa protein, which is consistent with its homodimerization (Fig. 1 ▸). The purity of the protein was analyzed by 12%(w/v) SDS–PAGE (Laemmli, 1970 ▸) and its concentration was measured by the Bradford assay (Sigma Bradford reagent, Sigma–Aldrich, USA). The yield of purified Tsprol-1 was 4 mg from 1 l of culture, with greater than 95% purity. The purified Tsprol-l demonstrated a prolidase activity of 690 µmol min−1 per milligram of protein in the enzymatic assay described by Yaron & Mlynar (1968 ▸), which was performed without the addition of any divalent metal cations to the reaction mixture. The macromolecule-production information is presented in Table 1 ▸.
Figure 1.

Size-exclusion chromatography of Tsprol-1 compared with a chromatogram of four molecular-weight (MW) markers. Peak 3, Tspro-l; peaks 1, 2, 4 and 5, MW standards (GE Healthcare; 1, blue dextran, MW 2 000 000; 2, rabbit muscle aldolase, MW 157 000; 4, chicken ovalbumin, MW 44 000; 5, bovine ribonuclease A, MW 13 700). Both chromatograms were obtained under the following conditions: column, GE Superdex G200 10/300; chromatography system, GE ÄKTAexplorer; temperature, room temperature; flow rate, 0.4 ml min−1; buffer, 40 mM Tris–HCl, 200 mM NaCl, 5%(v/v) glycerol pH 8.0.
Table 1. Macromolecule-production information.
Restriction sites are underlined; the N-terminal 6His tag is shown in italics and the TEV-protease digestion site is shown in bold.
| Source organism | T. sibiricus MM739 |
| DNA source | T. sibiricus MM739 |
| Forward primer | 5-GCCCATGGATTACAAGAGAAGGATTCATAAG-3 |
| Reverse primer | 5-TTTGTCGACCTATAGCGTGATCAATTCCCTATC-3 |
| Expression vector | pet-22b modified by the insertion of an N-terminal 6His tag followed by a tobacco etch virus (TEV) protease digestion site instead of the pelB signal peptide |
| Expression host | E. coli BL21-CodonPlus (DE3)-RIPL |
| Complete amino-acid sequence of the construct produced | MSYYHHHHHHDYDIPTTENLYFQGAMDYKRRIHKFQAHFGKKGFEGALVAPGSNFYYLTGFNPLGTLERLFVLILPSEGLLTAIAPRLYEKELEEFNGEVVLWSDSENPYKIFATKIKETFKEGEKLLIDDTMPVGVFLKAKDIFDKYSLHPISPVISELREIKDKDEIKAHKKAAEIVDKVFYRFIEGKLEGKSERELANRIEYMIKNEFGADDVSFEPIVASGPNGANPHHRPSHRKIRKGDVVIFDYGAKYLGYCSDVTRTVVVGPPSEEVKKVYEIVKEAQETAVQKVAEGIPAEVVDATARGIISKYGYGEYFIHRTGHGLGIDVHEEPYISPGNKKILKDGMVFTIEPGIYLQGKFGVRIEDDVALVDKKGIRLTNADRELITL |
2.2. Total reflection X-ray fluorescence spectroscopy (TXRF)
An S2 PICOFOX spectrometer (Bruker AXS Microanalysis GmbH, Germany), which is a portable benchtop TXRF instrument featuring an air-cooled low-power metal-ceramic X-ray tube with a molybdenum target, working at 50 W maximum power, and a liquid nitrogen-free silicon drift detector (SSD) were used to identify bound metals in purified Tsprol-l. 5 µl of the protein sample (5 mg ml−1 in 40 mM Tris–HCl, 200 mM NaCl) or buffer solution (background control) was dispensed onto a clean siliconized quartz glass sample carrier. The quartz disks were dried in a clean laminar flow hood and the samples were measured with the spectrometer in triplicate. The data-acquisition time was 10 s per sample. Interpretation of the TXRF spectra and data evaluation was performed using the SPECTRA 6.3 software (Bruker AXS Microanalysis GmbH). The resulting spectra (Fig. 2 ▸) showed that while the buffer alone had no metal contamination, Zn was detected in the Tsprol-l samples.
Figure 2.

TXRF spectra of Tsprol-l (top) and protein buffer (bottom) acquired with an S2 PICOFOX spectrometer. The positions of fluorescence peaks indicating the presence of metals and some other elements, including Ar (from air), Si and Br (from the sample carrier), Cl (from the buffer) and S (from the protein), are labelled.
2.3. Crystallization
Initial crystallization experiments were carried out using six commercial crystallization screens from Hampton Research (Index HT, Crystal Screen HT, Crystal Screen Cryo HT, PEG/Ion HT, PEGRx HT and SaltRx HT) in the Protein Factory department of the NBICS-Center of the National Research Centre ‘Kurchatov Institute’ in Moscow. All initial screens were performed using a protein concentration of 12 mg ml−1 and the sitting-drop vapour-diffusion method in 96-well CrystalMation plates (Rigaku, Japan) at a temperature of 293 K. 0.1 µl protein solution was mixed with an equal volume of reservoir solution and equilibrated against 50 µl reservoir solution. The initial crystals were grown in the following condition: 0.05 M zinc acetate dihydrate, 20%(w/v) PEG 3350. Further optimization was performed by the capillary counter-diffusion technique by changing the PEG and zinc acetate concentrations. The final reservoir solution consisted of 0.2 M zinc acetate dihydrate, 22%(w/v) PEG 3350. The crystals were grown over a period of 1–2 weeks.
The best crystals of Tsprol-1 chosen for X-ray data collection were grown in a microgravity environment onboard the International Space Station (ISS; Takahashi et al., 2010 ▸; Kuranova et al., 2011 ▸; Timofeev et al., 2012 ▸; Boyko et al., 2014 ▸); the crystallization facilities of the Japan Aerospace Exploration Agency (JAXA) and the support service were provided by JAXA (Sato et al., 2006 ▸) during the JAXA–Protein Crystal Growth (JAXA-PCG) experiment. The JAXA Crystallization Box (JCB), a modification of the original capillary counter-diffusion method (García-Ruiz & Moreno, 1994 ▸; García-Ruiz, 2003 ▸), was used as a crystallization device (Tanaka et al., 2004 ▸; Takahashi et al., 2010 ▸). A thick-walled glass capillary with 0.5 mm inner diameter and of 60 mm in length (Confocal Science, Japan) was loaded with protein solution in 40 mM Tris–HCl pH 8.0, 200 mM NaCl, 5%(v/v) glycerol, 0.1%(w/v) β-d-octylglucopyranoside to a height of 40 mm (about 8 µl protein solution). The top end of the capillary was sealed with plasticine. The bottom end was inserted into a silicone tube which was filled with 1% agarose gel and equilibrated in the protein buffer. The free end of the silicone tube was cut to a length of 20 mm and placed into a 180 µl volume cylinder half-filled with the precipitant solution. The bottom of the cylinder was closed with a cap with microholes in order to remove air and excess precipitant solution. All connections in the device were thoroughly sealed with Araldite glue. Each JCB containing the six items described above was hermetically packed into a plastic box and delivered to the ISS in a temperature-controlled bag by a Russian spacecraft. Crystals of the enzyme were grown from a solution of protein (at a concentration of 12 mg ml−1) in 40 mM Tris–HCl pH 8.0, 200 mM NaCl, 5%(v/v) glycerol, 0.1%(w/v) β-d-octylglucopyranoside and a precipitant solution composed of 0.2 M zinc acetate dihydrate, 22%(w/v) PEG 3350 at 293 K. Crystals of Tsprol-1 grown in the capillary are shown in Fig. 3 ▸. Final crystallization conditions are presented in Table 2 ▸.
Figure 3.

Crystals of Tsprol-1 in the capillary.
Table 2. Crystallization.
| Method | Counter-diffusion |
| Plate type | Capillary |
| Temperature (K) | 293 |
| Protein concentration (mgml1) | 12 |
| Buffer composition of protein solution | 40mM TrisHCl pH 8.0, 200mM NaCl, 5%(v/v) glycerol, 0.1%(w/v) -D-octylglucopyranoside |
| Composition of precipitant | 0.2M zinc acetate dihydrate, 22%(w/v) PEG 3350 |
| Volume of the protein solution in the capillary | 8l protein solution; no reservoir solution |
| Volume of reservoir solution (l) | 90 |
2.4. Data collection and processing
X-ray diffraction data were collected on the BL41XU beamline at the SPring-8 synchrotron, Japan using a Rayonix MX225HE CCD detector. Crystals were removed from the capillary, soaked for 5 s in the mother liquor containing 20%(v/v) glycerol as a cryoprotectant and flash-cooled using a cryoloop in a stream of cold nitrogen gas at 100 K. A total of 720 images were collected with a 0.5° oscillation range at a wavelength of 1.0 Å. The crystal-to-detector distance was 220 mm. Data collection was performed using the HKL-2000 software package (Otwinowski & Minor, 1997 ▸). Experimental details are summarized in Table 3 ▸. X-ray diffraction images were indexed, integrated and subsequently scaled using the XDS package (Kabsch, 2010 ▸). Data reduction was performed using the CCP4 package (Winn et al., 2011 ▸). The data-collection statistics are given in Table 3 ▸. The crystal belonged to space group P21221 and contained four subunits in the asymmetric unit.
Table 3. Data collection and processing.
Values in parentheses are for the outer shell.
| Diffraction source | BL41XU, SPring-8 |
| Wavelength () | 1.0 |
| Temperature (K) | 100 |
| Detector | Rayonix MX225HE CCD |
| Crystal-to-detector distance (mm) | 220 |
| Rotation range per image () | 0.5 |
| Total rotation range () | 360 |
| Exposure time per image (s) | 0.5 |
| Space group | P21221 |
| a, b, c () | 97.60, 123.72, 136.52 |
| , , () | 90, 90, 90 |
| Mosaicity () | 0.213 |
| Resolution range () | 30.002.60 (2.6672.600) |
| Total No. of reflections | 254105 (51103) |
| No. of unique reflections | 51207 (10136) |
| Completeness (%) | 99.3 (99.9) |
| Multiplicity | 5.04 |
| I/(I) | 15.1 (2.65) |
| R r.i.m. (%) | 8.6 (69.0) |
| Overall B factor from Wilson plot (2) | 52.9 |
| CC1/2 | 99.8 (78.4) |
2.5. Structure solution and refinement
The crystal structure of recombinant prolidase from T. sibiricus was solved at a resolution of 2.6 Å by the molecular-replacement method using Phaser (McCoy et al., 2005 ▸) with the atomic coordinates of another crystalline form of the prolidase (PDB entry 4fkc; Trofimov et al., 2012 ▸) as a search model. Structure refinement was carried out using REFMAC (Murshudov et al., 2011 ▸). Manual rebuilding of the models was performed using the Coot interactive graphics program (Emsley & Cowtan, 2004 ▸), and 2|F o| − |F c| and |Fo| − |F c| electron-density maps were calculated. Water molecules and zinc ions were located in difference electron-density maps. The refinement statistics are given in Table 4 ▸.
Table 4. Structure solution and refinement.
Values in parentheses are for the outer shell.
| Resolution range () | 30.002.60 |
| Completeness (%) | 99.3 (99.9) |
| Cutoff | 0 |
| No. of reflections, working set | 48609 |
| No. of reflections, test set | 2598 |
| Final R cryst (%) | 22.1 |
| Final R free (%) | 29.6 |
| Cruickshank DPI | 2.02 |
| No. of non-H atoms | |
| Protein | 11676 |
| Ion | 23 |
| Ligand | 0 |
| Water | 179 |
| Total | 11879 |
| R.m.s. deviations | |
| Bonds () | 0.011 |
| Angles () | 1.877 |
| Average B factors (2) | |
| Protein | 70.618 |
| Ion | 83.186 |
| Water | 78.563 |
| Ramachandran plot | |
| Most favoured (%) | 80.8 |
| Allowed (%) | 17.1 |
3. Results and discussion
The Tsprol-1 crystal belonged to space group P21221. The structure was solved by the molecular-replacement method. The previously reported structure of 6×His-tagged Tsprol obtained at 2.6 Å resolution (PDB entry 4fkc) was used as the starting model. Molecular-replacement calculations were performed using Phaser (McCoy et al., 2005 ▸) as implemented in the CCP4 program suite (Winn et al., 2011 ▸). X-ray diffraction data from 29.48 to 2.6 Å resolution were used to calculate the rotation and translation functions. The best solution gave a correlation coefficient of 0.612 for the top peak (Rf = 0.312). Several cycles of rigid-body refinement were performed using the maximum-likelihood refinement option in Phaser.
The obtained structure was refined using REFMAC and Coot to an R factor of 22.1% (R free = 29.6%) and a resolution of 2.6 Å. It was deposited in the Protein Data Bank as PDB entry 4rgz.
We should point out that we have previously published a structural analysis of the same protein produced in E. coli as a fusion with a 6×His-tag sequence at the N-terminus (Tsprol; PDB entry 4fkc) and demonstrated the pivotal role of cadmium ions from the crystallization solution and amino-acid residues from the 6×His tag in the formation of intersubunit contacts (Trofimov et al., 2012 ▸). In order to verify the role of the 6×His tag-dependent intermolecular interactions in the formation of the Tsprol crystal structure, we prepared a tag-free protein named Tsprol-1. To obtain tag-free Tsprol-1, we inserted a tobacco etch virus (TEV) protease digestion site after the 6×His tag, which allowed its removal during a purification procedure consisting of two subsequent Ni–NTA chromatography steps separated by TEV protease digestion and followed by size-exclusion chromatography. Crystals of tag-free Tsprol-1 grew in different conditions from those reported for 6×His-tagged Tsprol (Trofimov et al., 2012 ▸). In particular, in the case of Tsprol-1 the reservoir solution (precipitant) contained Zn2+ and PEG 3350 and the crystals grew over a period of about two weeks, whereas the Tsprol crystals grew in the presence of Cd2+ and PEG 400 in 2–3 d, which may indicate that the formation of Cd2+/His tag-dependent coordination bonds facilitated and accelerated the crystallization of the His-tagged prolidase. Finally, tag-free Tsprol-1 was successfully crystallized in microgravity using a modified capillary counter-diffusion method. Analysis of the spatial structure of Tsprol-1 showed that although the crystals of the tag-free protein grew in the presence of Zn2+, metal ions were only detected in metal-binding sites and no additional metal-dependent intermolecular interactions were observed in the crystal structure.
The Tsprol-1 crystals contained a tetramer consisting of four identical subunits in the asymmetric unit (Fig. 4 ▸). However, the results of size-exclusion chromatography performed during Tsprol-1 purification showed dimerization of the protein in solution (Fig. 1 ▸). All previously characterized archaeal prolidases [PDB entries 1pv9 (Maher et al., 2004 ▸) for PF1343, 2how (RIKEN Structural Genomics/Proteomics Initiative, unpublished work) for PH0974 and 1wy2 (RIKEN Structural Genomics/Proteomics Initiative, unpublished work) for PH1149] as well as 6×His-tagged Tsprol have been revealed to have homodimeric structures in the crystals. Analysis of interchain contacts in the Tsprol-1 crystal structure using PISA (Krissinel & Henrick, 2007 ▸) has shown that the observed tetramer results from the packing of two dimers in an asymmetric unit of the crystal. Thus, in spite of participating in the formation of intermolecular dimeric contacts, the N-terminal 6×His tag does not play a crucial role.
Figure 4.

Asymmetric unit content of the Tsprol-1 crystal. The molecules are coloured by chain.
Prolidases are metallopeptidases that require the binding of a metal cofactor near the active site in order for catalysis to occur; in particular, archaeal prolidases exhibit maximal activity with Co2+ or Mn2+ ions bound at the dinuclear metal centre (Lowther & Matthews, 2002 ▸). The metal-binding sites of prolidases are not highly selective with regard to the nature of the bound divalent ions (Alberto et al., 2011 ▸). In the previously published structure of 6×His-tagged Tsprol, Cd2+ ions from the crystallization solution were placed in the metal positions of the characteristic dinuclear metal-binding cluster. The activity of Tsprol-1 in the as-isolated form (without additional divalent cations in the reaction mixture) indicates the presence of a bound metal in the enzyme active site, which was identified by TXRF spectrometry to be Zn2+ (Fig. 2 ▸). In the Tsprol-1 crystal each of the active sites of the enzyme contains two Zn2+ ions, one of which is conserved and is coordinated by Glu117, Asp229 and Asp240 (Fig. 5 ▸), while the second ion location varies in the different protein subunits, with the distance between the metal ions being 2.3–4.5 Å (Fig. 6 ▸). This phenomenon may reflect an asynchronous operation of the active sites of the enzyme, as has previously been shown for archaeal prolidases (Ghosh et al., 1998 ▸; Theriot et al., 2010 ▸). Previously, Ghosh and coworkers demonstrated that the P. furiosus prolidase contains one tightly bound Co2+ ion per subunit and another loosely bound ion, which can be either Co2+ or Mn2+ and is required for activity. Theriot and coworkers subsequently showed that for full activation of P. horikoshii prolidases the dinuclear metal centre must be heterogeneous and must contain both Co2+ and Mn2+ ions rather than solely Co2+ ions. The differential role of various divalent cations for Tsprol-1 activation is being investigated (manuscript in preparation).
Figure 5.

Conserved Zn2+ ion (blue sphere) in the active centre of Tsprol-1.
Figure 6.

Different positions of the Zn2+ ions (small blue and pale blue spheres) in the Tsprol-1 structure (PDB entry 4rgz) and Cd2+ ions (large green spheres) in the Tsprol structure (PDB entry 4fkc). Nonconserved Zn2+ ions are shown as small pale blue spheres.
Like previously structurally studied prolidases, each subunit of Tsprol-1 consists of two domains: N-terminal (domain I, residues 1–146) and C-terminal (domain II, residues 147–370). By superimposing one subunit of Tsprol-1 on the other using Cα atoms, it was shown that domain I is much less ordered compared with domain II (Fig. 7 ▸). Analogous superimposition of the tag-free Tsprol-1 and the 6×His-tagged Tsprol subunits showed that the polypeptide chains are also differently folded, especially in the N-terminal domains (Fig. 8 ▸). There are several structures of hyperthermostable prolidases from P. horikoshii and P. furiosus which share 40–50% sequence identity to that described here (for example, PDB entries 2how, 1wy2 and 1pv9). The Cα-atom superposition of these structures on Tsprol-1 resulted in about the same extent of difference in the polypeptide chain folds as between the two different subunits of Tsprol-1 (data not shown). This indicates flexibility of the spatial structure, especially in the N-terminal domains, to be a common characteristic of prolidases.
Figure 7.
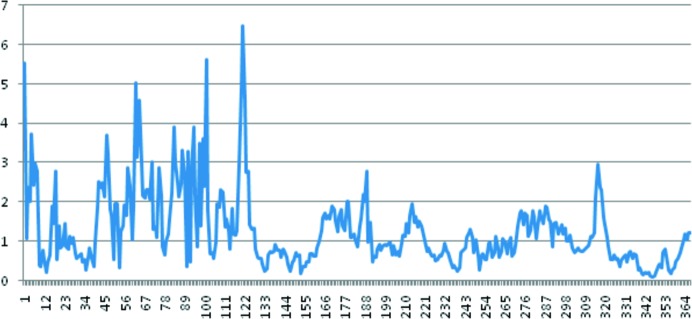
The overlap of two subunits of Tsprol-1 by the Cα atoms of all residues. The horizontal axis represents the amino-acid residue number and the vertical axis represents the root-mean-square deviation of Cα atoms (in Å) of these residues.
Figure 8.
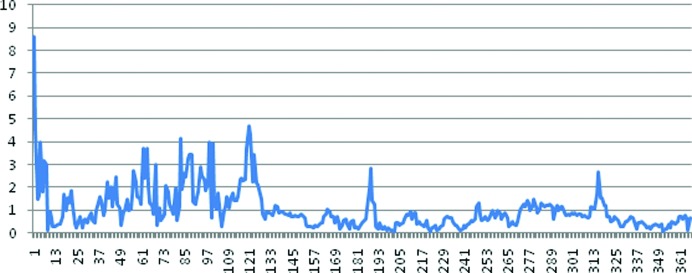
The overlap of the subunits of the two crystalline forms of the prolidase (PDB entries 4rgz and 4fkc) by the Cα atoms of all residues. The horizontal axis represents the amino-acid residue number and the vertical axis represents the root-mean-square deviation of Cα atoms (in Å) of these residues.
The structure of the tag-free prolidase from T. sibiricus (Tsprol-1) solved at 2.6 Å resolution and deposited in the PDB as entry 4rgz was compared with the previously published structure of the 6×His-tagged enzyme (Tsprol; PDB entry 4fkc). The newer structure lacking 6×His tag-related intermolecular interactions revealed a high variability in bound metal-ion positions, which could be a structural reflection of asynchronous operation of the enzyme active sites as well as very flexible folding of the N-terminal domain.
Supplementary Material
PDB reference: prolidase, 4rgz
Acknowledgments
This work was supported by the Russian Scientific Fund No. 14-24-00172 (cloning, protein expression and purification, and structure determination) and the NRC ‘Kurchatov Institute’ Internal Thematic Plan (crystallization screening), as well as the Russian Federal Space Agency No. 1313.13.04 (space crystallization and X-ray analysis). We thank our Japanese colleagues K. Ohta, H. Tanaka, K. Inaka and their coworkers for helpful advice, loading and assembling the JCB crystallization box and assistance in collecting X-ray diffraction data sets at the SPring-8 synchrotron-radiation facility and N. N. Novikova from the NRC ‘Kurchatov Institute’ for her help in conducting TXRF spectroscopy.
References
- Alberto, M. E., Leopoldini, M. & Russo, N. (2011). Inorg. Chem. 50, 3394–3403. [DOI] [PubMed]
- Boyko, K. M., Gorbacheva, M. A., Rakitina, T. V., Korzhenevsky, D. A., Dorovatovsky, P. V., Lipkin, A. V. & Popov, V. O. (2014). Dokl. Biochem. Biophys. 457, 121–124. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- García-Ruiz, J. M. (2003). Methods Enzymol. 368, 130–154. [DOI] [PubMed]
- García-Ruiz, J. M. & Moreno, A. (1994). Acta Cryst. D50, 484–490. [DOI] [PubMed]
- Ghosh, M., Grunden, A. M., Dunn, D. M., Weiss, R. & Adams, M. W. W. (1998). J. Bacteriol. 180, 4781–4789. [DOI] [PMC free article] [PubMed]
- Grunden, A. M., Ghosh, M. & Adams, M. W. W. (2001). Methods Enzymol. 330, 433–445. [DOI] [PubMed]
- Jeyakanthan, J., Takada, K., Sawano, M., Ogasahara, K., Mizutani, H., Kunishima, N., Yokoyama, S. & Yutani, K. (2009). J. Biophys. 2009, 434038. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kitchener, R. L. & Grunden, A. M. (2012). J. Appl. Microbiol. 113, 233–247. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Kuranova, I. P., Smirnova, E. A., Abramchik, Y. A., Chupova, L. A., Esipov, R. S., Akparov, V. K., Timofeev, V. I. & Kovalchuk, M. V. (2011). Crystallogr. Rep. 56, 884–891.
- Laemmli, U. K. (1970). Nature (London), 227, 680–685. [DOI] [PubMed]
- Lowther, W. T. & Matthews, B. W. (2002). Chem. Rev. 102, 4581–4608. [DOI] [PubMed]
- Maher, M. J., Ghosh, M., Grunden, A. M., Menon, A. L., Adams, M. W. W., Freeman, H. C. & Guss, J. M. (2004). Biochemistry, 43, 2771–2783. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Storoni, L. C. & Read, R. J. (2005). Acta Cryst. D61, 458–464. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Sato, M. et al. (2006). Microgravity Sci. Technol. 18, 184–189.
- Takahashi, S., Tsurumura, T., Aritake, K., Furubayashi, N., Sato, M., Yamanaka, M., Hirota, E., Sano, S., Kobayashi, T., Tanaka, T., Inaka, K., Tanaka, H. & Urade, Y. (2010). Acta Cryst. F66, 846–850. [DOI] [PMC free article] [PubMed]
- Tanaka, H., Inaka, K., Sugiyama, S., Takahashi, S., Sano, S., Sato, M. & Yoshitomi, S. (2004). J. Synchrotron Rad. 11, 45–48. [DOI] [PubMed]
- Theriot, C. M., Tove, S. R. & Grunden, A. M. (2010). Appl. Microbiol. Biotechnol. 86, 177–188. [DOI] [PubMed]
- Timofeev, V., Smirnova, E., Chupova, L., Esipov, R. & Kuranova, I. (2012). Acta Cryst. D68, 1660–1670. [DOI] [PubMed]
- Trofimov, A. A., Slutskaya, E. A., Polyakov, K. M., Dorovatovskii, P. V., Gumerov, V. M. & Popov, V. O. (2012). Acta Cryst. F68, 1275–1278. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Yaron, A. & Mlynar, D. (1968). Biochem. Biophys. Res. Commun. 32, 658–663. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: prolidase, 4rgz