

CoA binds in an extended conformation to the amino-terminal domain of the α-subunit of GTP-specific succinyl-CoA synthetase.
Keywords: ligase, ATP-grasp fold
Abstract
Pig GTP-specific succinyl-CoA synthetase is an αβ-heterodimer. The crystal structure of the complex with the substrate CoA was determined at 2.1 Å resolution. The structure shows CoA bound to the amino-terminal domain of the α-subunit, with the free thiol extending from the adenine portion into the site where the catalytic histidine residue resides.
1. Introduction
Succinyl-CoA synthetase (SCS; also known as succinate-CoA ligase) is responsible for breaking down succinyl-CoA to succinate and CoA, accompanied by the phosphorylation of NDP (ADP or GDP) to NTP (ATP or GTP) in the citric acid cycle (Sanadi et al., 1956 ▸): succinyl-CoA + NDP + Pi ⇌ succinate + CoA + NTP. The reaction catalyzed by SCS is reversible and its direction depends on the relative concentrations of the substrates and products. The reverse direction to that occurring in the citric acid cycle produces succinyl-CoA, which may be used for activating ketone bodies (Ottaway et al., 1981 ▸) and haem synthesis (Bishop et al., 2012 ▸).
In mammals, SCS is a heterodimer and is located within the mitochondria. Two different isoforms exist: one is ATP-specific (ATPSCS; EC 6.2.1.5) and the other is GTP-specific (GTPSCS; EC 6.2.1.4). It was proposed that the different isoforms would be functional in different metabolic pathways (Hamilton & Ottaway, 1981 ▸). The two isoforms have the same α-subunit but different β-subunits, and the β-subunit determines the nucleotide specificity (Johnson et al., 1998 ▸). In humans, the amount of ATP-specific and GTP-specific SCS varies in different tissues: ATPSCS is more highly expressed in heart, brain and skeletal muscle tissues, while GTPSCS predominates in kidney and liver (Lambeth et al., 2004 ▸). SCS deficiency can be caused by mutations of the genes coding for SCS. The three genes are SUCLG1, which codes for the α-subunit, SUCLG2, which codes for the β-subunit of GTPSCS, and SUCLA2, which codes for the β-subunit of ATPSCS. A mutation in SUCLA2 leading to encephalomyopathy and mitochondrial DNA depletion was first identified in 2005 (Elpeleg et al., 2005 ▸).
The interactions between GTP or GDP and the nucleotide-binding site of GTPSCS were revealed using crystals of the nucleotides complexed with pig GTPSCS (Fraser et al., 2006 ▸). The nucleotide binds in the ATP-grasp fold (Murzin, 1996 ▸) in the amino-terminal domain of the β-subunit, ∼30 Å from the histidine residue (His259α) that is phosphorylated transiently in the catalytic reaction. The phosphohistidine loop has been postulated to flip between the nucleotide-binding site and the site where CoA and succinate are proposed to bind, transporting the phosphoryl group between the two sites (Fraser et al., 1999 ▸). Although the CoA-binding site for GTPSCS has never been analyzed, it is predicted to lie in the amino-terminal domain of the α-subunit based on the structure of the complex of CoA with E. coli SCS (Wolodko et al., 1994 ▸). In this work, the crystal structure of the complex of CoA with pig GTPSCS, determined at 2.1 Å resolution, is described.
2. Materials and methods
2.1. Macromolecule production
The pT7-7 expression plasmid for GTPSCS has been described previously (Fraser et al., 2006 ▸). For ease of purification, a His6 tag was added to the carboxy-terminus of the α-subunit. In conjunction with this addition, a long untranslated region left from the cDNA cloning was removed from the plasmid. To do this, the plasmid was first modified to include an SpeI site (oligonucleotides JD1.1 and JD1.2; Table 1 ▸), and this site and an existing BamHI site were used for cleavage. The resulting fragments were separated on an agarose gel and the fragment that included the genes for GTPSCS was extracted from the gel. Pre-annealed oligonucleotides (JD1.3 and JD1.4; Table 1 ▸) containing the codons for six histidine residues and the stop codon TAA were ligated to the purified fragment. The sequence was confirmed and this modified expression plasmid was transformed into Escherichia coli BL21(DE3) cells. 100 ml Luria–Bertani broth (2.5 g mix from Life Technologies) containing 100 µg ml−1 ampicillin was inoculated with a single colony and the culture was grown overnight with shaking at 225 rev min−1 and 37°C. 1 l Terrific Broth (12 g tryptone, 24 g yeast extract, 4 ml glycerol, 2.31 g KH2PO4, 12.54 g K2HPO4 per litre) was inoculated with 50 ml overnight culture and the culture was grown at 37°C and 225 rev min−1 to an optical density OD600 nm of 1.6–2.0. Protein overproduction was induced with 1.0 mM isopropyl β-d-1-thiogalactopyranoside and the temperature was lowered to 25°C. After 16 h, the cells were harvested by centrifugation for 30 min at 2663g and 4°C. The cells were stored at −80°C in lysis buffer consisting of 20 mM imidazole, 300 mM NaCl, 50 mM NaH2PO4, 5 mM β-mercaptoethanol (β-ME) pH 8.0.
Table 1. Macromolecule-production information.
| Source organism | Sus scrofa |
| Oligonucleotide JD1.1 (SpeI site underlined) | GAAAAGAGGAAGATGCTACTAGTGCAAAACGAATGAG |
| Oligonucleotide JD1.2 (SpeI site underlined) | CTCATTCGTTTTGCACTAGTAGCATCTTCCTCTTTTC |
| Oligonucleotide JD1.3 (His6 tag underlined) | GATCCCGCCGGTCGCTTAAGTTTATTTATTTAATGGTGGTGATGGTGATGCA |
| Oligonucleotide JD1.4 (His6 tag underlined) | CTAGTGCATCACCATCACCACCATTAAATAAATAAACTTAAGCGACCGGCGG |
| Expression vector | pT7-7 |
| Expression host | E. coli BL21(DE3) |
| Complete amino-acid sequence of the construct produced | |
| Succinyl-CoA ligase (ADP/GDP-forming) subunit , mitochondrial | SYTASRKHLYVDKNTKVICQGFTGKQGTFHSQQALEYGTNLVGGTTPGKGGKTHLGLPVFNTVKEAKEQTGATASVIYVPPPFAAAAINEAIDAEVPLVVCITEGIPQQDMVRVKHRLLRQGKTRLIGPNCPGVINPGECKIGIMPGHIHKKGRIGIVSRSGTLTYEAVHQTTQVGLGQSLCVGIGGDPFNGTDFTDCLEIFLNDPATEGIILIGEIGGNAEENAAEFLKQHNSGPKSKPVVSFIAGLTAPPGRRMGHAGAIIAGGKGGAKEKITALQSAGVVVSMSPAQLGTTIYKEFEKRKMLLVHHHHHH |
| Succinyl-CoA ligase (ADP/GDP-forming) subunit , mitochondrial | MNLQEYQSKKLMSDNGVKVQRFFVADTANEALEAAKRLNAKEIVLKAQILAGGRGKGVFSSGLKGGVHLTKDPEVVGQLAKQMIGYNLATKQTPKEGVKVNKVMVAEALDISRETYLAILMDRSCNGPVLVGSPQGGVDIEEVAASNPELIFKEQIDIIGIKDSQAQRMAENLGFLGPLQNQAADQIKKLYNLFLKIDATQVEVNPFGETPEGQVVCFDAKINFDDNAEFRQKDIFAMDDKSENEPIENEAAKYDLKYIGLDGNIACFVNGAGLAMATCDIIFLNGGKPANFLDLGGGVKESQVYQAFKLLTADPKVEAILVNIFGGIVNCAIIANGITKACRELELKVPLVVRLEGTNVHEAQNILTNSGLPITSAVDLEDAAKKAVASVTKK |
GTPSCS was purified using affinity and gel-filtration chromatography. With the exception of the gel-filtration step, the sample was maintained at 4°C during purification. Frozen cells were thawed on ice and lysed by sonication. Cell debris was separated by centrifugation at 11 235g for 30 min. The soluble fraction was loaded onto a column containing 10 ml nickel–nitrilotriacetic acid agarose (Qiagen) and washed with 20 mM imidazole, 300 mM NaCl, 50 mM NaH2PO4, 5 mM β-ME pH 8.0. The bound protein was eluted with 100 mM imidazole, 300 mM NaCl, 50 mM NaH2PO4, 5 mM β-ME pH 8.0 and precipitated with 0.5 g ammonium sulfate per millilitre of sample. The precipitate was collected by centrifugation at 7802g for 30 min, redissolved in a minimal volume of buffer and then injected onto a Superdex 200 prep-grade column (GE Healthcare; 1.6 × 62 cm) pre-equilibrated with the same buffer: 50 mM NaH2PO4, 150 mM NaCl, 5 mM β-ME pH 8.0. Pooled fractions were concentrated and exchanged into buffer consisting of 10 mM Tris–HCl, 5 mM β-ME pH 8.0. Previous work showed that the starting methionine is cleaved from the α-subunit (Table 1 ▸; Fraser et al., 2006 ▸). The concentration of purified GTPSCS was determined using an A 280 nm(0.1%) value of 0.35 A mg−1 cm−1 (Murakami & Nishimura, 1974 ▸). 20 µl aliquots were flash-frozen in thin-walled PCR tubes in liquid nitrogen and stored at −80°C (Deng et al., 2004 ▸). The final yield was ∼2 mg purified GTPSCS per litre of culture.
2.2. Crystallization
The hanging-drop vapour-diffusion technique was used for co-crystallization of the complex of GTPSCS and CoA. Crystallization conditions were based on those used to crystallize GTPSCS in the absence of CoA (Fraser et al., 2000 ▸). A bundle of crystals was visible within days under the conditions described in Table 2 ▸. Crystals from the bundle were crushed to form a seed stock. A single crystal was grown by microseeding after a 103-fold dilution of the stock.
Table 2. Crystallization.
| Method | Hanging-drop vapour diffusion |
| Plate type | VDX |
| Temperature (K) | 294 |
| Protein concentration (mgml1) | 7 |
| Buffer composition of protein solution | 0.6mM CoA, 10mM TrisHCl, 5mM -ME pH 8.0 |
| Composition of reservoir solution for initial microcrystals | 1.8M ammonium sulfate, 1mM -ME, 50mM sodium/potassium phosphate pH 7.0 |
| Composition of reservoir solution for seeding | 1.6M ammonium sulfate, 1mM -ME, 50mM sodium/potassium phosphate pH 7.0 |
| Volume and ratio of drop | 1l, 1:1 |
| Volume of reservoir (ml) | 1 |
2.3. Data collection and processing
Diffraction data were collected on beamline 08ID-1 (CMCF-ID) at the Canadian Light Source (CLS), Saskatoon, Saskatchewan, Canada. Crystals were cryoprotected in solution consisting of 30%(v/v) glycerol, 1.6 M ammonium sulfate, 80 mM sodium/potassium phosphate pH 7.0 and mounted in loops before flash-cooling in nitrogen gas at 100 K and storage in liquid nitrogen for shipment to the synchrotron. Diffraction data were processed using iMosflm (Battye et al., 2011 ▸) and other programs from the CCP4 package (Winn et al., 2011 ▸; Table 3 ▸).
Table 3. Data collection and processing.
Values in parentheses are for the outer shell.
| Diffraction source | 20mm asymmetric hybrid small-gap in-vacuum undulator, CLS beamline 08ID-1 |
| Wavelength () | 0.97795 |
| Temperature (K) | 100 |
| Detector | Rayonix MX-300 CCD |
| Crystal-to-detector distance (mm) | 250 |
| Rotation range per image () | 0.5 |
| Total rotation range () | 150 |
| Exposure time per image (s) | 1 |
| Space group | P21 |
| a, b, c () | 86.34, 82.50, 49.38 |
| , , () | 90, 104.25, 90 |
| Mosaicity () | 1.2 |
| Resolution range () | 41.402.10 (2.212.10) |
| Total No. of reflections | 114697 (15619) |
| No. of unique reflections | 36903 (5192) |
| Completeness (%) | 94.6 (91.6) |
| Multiplicity | 3.1 (3.0) |
| I/(I) | 5.9 (2.6) |
| R r.i.m. † | 0.140 (0.556) |
| Overall B factor from Wilson plot (2) | 25.37‡ |
Estimated R r.i.m. = R merge[N/(N 1)]1/2, where N is the data multiplicity.
There are two spikes at 2.25 and 2.17. The fit of the curve used to estimate the B factor is better at lower resolution, >2.8, than at high resolution.
2.4. Structure solution and refinement
The structure of the complex of GTPSCS with CoA was solved by molecular replacement using the structure of dephosphorylated GTPSCS (PDB entry 1euc; Berman et al., 2000 ▸; Fraser et al., 2000 ▸) as a model. PHENIX (Adams et al., 2010 ▸) and Coot (Emsley et al., 2010 ▸) were used to refine the structure, and the model quality was judged using MolProbity (Chen et al., 2010 ▸; Table 4 ▸).
Table 4. Structure refinement.
Values in parentheses are for the outer shell.
| Resolution range () | 41.402.10 (2.1752.100) |
| Completeness (%) | 93.92 (90.68) |
| Cutoff | None |
| No. of reflections, working set | 33256 |
| No. of reflections, test set | 3584 |
| Final R cryst | 0.185 (0.274) |
| Final R free | 0.230 (0.344) |
| No. of non-H atoms | |
| Protein | 5211 |
| CoA | 48 |
| Other ligands (phosphate, sulfate, glycerol) | 53 |
| Solvent | 193 |
| Total | 5505 |
| R.m.s. deviations | |
| Bonds () | 0.002 |
| Angles () | 0.611 |
| Average B factors (2) | |
| Protein | 31.8 |
| CoA | 26.4 |
| Other ligands (phosphate, sulfate, glycerol) | 42.4 |
| Solvent | 31.3 |
| Ramachandran plot | |
| Favoured regions (%) | 98 |
| Outliers (%) | 0.14 |
3. Results and discussion
Electron density for CoA was evident in the initial electron-density maps (Fig. 1 ▸). CoA binds to the amino-terminal domain of the α-subunit of GTPSCS in an extended conformation (Fig. 2 ▸). Seven hydrogen bonds were identified between CoA and residues of GTPSCS, and four water molecules bridge between CoA and the α-subunit (Fig. 3 ▸ a). The adenine base binds to two of these water molecules, but also makes hydrophobic interactions with Pro48α and Val80α packing against the two faces. A charged interaction has been identified between Lys50α and the 3′-phosphoryl group of CoA.
Figure 1.

Initial electron density for CoA. The electron-density map (F o − F c, αc) was calculated using the model of the αβ-dimer after molecular replacement and is contoured in green at the 3 r.m.s.d. level. The yellow ribbon diagram shows parts of the α-subunit, with N denoting its amino-terminus. The final model of CoA is shown as a stick model coloured according to atom type (purple, C; blue, N; red, O; yellow, S; orange, P) for verification.
Figure 2.
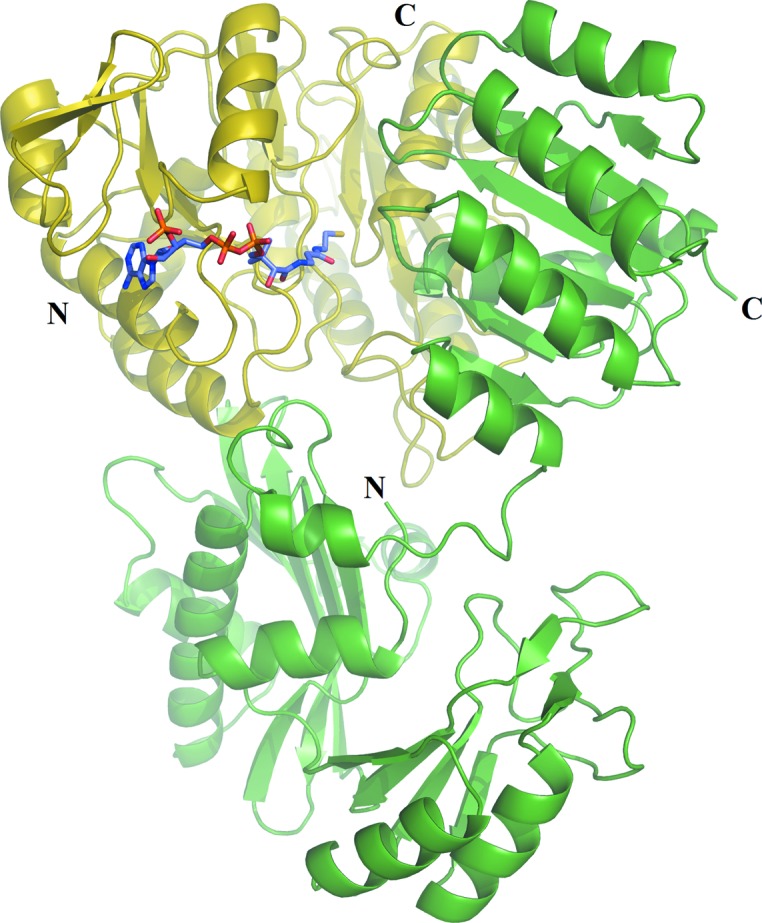
CoA bound to pig GTPSCS. CoA is shown as a stick model coloured as in Fig. 1 ▸. The α- and β-subunits are drawn as ribbon diagrams in yellow and green, respectively, with their termini labelled.
Figure 3.

CoA interactions. (a) Superposition of GTPSCS bound to CoA with GTPSCS in the absence of CoA. (b) Superposition of GTPSCS bound to CoA with E. coli SCS bound to CoA. The C atoms of GTPSCS bound to CoA are yellow, while those of GTPSCS without CoA are magenta and those of E. coli SCS and its bound CoA are green. CoA is coloured as in Fig. 1 ▸.
The adenosine and phosphate portions of CoA are well ordered, with clear electron density, but poorer electron density for the free thiol portion indicates that this portion is not well ordered. The conformation modelled is similar to that originally determined for the complex of E. coli SCS with CoA (Wolodko et al., 1994 ▸). The electron-density maps suggest that a second conformation might exist, with the thiol of Cys132α flipped to the exterior and disulfide-linked to CoA. This conformation was observed in the complex of E. coli SCS with CoA and ADP–Mg2+ (Joyce et al., 2000 ▸). Since the mitochondrial matrix would generally be maintained as a reducing environment, β-ME was used in the purification and the crystallization of GTPSCS to mimic this reducing environment. However, the oxidation of β-ME during crystallization may have led to formation of the disulfide between Cys132α and the thiol of CoA in some molecules of the crystal.
The protein has a very similar conformation to previous structures of phosphorylated and dephosphorylated pig GTPSCS without CoA (PDB entries 1euc and 1eud; Fraser et al., 2000 ▸). Superposition with dephosphorylated GTPSCS without CoA (PDB entry 1euc) gives a root-mean-squared deviation (r.m.s.d.) of 0.44 Å based on 696 Cα atoms from the α- and β-subunits. When CoA binds, it displaces eight water molecules that have similar locations to the N6A, C2A, O2A, P2A, O9P, N4P and S1P atoms of CoA (Fig. 3 ▸ a). Also, the side chain of Gln27α rotates away from the CoA-binding site. As in the structure with PDB code 1euc, the active-site histidine residue His259α is not phosphorylated, presumably because the protein was not phosphorylated with GTP–Mg2+ before crystallization.
The interactions of CoA with GTPSCS are similar to those seen for CoA and E. coli SCS (PDB entry 2scu; Fraser et al., 1999 ▸; Fig. 3 ▸ b). Superposition with the complex of CoA bound to E. coli SCS gives an r.m.s.d. of 1.54 Å for 648 Cα atoms. Identical residues interact with CoA, with the exception that Ser18α in E. coli SCS is substituted by Lys26α in GTPSCS. Since CoA interacts with the amide NH of Ser18α or Lys26α, the difference in side chains has little impact.
In addition to succinyl-CoA synthetases, one other family of proteins has been found to bind CoA in an extended conformation at a Rossmann fold. This family consists of single-domain CoA-binding proteins (Hiyama et al., 2006 ▸) that are similar to E. coli YccU (Wasinger & Humphery-Smith, 1998 ▸). Isothermal titration calorimetry has been used to measure the binding of CoA to one member of this family, TT1466 from Thermus thermophilus HB8 (Wada et al., 2003 ▸). The value of 1.2 ± 0.5 × 105 M −1 is comparable to the apparent Michaelis constant of E. coli SCS for CoA: 2.5 × 105 M −1 (Joyce et al., 1999 ▸). X-ray crystallography showed that these single-domain CoA-binding proteins bind CoA at the Rossmann fold, although the path of the CoA is different from that in the complex with SCS from the 5′-phosphates to the free thiol (Hiyama et al., 2006 ▸). Although the structural comparison of Hiyama and coworkers suggested that E. coli SCS uses both the α- and β-subunits to bind CoA, the interaction with the β-subunit is a crystal-packing interaction, not one that exists in solution. The fact that CoA only binds to the α-subunit is evident in the complex of CoA with GTPSCS.
Supplementary Material
PDB reference: GTP-specific succinyl-CoA synthetase, CoA complex, 4xx0
Acknowledgments
This research was supported by a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada (NSERC). Diffraction data were collected on beamline 08ID-1 at the Canadian Light Source (CLS), which is supported by the NSERC, the National Research Council Canada, the Canadian Institutes of Health Research, the Province of Saskatchewan, Western Economic Diversification Canada and the University of Saskatchewan. We acknowledge Pawel Grochulski and other instructors at the CLS Mx Data Collection School.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Battye, T. G. G., Kontogiannis, L., Johnson, O., Powell, H. R. & Leslie, A. G. W. (2011). Acta Cryst. D67, 271–281. [DOI] [PMC free article] [PubMed]
- Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N. & Bourne, P. E. (2000). Nucleic Acids Res. 28, 235–242. [DOI] [PMC free article] [PubMed]
- Bishop, D. F., Tchaikovskii, V., Hoffbrand, A. V., Fraser, M. E. & Margolis, S. (2012). J. Biol. Chem. 287, 28943–28955. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Deng, J., Davies, D. R., Wisedchaisri, G., Wu, M., Hol, W. G. J. & Mehlin, C. (2004). Acta Cryst. D60, 203–204. [DOI] [PubMed]
- Elpeleg, O., Miller, C., Hershkovitz, E., Bitner-Glindzicz, M., Bondi-Rubinstein, G., Rahman, S., Pagnamenta, A., Eshhar, S. & Saada, A. (2005). Am. J. Hum. Genet. 76, 1081–1086. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Fraser, M. E., Hayakawa, K., Hume, M. S., Ryan, D. G. & Brownie, E. R. (2006). J. Biol. Chem. 281, 11058–11065. [DOI] [PubMed]
- Fraser, M. E., James, M. N. G., Bridger, W. A. & Wolodko, W. T. (1999). J. Mol. Biol. 285, 1633–1653. [DOI] [PubMed]
- Fraser, M. E., James, M. N. G., Bridger, W. A. & Wolodko, W. T. (2000). J. Mol. Biol. 299, 1325–1339. [DOI] [PubMed]
- Hamilton, M. L. & Ottaway, J. H. (1981). FEBS Lett. 123, 252–254. [DOI] [PubMed]
- Hiyama, T. B., Zhao, M., Kitago, Y., Yao, M., Sekine, S.-I., Terada, T., Kuroishi, C., Liu, Z.-J., Rose, J. P., Kuramitsu, S., Shirouzu, M., Watanabe, N., Yokoyama, S., Tanaka, I. & Wang, B.-C. (2006). J. Struct. Funct. Genomics, 7, 119–129. [DOI] [PubMed]
- Johnson, J. D., Muhonen, W. W. & Lambeth, D. O. (1998). J. Biol. Chem. 273, 27573–27579. [DOI] [PubMed]
- Joyce, M. A., Fraser, M. E., Brownie, E. R., James, M. N. G., Bridger, W. A. & Wolodko, W. T. (1999). Biochemistry, 38, 7273–7283. [DOI] [PubMed]
- Joyce, M. A., Fraser, M. E., James, M. N. G., Bridger, W. A. & Wolodko, W. T. (2000). Biochemistry, 39, 17–25. [DOI] [PubMed]
- Lambeth, D. O., Tews, K. N., Adkins, S., Frohlich, D., & Milavetz, B. I. (2004). J. Biol. Chem. 279, 36621–36624. [DOI] [PubMed]
- Murakami, Y. & Nishimura, J. S. (1974). Biochem. Biophys. Acta, 336, 252–263.
- Murzin, A. G. (1996). Curr. Opin. Struct. Biol. 6, 386–394. [DOI] [PubMed]
- Ottaway, J. H., McClellan, J. A. & Saunderson, C. L. (1981). Int. J. Biochem. 13, 401–410. [DOI] [PubMed]
- Sanadi, D. R., Gibson, D. M., Ayengar, P. & Jacob, M. (1956). J. Biol. Chem. 218, 505–520. [PubMed]
- Wada, T., Shirouzu, M., Terada, T., Ishizuka, Y., Matsuda, T., Kigawa, T., Kuramitsu, S., Park, S.-Y., Tame, J. R. H. & Yokoyama, S. (2003). Acta Cryst. D59, 1213–1218. [DOI] [PubMed]
- Wasinger, V. C. & Humphery-Smith, I. (1998). FEMS Microbiol. Lett. 169, 375–382. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Wolodko, W. T., Fraser, M. E., James, M. N. G. & Bridger, W. A. (1994). J. Biol. Chem. 269, 10883–10890. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: GTP-specific succinyl-CoA synthetase, CoA complex, 4xx0