

Abstract
Y chromosomes are challenged by a lack of recombination and are transmitted to the next generation only via males. Sequencing of the mouse Y reveals how these properties drive opposing evolutionary processes: massive decay of ancestral genes and convergent acquisition and amplification of spermatid-expressed gene families on the X and Y chromosome. The convergent acquisition and amplification of X-linked paralogs on the Y maintains a surprisingly gene-rich, euchromatic mammalian male chromosome.
Sex chromosomes evolved independently many times from autosomes in different lineages; for example, the mammalian X and Y originated ~150 million years ago (Bellott and Page, 2009). Two features set sex chromosomes apart from the rest of the genome and drive their unique evolution. These are a lack of recombination on the male-specific region on the Y (MSY) and sex-biased or sex-limited transmission. The lack of recombination renders natural selection inefficient on the Y, resulting in the accumulation of detrimental mutations and, in the long term, massive gene decay (i.e., Y chromosome degeneration). Indeed, old Y chromosomes have lost most of their ancestral genes in many taxa, are often highly repetitive, and have become partly or fully heterochromatic. Sex-biased transmission of sex chromosomes, on the other hand, makes the X and Y chromosome a battleground for sexual conflict. This can result in the accumulation of mutations with sex-specific fitness effects and the invasion of selfish elements on sex chromosomes that cheat meiosis to gain preferential transmission at the expense of homologous loci and bias the sex ratio of progeny. Skewed population sex ratios strongly favor suppressor genes that re-establish fair meiosis and an equal sex ratio.
In this issue of Cell, Soh et al. (2014) present the genome sequence of the mouse Y chromosome, which bears the signatures of both a lack of recombination and genomic conflict between sex chromosomes to an extreme extent. Sequencing of Y chromosomes is challenging due to their high content of repetitive sequences. David Page and colleagues have devised a labor-intensive and careful strategy, termed single-haplotype iterative mapping and sequencing (SHIMS), to assemble ampliconic and highly repetitive sequences. This strategy was successfully employed by Page’s lab to sequence several primate sex chromosomes (Bellott and Page, 2009), but none of the previously sequenced Y’s provided such a technical challenge as the mouse Y due to its highly repetitive nature.
In contrast to the classical view of Y chromosomes being heterochromatic and gene poor, the mouse MSY is almost entirely euchromatic and contains about 700 protein-coding genes. The mouse Y chromosome consists of two sequence classes: ancestral sequence, which originated from the autosomal ancestor of the mammalian sex chromosomes, and acquired sequence not originally present on the ancestral autosomes. Ancestral sequence occupies only 2 Mb and is entirely located within the short arm of the chromosome. Its evolutionary history follows the classical trajectory of Y chromosome degeneration. Of 639 genes present on the ancestral autosomes, only 9 remain on the mouse Y, which is fewer than on other mammalian Y chromosomes (Bellott et al., 2014). Thus, the mouse MSY has experienced greater degeneration than the MSY of other mammals studied to date. The remaining, acquired sequence consists almost entirely of repeated sequences on the chromosome’s long arm. The ampliconic sequence is made up of ~200 copies of a half-megabase unit that contains three rodent-specific protein-coding gene families. Members of the gene families are consequently massively amplified on the mouse Y (132, 197, and 317 copies of Sly, Srsy, Ssty, respectively, with intact ORFs), and their sequences are highly similar to each other. Ampliconic sequences have also been found to a lesser extent on the Y chromosome of other primates, including humans, and are thought to be a Y-specific adaptation that allows for intrachromosomal recombination and gene conversion to retard Y degeneration (Rozen et al., 2003). Like most Y-linked genes, members of these gene families are highly transcribed in testis, and their high copy number may be needed for high levels of gene expression (Mueller et al., 2013).
However, several observations suggest that the picture of gene amplification on the Y is not that simple. Intriguingly, the same gene families found on the mouse Y have been convergently acquired and amplified on the X chromosome, and both the X and Y acquired and amplified genes are expressed predominantly in the male germline (Mueller et al., 2013). Because the X undergoes normal recombination in female meiosis, accumulation of amplicons on the X is unlikely for the benefit of allowing gene conversion among gene family members. In addition, gene amplification of these gene families had comparatively little effect on transcript abundance among species that differ in their copy number of gene families (Ellis et al., 2011). What then would be driving the convergent coamplification of testis-expressed gene families on both the X and the Y?
Genomic conflict between the X and the Y chromosome during spermatogenesis to increase their transmission (i.e., meiotic drive) might be responsible for the observed expansion of sex-linked spermatid-expressed genes in mouse. Males with a partial deletion of the long arm of the Y produce offspring with a sex ratio skewed toward females, suggesting that this arm encodes a factor suppressing sex ratio distortion (Cocquet et al., 2009). Detailed molecular analysis of the Sly and closely related X-linked Slx/Slxl1 gene family provides strong support for segregation distortion as a major force driving gene acquisition and amplification on the mouse X and Y. In particular, Sly is essential for normal spermatogenesis, and Sly-deficient male lab mice have reduced fertility, show widespread overexpression of X-linked postmeiotic genes, including Slx/Slxl1, and produce female-biased litters (Cocquet et al., 2009). In contrast, deficiency of Slx/Slxl1 reduces male fertility and results in male-biased litters (Cocquet et al., 2012), and simultaneous knockdown of all three genes significantly improves fertility, returns expression of sex-linked genes to wild-type levels, and restores equal sex ratio (Cocquet et al., 2012). These antagonistic interactions suggest that Slx/Slxl1 and Sly are involved in an intragenomic conflict, and an evolutionary arms race between segregation distorter and repressor may drive their dramatic copy number expansion and perhaps amplification of several other X- and Y-linked genes in mice (Figure 1A). The phylogenetic pattern of Slx/Slxl1 and Sly amplification is consistent with this meiotic drive scenario, i.e., they were first convergently acquired on sex chromosomes in the Palaearctic mouse clade, and closely related species differ dramatically in copy number (Ellis et al., 2011).
Figure 1.
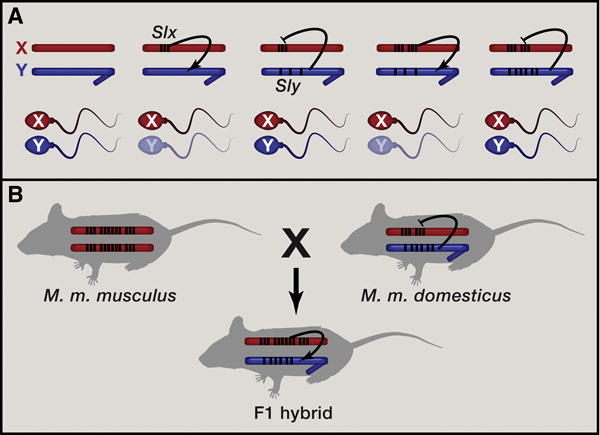
Why Convergent Acquisition and Amplification of Genes on Mouse X and Y Chromosomes May Occur, Yielding a Gene-Rich Euchromatic Mammalian Y Chromosome
(A) Model of recurrent bouts of coevolution between sex ratio distorters and suppressors. A sex ratio distorter on the X chromosome (Slx) that incapacitates Y-bearing sperm (indicated by arrow and transparent sperm) invades the population, skewing the population sex ratio. This creates a selective advantage to evolve a Y-linked suppressor (Sly) that is resistant to the distorter (indicated by repression line). Increased copy number of Slx can increase its ability to drive against the Y chromosome, and the Y will respond by increasing the copy number of Sly to neutralize the effects of increased Slx-dose.
(B) Coevolution of sex ratio distorters and suppressors may contribute to F1 male hybrid sterility in crosses between M. m. musculus mothers and M. m. domesticus fathers, which differ in their copy number for Slx/Slxl1 and Sly genes.
The occurrence of sex ratio distortion in a population can often be transient and can easily escape notice, but recurrent bouts of sex ratio meiotic drive and its subsequent suppression can have important consequences on sex chromosome biology and speciation (Meiklejohn and Tao, 2010). For example, they can lead to incompatibilities that cause sterility in hybrids and thus might contribute to reproductive isolation between species. Intriguingly, F1 hybrid sterile males produced by asymmetric crosses between M. m. musculus mothers and M. m. domesticus fathers display sperm differentiation defects and widespread overexpression of X-linked genes, mimicking the phenotype of Sly-deficient mice (Good et al., 2010); males from the reciprocal cross are fertile. Sterile F1 males with a M. m. musculus X chromosome and M. m. domesticus Y have an excess of Slx/Slxl1 copies compared to Sly copies (i.e., estimated copy number for Slx/Slxl1 and Sly on the X and Y are about 50 copies for M. m. domesticus and about 100 copies for M. m. musculus; Ellis et al., 2011), and the relative deficiency in the number of Sly copies may contribute to F1 male hybrid sterility (Figure 1B) and could explain the observed overexpression of X-encoded genes and associated sperm defects and infertility.
Having the full mouse Y chromosome sequence in hand will aid in the molecular dissection of function of other gene families on the mouse sex chromosomes, and further sequencing of other species’ sex chromosomes should allow reconstruction of their evolutionary history. Detailed molecular and evolutionary analysis should shed light on the role that genetic conflict over sex chromosome transmission has played to shape the evolution of sex chromosome gene content and epigenetic regulation, as well as its contribution to hybrid sterility between species.
References
- Bellott DW, Page DC. Cold Spring Harb Symp Quant Biol. 2009;74:345–353. doi: 10.1101/sqb.2009.74.048. [DOI] [PubMed] [Google Scholar]
- Bellott DW, Hughes JF, Skaletsky H, Brown LG, Pyntikova T, Cho TJ, Koutseva N, Zaghlul S, Graves T, Rock S, et al. Nature. 2014;508:494–499. doi: 10.1038/nature13206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocquet J, Ellis PJ, Yamauchi Y, Mahadevaiah SK, Affara NA, Ward MA, Burgoyne PS. PLoS Biol. 2009;7:e1000244. doi: 10.1371/journal.pbio.1000244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocquet J, Ellis PJ, Mahadevaiah SK, Affara NA, Vaiman D, Burgoyne PS. PLoS Genet. 2012;8:e1002900. doi: 10.1371/journal.pgen.1002900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis PJ, Bacon J, Affara NA. Hum Mol Genet. 2011;20:3010–3021. doi: 10.1093/hmg/ddr204. [DOI] [PubMed] [Google Scholar]
- Good JM, Giger T, Dean MD, Nachman MW. PLoS Genet. 2010;6:e1001148. doi: 10.1371/journal.pgen.1001148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiklejohn CD, Tao Y. Trends Ecol Evol. 2010;25:215–223. doi: 10.1016/j.tree.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller JL, Skaletsky H, Brown LG, Zaghlul S, Rock S, Graves T, Auger K, Warren WC, Wilson RK, Page DC. Nat Genet. 2013;45:1083–1087. doi: 10.1038/ng.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H, Marszalek JD, Minx PJ, Cordum HS, Waterston RH, Wilson RK, Page DC. Nature. 2003;423:873–876. doi: 10.1038/nature01723. [DOI] [PubMed] [Google Scholar]
- Soh YQS, Alföldi J, Pyntikova T, Brown LG, Graves T, Minx PJ, Fulton RS, Kremitzki C, Koutseva N, Mueller JL, et al. Cell. 2014;159:800–813. doi: 10.1016/j.cell.2014.09.052. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]