

The cloning, expression, purification, crystallization and crystallographic studies of ruminant-specific galectin-11 are described.
Keywords: galectin, parasite, glycan, livestock
Abstract
Galectins are an evolutionarily conserved family of proteins that translate glycan recognition into cellular effects. Galectin-11 is a unique member of the galectin family that is only expressed in ruminants such as sheep, goat and cattle and that plays a critical role in several important biological processes, such as reproduction and parasite-mediated innate immune responses. Currently, these two areas are of major importance for the sustainability of ruminant livestock production. Despite the emerging biological significance of galectin-11, no structural information is available. It is expected that structural studies will unravel the functional mechanisms of galectin-11 activity. Here, the expression, purification and crystallization of the ruminant-specific galectin-11 from domestic sheep and the collection of X-ray data to 2.0 Å resolution are reported.
1. Introduction
The sustainability of ruminant livestock production is being threatened by several factors, including climate change, disease control and reproductive success (Singh et al., 2012 ▸). Considerable research has led to the identification of several proteins that play important roles in immune responses and reproductive processes. One such protein is termed galectin-11 and belongs to the evolutionarily conserved galectin family that modulates various cellular communications, cell adhesion and pathogen recognition (Vasta, 2012 ▸). Galectins are characterized by the presence of a conserved carbohydrate-recognition domain (CRD) and exhibit a preference towards β-galactosides. While other galectins appear to be highly conserved in all higher vertebrates, galectin-11 has only been found in ruminants such as sheep, goats and cattle (Dunphy et al., 2000 ▸). Galectin-11 has the predicted CRD as well as LDV and RGD recognition sequences for proposed integrin binding (Farmer et al., 2008 ▸). Integrins recognize the RGD and LDV motifs within ligands such as galectin-11 and the binding mediates various cell processes (D’Souza et al., 1991 ▸). Integrin binding by galectin-11 in the uterus is important for trophectoderm cell migration, attachment and activation, which are critical for proper blastocyst elongation and implantation (Farmer et al., 2008 ▸). Apart from reproduction, galectin-11 also has a proposed role in immune responses against parasites (Meeusen et al., 2005 ▸). Expression of galectin-11 in the abomasal mucosa leads to the development of a mucosal microenvironment that discourages parastic larval establishment in the abomasal crypts (Jackson et al., 2004 ▸). It has been proposed that galectin-11 changes the mucosal microenvironment by increasing the viscosity via carbohydrate binding (Meeusen et al., 2005 ▸). In spite of the emerging biological significance of galectin-11 in livestock production, no structural study on this protein has been reported. This lack of structural knowledge has hampered research efforts in the development of galectin-11 for the treatment of parasitic diseases or for increasing reproductive success. Here, we report our progress on the expression, purification and preliminary crystallographic study of galectin-11.
2. Materials and methods
2.1. Macromolecule production
2.1.1. Cloning
The Ovis aries galectin-11-encoding sequence was amplified using cDNA prepared from sheep abomasum tissue by polymerase chain reaction (PCR) with galectin-specific primers (Table 1 ▸) using Phusion High-Fidelity DNA polymerase (New England Biolabs). The galectin-11 gene was cloned into a modified pET-28 vector (Luna-Vargas et al., 2011 ▸), giving a protein with an HRV 3C-cleavable N-terminal hexahistidine tag using a ligation-independent cloning method as described in Doyle (2005 ▸) (Table 1 ▸). The ligated plasmid was transformed into Escherichia coli XL-1 Blue cells and plated onto a Luria–Bertani (LB) agar plate containing kanamycin (50 µg ml−1). The clone obtained was confirmed by DNA sequencing.
Table 1. Macromolecule-production information.
| Source organism | O. aries |
| DNA source | O. aries cDNA |
| Forward primer | 5-CAGGGACCCGGTATGGACTCCTTGCCG-3 |
| Reverse primer | 5-CGAGGAGAAGCCCGGTTATAACGTATCCACT-3 |
| Cloning vector | pET-28 plasmid |
| Expression vector | pET-28 plasmid |
| Expression host | E. coli BL21 (DE3) |
| Complete amino-acid sequence of the full-length construct produced (galectin-111140) | MAHHHHHHSAALEVLFQGPGMDSLPNPYLQSVSLTVCYMVKIKANLLSPFGKNPELQVDFGTGTGQGGDIPFRFWYCDGIVVMNTLKDGSWGKEQKLHTEAFVPGQPFELQFLVLENEYQVFVNNKPICQFAHRLPLQSVKMLDVRGDIVLTSVDTL |
| Amino-acid sequence of crystallized galectin-11 after HCV-3C protease treatment (galectin-1118140) | GPGMDSLPNPYLQSVSLTVCYMVKIKANLLSPFGKNPELQVDFGTGTGQGGDIPFRFWYCDGIVVMNTLKDGSWGKEQKLHTEAFVPGQPFELQFLVLENEYQVFVNNKPICQFAHRLPLQSVKMLDVRGDIVLTSVDTL |
2.1.2. Recombinant protein production of galectin-11
Recombinant protein expression of full-length galectin-11 (encoding amino acids 1–140; galectin-111–140) was performed in the BL21 (DE3) E. coli strain (Table 1 ▸). A starter culture was grown from a single colony overnight in 100 ml LB medium containing kanamycin (50 µg ml−1). The starter culture was used at 1:100 dilution to inoculate 800 ml fresh LB medium containing 50 µg ml−1 kanamycin and grown at 310 K until the OD600 reached 0.6. Expression of galectin-111–140 was induced with 0.5 mM IPTG and the cells were allowed to grow for a further 16 h at 291 K. The cells were collected following centrifugation at 4000g for 30 min at 277 K and were stored at 193 K.
2.1.3. Purification of recombinant protein
The frozen cell pellet was thawed on ice and resuspended in 20 ml lysis buffer [20 mM Tris–HCl, 150 mM NaCl, 0.1%(v/v) Triton X-100] per gram in the presence of 0.2 µM phenylmethanesulfonylfluoride. The bacterial suspension was lysed by sonication, the cell debris was removed by centrifugation and fine particles were filtered. The crude E. coli extract was applied onto 5 ml nickel Sepharose beads (GE Healthcare) under native conditions. The unbound proteins was washed with three column volumes of wash buffer [50 mM Tris–HCl pH 8.0, 500 mM NaCl, 5%(v/v) glycerol, 30 mM imidazole] and eluted with 15 ml elution buffer [50 mM Tris–HCl pH 8.0, 500 mM NaCl, 5%(v/v) glycerol, 300 mM imidazole] and collected as 2 ml fractions. The eluted protein fractions were analysed by SDS–PAGE.
Fractions containing galectin-111–140 were pooled and concentrated to 5 ml using Amicon ultracentrifugal filters (3000 kDa molecular-weight cutoff; Millipore). After the addition of 50 mM arginine and 50 mM glutamic acid, the purified protein fractions were concentrated to 5 mg ml−1 and the hexahistidine tag was cleaved by incubation at 277 K overnight with HRV-3C protease in a 1:100 ratio (protease:protein). Uncleaved protein was purified using nickel Sepharose beads and the cleaved protein was confirmed by Western blotting with an anti-hexahistidine antibody. The cleaved galectin-11 (containing amino acids 18–140; galectin-1118–140; Table 1 ▸) was further purified by size-exclusion chromatography with a Superdex S75 16/60 gel-filtration column (GE Healthcare Life Sciences) equilibrated in TBS (10 mM Tris–HCl, 125 mM NaCl, 5 mM TCEP [tris(2-carboxyethyl)phosphine] and 10%(v/v) glycerol using an ÄKTA Basic fast protein liquid-chromatography (FPLC) system (Fig. 1 ▸ a). The molecular weight, purity and identity of the protease-treated galectin-1118–140 were confirmed by SDS–PAGE (Fig. 1 ▸ b), Western blotting with anti-Gal-11 antibody (Dunphy et al., 2000 ▸) and N-terminal sequencing. The purified protein was concentrated to 5.0 mg ml−1 and stored at 277 K for use in crystallization trials. The concentration of galectin-1118–140 was determined spectrophotometrically (NanoDrop 1000, Thermo Scientific) at 280 nm and calculated using an extinction coefficient of 16 960 M −1 cm−1 as determined using the ProtParam tool (http://web.expasy.org/protparam)
Figure 1.

(a) Size-exclusion chromatography trace of recombinant galectin-1118–140. Arrows indicate the elution volumes of proteins of known molecular weight (labelled in kDa). (b) SDS–PAGE analysis of purified protease-treated recombinant galectin-1118–140 (lane 2). Molecular weights (lane 1) are indicated on the left in kDa. (c) Recombinant galectin-111–140 is shown to be functional by elution from a galactose Sepharose column with different sugars. Lanes 2, 3, 4 and 5 show galectin-111–140 eluted from a column with galactose, lactose, mannose and fructose, respectively. The positions of molecular-weight markers (labelled in kDa) are shown in lane 1.
2.1.4. Sugar binding by recombinant galectin-11
To determine whether recombinant galectin-111–140 is functional, the following procedure was followed. 500 µl recombinant galectin-111–140 (2.5 mg ml−1) was incubated with 200 µl galactose Sepharose (Pierce) equilibrated in sugar-binding buffer (20 mM Tris–HCl pH 8.2, 200 mM NaCl, 5 mM β-mercaptoethanol) overnight at 277 K using an end-to-end shaker. Unbound protein was washed with three column volumes of sugar-binding buffer and eluted with 2 ml 1 M galactose, lactose, mannose and fructose in sugar-binding buffer. The eluted protein was analyzed by SDS–PAGE and Coomassie Blue staining (Fig. 1 ▸ c).
2.2. Crystallization
The initial crystallization conditions of protease-treated galectin-1118–140 (Table 1 ▸) were screened using 288 conditions from commercially available kits from Hampton Research (Crystal Screen, Crystal Screen 2 and PEG/Ion) and Qiagen (The JCSG+ Suite) with a CrystalMation integrated robotic workstation (Rigaku). Over 30 different conditions produced crystals of galectin-1118–140 with various morphologies within 4 d. The crystals with the best morphology were obtained in 24-well Linbro plates (Hampton Research) with drops consisting of 1 µl protein solution and 1 µl precipitant solution and a reservoir volume of 500 µl (Fig. 2 ▸). Crystallization information is summarized in Table 2 ▸.
Figure 2.

Crystal of galectin-1118–140 from O. aries. The approximate dimensions of the crystal used for data collection were 1.8 × 0.8 × 0.1 mm. The scale bar represents 0.1 mm.
Table 2. Crystallization.
| Method | Hanging drop |
| Plate type | 24-well Linbro plate |
| Temperature (K) | 293 |
| Protein concentration (mgml1) | 5 |
| Buffer composition of protein solution | 10mM TrisHCl, 125mM NaCl, 5mM TCEP, 10%(v/v) glycerol |
| Composition of reservoir solution | 2%(v/v) Tacsimate pH 7.0, 0.1M HEPES pH 7.5, 20%(w/v) PEG 3350 |
| Volume and ratio of drop | 1l protein solution:1l reservoir solution |
| Volume of reservoir (l) | 500 |
2.3. Data collection and processing
Crystals were soaked for 30 s in cryoprotectant solution consisting of 20%(v/v) glycerol, 2%(v/v) Tacsimate pH 7.0, 0.1 M HEPES–HCl pH 7.5, 20%(w/v) PEG 3350 before cooling at 100 K in a stream of nitrogen gas. A complete data set was collected from a single crystal on the MX1 beamline at the Australian Synchrotron (Cowieson et al., 2015 ▸) using an ADSC Quantum 210r CCD detector (Fig. 3 ▸). The data were processed with MOSFLM (Leslie & Powell, 2007 ▸) and various programs from the CCP4 program suite (Winn et al., 2011 ▸). The final statistics of data collection and processing are summarized in Table 3 ▸.
Figure 3.

Example of a diffraction image from a galectin-1118–140 crystal obtained on beamline MX1 at the Australian Synchrotron. The outer black circle corresponds to a resolution of 2.0 Å.
Table 3. Data collection and processing.
Values in parentheses are for the outer shell.
| Diffraction source | MX1, Australian Synchrotron |
| Wavelength () | 0.9537 |
| Temperature (K) | 100 |
| Detector | ADSC Quantum 210r CCD |
| Crystal-to-detector distance (mm) | 200 |
| Rotation range per image () | 0.5 |
| Total rotation range () | 180 |
| Exposure time per image (s) | 2 |
| Space group | P212121 |
| Unit-cell parameters (, ) | a = 96.2, b = 127.5, c = 141.4, = = = 90.0 |
| Resolution range () | 38.862.00 (2.032.00) |
| Total No. of reflections | 915448 (35018) |
| No. of unique reflections | 115982 (5501) |
| Completeness (%) | 98.6 (95.4) |
| I/(I)† | 6.6 (1.9) |
| R p.i.m. ‡ (%) | 10.8 (59.3) |
| Wilson B factor (2) | 14.26 |
I is the integrated intensity and (I) is the estimated standard deviation of that intensity.
R
p.i.m. = 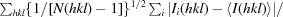
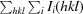 , where Ii(hkl) is the scaled intensity of the ith measurement and I(hkl) is the mean intensity for that reflection.
, where Ii(hkl) is the scaled intensity of the ith measurement and I(hkl) is the mean intensity for that reflection.
3. Results and discussion
The coding region of sheep galectin-11 was successfully cloned by ligation-independent cloning and expressed in E. coli strain BL21 (DE3). The recombinant galectin-11 was purified purified to homogeneity by two chromatographic steps: nickel-affinity chromatography and His-tag removal by HRV-3C protease followed by size-exclusion chromatography. The major elution volume peak in the gel-filtration column corresponds to a dimer in solution with an approximate molecular weight of 30 kDa (Fig. 1 ▸ a). Purified protein was analysed by SDS–PAGE (Fig. 1 ▸ b) and Western blotting to confirm its identity and purity. This expression and purification strategy routinely produced yields of 9 mg per litre of culture. Recombinant galectin-111–140 was successfully eluted from galactose Sepharose beads by various sugars, suggesting that recombinant galectin-11 is functional and that the glycan specificity of galectin-11 may be very broad (Fig. 1 ▸ b). Crystals of galectin-1118–140 were initially obtained by the sitting-drop vapour-diffusion method and were optimized manually using the hanging-drop vapour-diffusion method. Crystals of galectin-1118–140 were obtained in many crystallization conditions; however, a complete diffraction data set was collected at 100 K from crystals grown in 2%(v/v) Tacsimate pH 7.0, 0.1 M HEPES–HCl pH 7.3, 14%(w/v) PEG 3350 (Fig. 3 ▸). The diffraction quality of the crystals was strictly dependent on maintaining the recombinant galectin-11 in a reduced state. Preliminary crystallographic analysis indicated that the crystals belonged to space group P212121, with unit-cell parameters a = 96.2, b = 127.5, c = 141.4 Å, α = 90.0, β = 90.0, γ = 90.0°. Based on Matthews coefficient calculations, 12 molecules of galectin-11 (52% solvent content) could be accommodated in the asymmetric unit, with an acceptable V M of around 2.58 Å3 Da−1 (Matthews, 1968 ▸). The data-collection and processing statistics are summarized in Table 3 ▸. Structure determination by means of molecular replacement was successful using the known structure of human galectin-10 (Charcot–Leyden crystal protein; PDB entry 1g86; Ackerman et al., 2002 ▸), which shares 40% sequence identity with galectin-11. Structure verification and model rebuilding are currently in progress, as well as co-crystallization experiments with the identified sugars.
Acknowledgments
We thank the staff of Monash Macromolecular Crystallization Facility for their assistance in crystallization and the Australian Synchrotron for X-ray data collection and the support provided. We are grateful to Professor Els Meeusen, Department of Physiology, Monash University for kindly providing the sheep cDNA. DS thanks the Department of State Development, Business and Innovation (DSDBI), Victoria, Australia and the Australia India Institute (AII) for supporting DS through a Victoria India Doctoral Scholarship (VIDS). JR is an NHMRC Australia Fellow.
References
- Ackerman, S. J., Liu, L., Kwatia, M. A., Savage, M. P., Leonidas, D. D., Swaminathan, G. J. & Acharya, K. R. (2002). J. Biol. Chem. 277, 14859–14868. [DOI] [PubMed]
- Cowieson, N. P., Aragao, D., Clift, M., Ericsson, D. J., Gee, C., Harrop, S. J., Mudie, N., Panjikar, S., Price, J. R., Riboldi-Tunnicliffe, A., Williamson, R. & Caradoc-Davies, T. (2015). J. Synchrotron Rad. 22, 187–190. [DOI] [PMC free article] [PubMed]
- Doyle, S. A. (2005). Methods Mol. Biol. 310, 107–113. [DOI] [PubMed]
- D’Souza, S. E., Ginsberg, M. H. & Plow, E. F. (1991). Trends Biochem. Sci. 16, 246–250. [DOI] [PubMed]
- Dunphy, J. L., Balic, A., Barcham, G. J., Horvath, A. J., Nash, A. D. & Meeusen, E. N. T. (2000). J. Biol. Chem. 275, 32106–32113. [DOI] [PubMed]
- Farmer, J. L., Burghardt, R. C., Jousan, F. D., Hansen, P. J., Bazer, F. W. & Spencer, T. E. (2008). FASEB J. 22, 548–560. [DOI] [PubMed]
- Jackson, F., Greer, A. W., Huntley, J., McAnulty, R. W., Bartley, D. J., Stanley, A., Stenhouse, L., Stankiewicz, M. & Sykes, A. R. (2004). Vet. Parasitol. 124, 73–89. [DOI] [PubMed]
- Leslie, A. G. W. & Powell, H. R. (2007). Evolving Methods for Macromolecular Crystallography, edited by R. J. Read & J. L. Sussman, pp. 41–51. Dordrecht: Springer.
- Luna-Vargas, M. P., Christodoulou, E., Alfieri, A., van Dijk, W. J., Stadnik, M., Hibbert, R. G., Sahtoe, D. D., Clerici, M., Marco, V. D., Littler, D., Celie, P. H., Sixma, T. K. & Perrakis, A. (2011). J. Struct. Biol. 175, 113–119. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Meeusen, E. N., Balic, A. & Bowles, V. (2005). Vet. Immunol. Immunopathol. 108, 121–125. [DOI] [PubMed]
- Singh, S. K., Meena, H. R., Kolekar, D. V. & Singh, Y. P. (2012). J. Vet. Adv. 2, 407–412.
- Vasta, G. R. (2012). Adv. Exp. Med. Biol. 946, 21–36. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.