

Abstract
Adaptable hydrogels have recently emerged as a promising platform for three-dimensional (3D) cell encapsulation and culture. In conventional, covalently crosslinked hydrogels, degradation is typically required to allow complex cellular functions to occur, leading to bulk material degradation. In contrast, adaptable hydrogels are formed by reversible crosslinks. Through breaking and re-forming of the reversible linkages, adaptable hydrogels can be locally modified to permit complex cellular functions while maintaining their long-term integrity. In addition, these adaptable materials can have biomimetic viscoelastic properties that make them well suited for several biotechnology and medical applications. In this review, adaptable hydrogel design considerations and linkage selections are overviewed, with a focus on various cell compatible crosslinking mechanisms that can be exploited to form adaptable hydrogels for tissue engineering.
Keywords: adaptable hydrogel, reversible linkage, cell encapsulation
1. Motivation
Cells in vivo are embedded within a complex, bioactive microenvironment composed of extracellular matrix (ECM) biopolymers, soluble factors, and neighboring cells.[1, 2] In living organisms, cells are constantly interacting with and remodeling their surrounding ECM to enable various cell behaviors, including spreading, migration, proliferation, and differentiation.[2–8] Building three-dimensional (3D) biomaterials that can recapitulate aspects of the native ECM is of great importance both to better investigate cell and tissue physiology in healthy and diseased states and to facilitate the functional restoration of dysfunctional tissues for regenerative medicine.[9, 10] Hydrogels are attractive candidates for building 3D ECM mimics because of their high water content, compliant elasticity, and facile diffusion of biomacromolecules.[11–13] Various crosslinking mechanisms, including both chemical and physical crosslinking, have been exploited to form cell-compatible hydrogels. The initial mesh sizes of the hydrogels for cell encapsulation are typically engineered to be at the nanometer-scale,[14, 15] which has been shown to restrict the spreading, proliferation, and migration of encapsulated cells.[16–19] In most chemically crosslinked hydrogels, the junction points are stable, permanent, covalent bonds. As a result, a hydrogel degradation mechanism, such as hydrolytic degradation [20–24] or cell-mediated enzymatic degradation [25–29], is required to permit cell spreading and migration and potentially to enable other cellular functions such as differentiation.[18]
However, several drawbacks exist in conventional, permanently crosslinked, degradable hydrogel systems when they are used for cell culture (Figure 1A). First, the bulk material degradation will lead to hydrogel disappearance overtime. This is not desirable for many tissue engineering applications, especially when long-term cell culture is required.[30, 31] Second, the mechanical properties deteriorate over time in these systems, and the evolving mechanical properties make it difficult to decouple chemical cues from mechanical cues and draw effective conclusions about how the local biochemical environment affects cell behavior.[32–34] Third, there is a spatial inhomogeneity in mechanical properties that local material properties cannot be represented by bulk properties, posing challenges on local mechanics measurements and the study of how cells respond to local biophysical cues.[35]
Figure 1.

A) Schematic of a permanently crosslinked hydrogel where irreversible degradation occurs. B) Schematic of an adaptable hydrogel built from reversible crosslinks. C) Reversible linkages can be formed either by physical associations or reversible chemical reactions. The dynamics of crosslink breaking and re-forming are related to the kinetic constants of the reactions.
To overcome these limitations, new approaches are needed to build biomaterials that enable normal cellular functions without requiring irreversible hydrogel degradation. In other words, both long-term bulk stability and local adaptability need to be satisfied. One emerging strategy to achieve this goal is to create hydrogels with reversible linkages, which we refer to as ‘adaptable’ linkages in this review. Adaptable hydrogels are polymer networks with adaptable linkages that can be broken and re-formed in a reversible manner without external triggers (Figure 1B). While many dynamic hydrogels and smart hydrogels have been reported previously, these materials typically rely on high temperatures, low pH, ionic strength, or UV light to trigger changes in crosslinking.[36–42] When used for cell culture, to minimize adverse effects on the cultured cells, it is preferable that formation and breaking of adaptable linkages can occur under physiological conditions and without external stimuli.
The mechanisms to form adaptable linkages include physical associations and dynamic covalent chemistry (Figure 1C). Physically crosslinked hydrogels, sometimes called ‘reversible’ gels,[43] are networks held together by molecular entanglements and/or secondary forces, such as ionic, hydrogen-bonding or hydrophobic interactions. Only those showing their reversible nature of physical interactions at physiological conditions are regarded as adaptable hydrogels in our criteria. Dynamic covalent chemistry is the study of covalent bonds that are able to be formed, broken, and re-formed reversibly under equilibrium control.[44, 45] While dynamic covalent chemistry has been utilized to design many interesting self-healing materials,[38, 46–49] only more recently researchers are beginning to apply it to build materials under physiological conditions to keep cells viable for cell culture.[50] Compared to physical associations, dynamic covalent reactions usually have slower kinetics of bond cleavage and formation, and the majority of them may require the assistance of catalysts to achieve rapid equilibrium.[51] Through careful selection, dynamic covalent chemistry has been successfully applied to form adaptable hydrogels for cell encapsulation and culture.[52]
These new adaptable hydrogels, built through physical associations or dynamic covalent bonds, are promising materials for several applications in the field of biomedicine and tissue engineering. The dynamic nature of the linkages enables adaptable hydrogels to have shear-thinning (viscous flow under shear stress) and self-healing (time dependent recovery upon relaxation) characteristics.[53] Therefore, adaptable hydrogels can be used as injectable materials to protect sensitive biological cargo and to deliver them to target sites in a minimally invasive way.[54–56] Furthermore, hydrogels with shear-thinning properties have been used as bio-ink for 3D bioprinting to preserve high cell viability during printing process.[57–60] Adaptable hydrogels also have been shown to display viscoelastic behavior similar to natural tissue,[52, 61, 62] making them suitable to function as in vitro 3D cell culture model for better understanding of how matrix biophysical signaling affects cell behavior.[35, 62, 63] Currently hydrogel elasticity is being intensely investigated in biophysical and biomaterials science while studies on viscous and dissipative contributions are underrepresented and their impacts are largely unknown.[64] Several recent studies have demonstrated that the viscous component of a substrate can affect cell behavior, including cell morphology, proliferation, and differentiation potential.[65–67] The unique features of adaptable hydrogels may shed some light in this emerging field of study.
In this review, we aim to summarize recent advances in the design of adaptable hydrogels for cell encapsulation, cell culture and cell transplantation applications. First, we discuss the critical design criteria for adaptable hydrogels used in tissue engineering applications. Following this, we highlight case studies of recently reported adaptable hydrogels, with a focus on the physical and chemical crosslinking mechanisms that can be used. The review closes with a discussion of the challenges involved in using adaptable linkages to build hydrogels and potential future applications of adaptable hydrogels.
2. Design Criteria for Adaptable Hydrogels
In this section, we will provide perspective on the important design criteria for building adaptable hydrogels that mimic the natural cell microenvironment and permit multiple cell functions to occur through dynamic matrix remodeling. To function effectively as a 3D cell culture scaffold, adaptable hydrogels first need to satisfy the conventional basic requirements for ECM-mimetic materials including cytocompatibility of the gel precursors and the gel crosslinking reaction, proper hydrogel formation rate for cell encapsulation without sedimentation, and appropriate mass transport to support exchange of nutrients and waste products (Figure 2A).[1, 56] Traditional design parameters including polymer molecular weight, polymer concentration, stoichiometry of reactive groups, and final crosslink density have been found to be important parameters in tuning hydrogel properties for cell culture.[12, 68] Beyond this, further considerations are required for adaptable hydrogels due to the unique properties of the reversible linkages. The thermodynamic equilibrium constant (Keq) as well as the kinetic rate constants of the physical interactions or the dynamic covalent chemical reactions are important parameters to consider (Figure 2B). Although there is a complex interplay among these parameters (Figure 1C), we will separate our discussion of the thermodynamic and kinetic design criteria for clarity.
Figure 2.

A) Basic design considerations of all hydrogels used as ECM-mimetic materials for cell encapsulation and 3D culture. B) Additional selection criteria for the design of hydrogels with adaptable linkages.
The delicate balance between network lability and stability must be carefully considered to enable cell spreading and migration while still providing physical cell support. First, to create an adaptable hydrogel, the reversible linkages should be labile enough in order to enable the cell to spread, migrate, proliferate, and carry out other complex bioactive functions. If the interaction strength between the moieties is too strong, i.e. Keq is too large, then the linkages do not undergo significant breakage, and the hydrogel will function similarly to a non-degradable, permanently-crosslinked hydrogel in which cells are entrapped and unable to adjust their morphology, spread, or migrate. On the other hand, the number of reversible linkages must be sufficient to permit the formation of a stable hydrogel that can maintain its bulk integrity during the cell culture period. To form a stable hydrogel network, the probability of forming a crosslink, p, must be above the gel-point threshold to form a percolating polymeric network, pc. According to the mean-field model of gelation, pc decreases as the functionality, f (i.e., number of potential crosslinking sites per molecule), increases.[69, 70] In adaptable hydrogels at equilibrium, some fraction of all possible crosslinks are ‘inactive’, i.e. disassociated, while the remaining crosslinks are ‘active’, i.e. connected.[71] The fraction of ‘active’ crosslinks, is determined by the thermodynamic equilibrium constant and the concentration of the binding or reactive moieties in solution.[71–73] Therefore, very weak interactions between the moieties, i.e. a low Keq, may pose a difficulty in stable hydrogel formation. This is a particularly important concern for physically crosslinked hydrogels, which can have relatively fast erosion rates when exposed to an excess of fluid medium.[74] This might be compensated for by increasing the moiety concentration either through increasing the functionality per molecule, f, or through increasing the molecule concentration.[75, 76] Alternatively, hydrogels that combine two or more different crosslinking mechanisms within a single formulation can also be designed to achieve adaptable hydrogels with decreased erosion rates. [61, 74, 77, 78]
Noticeably, the mechanical properties of a hydrogel are closely related to these design parameters, since the hydrogel elastic modulus is proportional to the effective crosslinking density.[79, 80] Therefore, hydrogels with permanent covalent crosslinks will typically be stiffer than those with dynamic covalent crosslinks, while physically crosslinked hydrogels will have the most compliant mechanical properties. It was shown that within the same gel design, stronger binding moieties, indicated by a lower Kd and higher Keq, can result in higher moduli compared with weaker binding moieties.[75] In addition, the stoichiometry of the binding or reaction moieties may also affect the hydrogel mechanical properties by shifting equilibrium and resulting in different amounts of ‘active’ crosslinks.[81] For example, in a covalently adaptable bis-aliphatic hydrazone crosslinked PEG hydrogel, hydrogel formed at 2:1 stoichiometry of hydrazine to aldehyde functional groups resulted in a much lower shear moduli compared to the stoichiometric hydrogel.[35]
In addition to the requirements of the thermodynamic equilibrium constant, the kinetic rate constants of the adaptable linkages also have important requirements to consider. The adaptable linkages should enable the hydrogel to form and rearrange on an appropriate time scale that is compatible with the cell dynamics. The kinetics of the physical association or dynamic covalent chemical reactions, are characterized by the rate constants kon/koff or k1/k−1 respectively. The rate of gelation was found to correspond closely with the forward rate constant (kon, k1), and the rate of gel relaxation was found to correspond closely with reverse rate constant (koff, k−1).[50] Generally speaking, a fast gelation rate is desirable to achieve a homogeneous cell distribution during encapsulation and to prevent cell sedimentation or aggregation.[82] If the rate constants are too low, network formation, adaption, and recovery may be too slow for effective 3D cell encapsulation and study. Since different processes that control cell fate can occur at varying time scales,[83] the adaptable hydrogel may require a rearrangement rate that is compatible with the cell dynamics of interest. For example, adaptable linkages have been thought as a ‘revolving door’ through which a cell can migrate.[84] If the rate constants are too high, then the cell may fail to go through the fast rotating, revolving door as the adaptable linkages keep breaking and reforming very quickly relative to the rate of cell migration.
It might be possible that cells can alter thermodynamics (Keq) and kinetics (kon/koff or k1/k−1) of the reversible linkages by exerting forces. It has been proposed that the conformational structure of biomolecules and their binding affinities for ligands can be altered by force.[85–88] Therefore, it is important to consider how cell-exerted stress affects Keq and the mechanical properties when designing adaptable hydrogels for cell encapsulation and culture. Similar to Keq, rate constants of the reversible linkage can also be altered by stress. For example, in the catch-bond mechanism found in many receptor-ligand systems, bond lifetime can be altered by the applied mechanical force.[89–91] A better understanding of how cell-exerted stress affects the thermodynamics and kinetics of the selected reversible linkages can be very helpful when designing adaptable hydrogels for cell encapsulation and culture. The powerful technique of single-molecule force spectroscopy can be a useful tool to study this complicated scenario.[92–94]
Take it together, in this section, we have discussed the design criteria to develop adaptable hydrogels with reversible linkages to function as an ECM-mimetic 3D cell culture scaffold, with a focus on the thermodynamic and kinetic considerations of adaptable linkage.
3. Case Studies of Adaptable Hydrogels with Reversible linkages
In this section, we highlight recent examples of candidate reversible linkages that can be exploited to form adaptable hydrogels for cell encapsulation. The case studies are categorized under two main groups: (i) physical interactions, (ii) dynamic covalent chemical reactions (Table 1). For each group, we further divide the discussion based on the interaction/reaction type.
Table 1.
Physical associations and dynamic covalent reactions that can be utilized in building adaptable hydrogels for cell encapsulation. Selected references are provided.
| Physical Linkage Category | Specific Binding Mechanism | Section | References |
|---|---|---|---|
| Host-guest Interaction | Cyclodextrins (CDs) | 3.1.1. | [61,108–112] |
| Cucurbit[n]urils (CB[n]s) | 3.1.2. | [114,117–121] | |
| Biorecognition: Domain/Protein +Ligand | WW domain + proline-rich peptide | 3.2.1. | [74–76,127–129] |
| PDZ domain-containing-TIP1 + peptide | 3.2.2. | [133–138] | |
| DDD + AD | 3.2.3. | [77,156] | |
| TPR domains + peptide | 3.2.4. | [163–164] | |
| Other possible biorecognition mechanisms | 3.2.5. | [174,189] | |
| Hydrophobic Interaction | Coiled-coil peptides | 3.3.1. | [84,202] |
| Amphiphilic block copolymers | 3.3.2. | [78,209–219] | |
| Hydrogen Bonding | Nucleobase pairing | 3.4.1. | [229–233] |
| UPy | 3.4.2. | [236–238] | |
| Electrostatic (Ionic) Interactions | Electrostatic (Ionic) Interactions | 3.5. | [62–63,252–259] |
| Chemical Linkage Category | Specific Reaction Type | Section | References |
|---|---|---|---|
| Dynamic Covalent Linkage | Schiff base (imine) bonds | 3.6.1. | [261–263,266–268] |
| Reversible hydrazone bonds | 3.6.2. | [35,52,273–277] | |
| Oxime bonds | 3.6.3. | [280–281] | |
| Difulfide bonds | 3.6.4. | [292–293] | |
| Reversible Diels-Alder reaction | 3.6.5. | [302–303] |
3.1. Host-guest Interaction
Macrocyclic host-guest complexation is one type of non-covalent interaction that has great potential in the building of adaptable hydrogels. The macrocyclic host molecules externally interact with solvent and internally facilitate the binding of a guest molecule to form an inclusion complex.[71] In this section, we will focus our discussion on two types of hosts, cyclodextrins (CDs) and cucurbit[n]urils (CB[n]s), due to their nontoxicity and biocompatibility. Macrocyclic host-guest systems that rely on external stimuli, such as temperature, light, pH and redox potentials, are not discussed here, and the interested reader is directed to several excellent reviews on this topic.[95–97]
3.1.1. Cyclodextrins (CDs)
Cyclodextrins (CDs) are naturally derived, water-soluble, cyclic oligosaccharides consisting of D-glucose units linked by α-1,4-glucosidic linkages and named based on the number of anhydroglucose units, such as α-,β-,and γ-CDs for 6,7, and 8 D-glucose repeating units, respectively (Figure 3A).[71, 98, 99] The 3D structure of CDs can be represented by a truncated cone in which the secondary and primary hydroxyl groups are located at the solvent-exposed outer surface, creating a hydrophobic inner cavity and favoring the hosting of hydrophobic molecules. Two different strategies have been used to build hydrogels with CDs: a ‘threading’ design and a ‘pendant’ design. In threading systems, CDs slide along linear polymer chains, such as poly(ethylene oxide) (PEO), poly (ε-caprolactone) (PCL), or poly(vinyl alcohol) (PVA).[100–102] The formation of inclusion complexes between CD and the linear polymer is largely dependent on the dimensions of the CD as well as the cross-sectional area of the polymer chain.[53, 99, 103] In some cases, polymer chains can penetrate the inner cavity of β- and γ-CDs, but their cross-sectional areas are often too large to penetrate that of α-CDs.[104, 105] In other cases, two polymer chains can be threaded through a single γ-CD cavity of (Figure 3B), which results in hydrogels with strong entanglements. [106, 107] In pendant systems, the host CDs and guest moieties are presented as decorating functional groups along the polymer chains (Figure 3C). For example, polymers with pendant adamantane and cholesterol have been reported to form effective hydrogels when mixed with polymer chains modified with CDs.[61, 108–112] These materials have shear-thinning properties for ease of injection and can be combined with a secondary covalent crosslinking to further modify the materials for potential clinical applications. [61]
Figure 3.

A) Chemical structures, schematic, and physical dimensions of the cyclodextrin (CD) family. Reproduced with permission.[71] Copyright 2012, Royal Society of Chemistry. B) Two guest polymer chains were threaded through γ-CD while only one chain was threaded through β-CD due to differences in CD size. Reproduced with permission.[107] Copyright 2007, American Chemical Society. C) Schematic representation of a shear-thinning hyaluronic acid (HA) hydrogel based on the pendant design of guest-host interactions between adamantine-modified HA and β-cyclodextrin-modified HA. Reproduced with permission.[110] Copyright 2013, American Chemical Society.
3.1.2. Cucurbit[n]urils (CB[n]s)
Cucurbit[n]urils (CB[n], n = 5–8, 10), are another kind of macrocyclic oligomer, where n indicates the number of repeating monomer units of glycoluril.[113] CB[n]s have a hydrophobic cavity and a symmetric ‘barrel’ shape with two identical portal regions laced by ureido-carbonyl oxygens.[114] Recently, the CB[n] family has been screened for toxicity and was found to be generally compatible with cell cultures.[115, 116] Several hydrogel systems have been designed with CB[n], mainly utilizing CB[8] and CB[6]. For example, CB[8] was used as a small crosslinker to form a hydrogel upon mixing with polymers that displayed pendant guest moieties (Figure 4A).[114, 117, 118] The rapid dynamics of CB[8] ternary inclusion complex formation endowed the three-component hydrogel system with shear-thinning and self-healing properties. In another example, the pendant design strategy was used to form a hydrogel upon the mixing of two hyaluronic acid (HA) polymers modified with CB[6] and its guest (Figure 4B).[119–121] This system forms a biocompatible hydrogel in situ and can be further modified with CB[6] displaying bioactive drugs and growth factors to control the differentiation of mesenchymal stem cells (MSCs).
Figure 4.

A) Schematic of a supramolecular hydrogel prepared through addition of cucurbit[8]uril (CB[8]) to a mixture of two multivalent guest-functionalized polymers. Reproduced with permission.[117] Copyright 2012, American Chemical Society. B) Schematic of a supramolecular hydrogel and its modular modification using host-guest interactions with cucurbit[6]uril (CB[6]) to display bioactive and fluorescent molecules. Reproduced with permission.[119] Copyright 2012, American Chemical Society.
3.2. Biorecognition: Domain/Protein + Ligand
Transient protein interactions, which are important in many aspects of cellular function,[122] can be adopted for the design of biorecognition based adaptable hydrogels. Several important examples of this type of adaptable linkage that have been used to create hydrogels for cell encapsulation and culture are discussed below.
3.2.1. WW Domain + Proline-Rich Pepetide
The WW domain is a naturally evolved peptide (30–50 amino acid long) characterized by an anti-parallel, triple-stranded β-sheet structure with two highly conserved trytophans.[123] WW domains can hetero-dimerize with proline-rich peptides with a range of binding affinities (dissociation equilibrium constant Kd : 5–700 uM).[123–126] This protein-peptide binding was utilized to develop a mixing-induced, two-component hydrogel (MITCH) for cell encapsulation and growth factor delivery (Figure 5A).[74, 75, 127–129] Due to the transient nature of the molecular-recognition based crosslinks, these hydrogels are shear-thinning, injectable, and self-healing. [75] Careful selection of the protein-peptide binding affinity, the number of interacting repeat units, and the relative compositional ratio of the interacting units was shown to directly control the rheological properties of the hydrogels.[75, 76] For example, the authors found that hydrogels formed with a stronger peptide-binding pair (C7/P9) had a storage shear modulus (G′) five times higher than that for a gel formed with a weaker binding pair (N7/P9) (Figure 5B). Successful proliferation and differentiation of encapsulated neural stem cells were observed in these 3D MITCH materials (Figure 5C). Many variant structures of this well-studied protein-peptide pair have been evolved in nature, and additional variants have been derived computationally.[124, 130–132] Therefore, the WW domain and the proline-rich peptide represent a large library of potential association domains with varying degrees of binding specificity and strength.
Figure 5.

A) Schematic of a mixing-induced, two-component hydrogel formed through biorecognition. Two WW domains (CC43 and a Nedd4.3 variant) bind the same proline-rich peptide (PPxY) with different binding affinities. B) Strain sweeps of C7:P9 and N7:P9 hydrogels demonstrate that tuning of Kd leads to changes in gel stiffness. C) Confocal z-stack projection of neural stem cells differentiated within C7:P9. (red, glial marker, GFAP; green, neuronal marker, MAP2; yellow, progenitor marker, nestin; blue, nuclei, DAPI). Reproduced with permission.[75] Copyright 2009, National Academy of Sciences, U.S.A.
3.2.2. PDZ Domain-Containing Tax-Interacting Protein-1(TIP1) + Peptide Ligand
Another type of biorecognition hydrogel formed through protein-peptide interactions relies on Tax-interacting protein-1 (TIP1).[133–138] TIP1 was originally identified as one of the binding partners of the T-cell leukemia viral Tax oncoprotein,[139] and protein-peptide binding occurs through a PDZ domain.[140–142] Mixing of PDZ-domain-recognizable peptides with TIP1 motifs can induce the formation of hydrogel (Figure 6A) with shear-thinning properties.[138] Hydrogels built based on this recognition mechanism were found suitable for 3D culture of MSCs and chondrocytes.[133, 138] Moreover, different PDZ-binding peptide sequences, including NISYRRESAI,[143] RRESAI,[136] and CGGGRGDWRESAI,[133] were found to have different binding affinities with TIP1. PDZ-domain-containing proteins are also involved in many different functions, including trafficking, endocytic recycling, and membrane retention of transmembrane proteins,[144–146] which suggests that there is a large potential library of TIP1-binding peptides for use in adaptable hydrogels. Additionally, multiple computational methods have been developed to predict these domain-peptide interactions.[147–151] The prediction of the domain-peptide binding affinity will be very helpful for sequence selection during adaptable hydrogel design.
Figure 6.

A) Schematic of a hydrogel formed through biorecognition between the PDZ domain in TIP1 protein (CutA-TIP1) and the TIP1-binding peptide. Reproduced with permission.[138] Copyright 2009, Elsevier. B) Proposed structure of the tetrameric ULD-TIP-1 protein (modified from the crystal structures of ULD and TIP-1). Reproduced with permission.[136] Copyright 2012, John Wiley and Sons. C) Schematic of Dock-and-Lock self-assembly mechanism. Docking domains dimerize and lock with the anchoring domains to form shear-thinning hydrogels when mixed. Reproduced with permission.[77] Copyright 2011, Elsevier. D) Schematic of a hydrogel assembled through biorecognition between peptide-binding modules (red) in TPR arrays and peptides coupled to multiarm PEG. Reproduced with permission.[163] Copyright 2012, John Wiley and Sons.
In addition to assembly between the PDZ domain and its peptide ligand, these networks often involve the self-assembly of protein crosslinker to multimerize the PDZ domain (Figure 6A–B). For example, CutA protein, a stable triangular-shaped trimer from Pyrococcus horikoshii,[152] has been used as a crosslinker.[137, 138, 153] Because the CutA self-assembled complex has ultrahigh stability,[154] it can be used to maintain the stability and inhibit erosion of an adaptable hydrogel system.[155] Similarly, self-assembly of the ubiquitin-like domain (ULD) (Figure 6B) has been used as an effective crosslink to form TIP1 tetramers.[133–136]
3.2.3. Docking and Dimerization Domain (DDD) + Anchoring Domain (AD)
Another example of a hydrogel that forms due to protein-protein interactions is the self-assembling Dock-and-Lock (DnL) system (Figure 6C).[77, 156] In these materials, biorecognition occurs between the docking and dimerization domain (DDD) of cAMP-dependent protein kinase A (PKA) and the anchoring domain (AD) of A-kinase anchoring proteins (AKAP) to create self-assembled hydrogels. The selected DDD sequences form a type-X four-helix bundle which binds to the α-helical and amphipathic AD sequence with strong affinity.[157, 158] The rapidly associating and non-covalent interactions between DDD and AD peptides can be disrupted by physical forces,[156] endowing the hydrogels with shear-thinning and self-healing characteristics. Encapsulated MSCs were homogeneously dispersed within DnL gels in vitro and retained a high cell viability (>90%) after injection studies.[156] It was found that light-initiated, radical polymerization of methacrylate functional groups could be used in combination with the DDD-AD biorecognition interactions to form hydrogels with moduli up to ~10 times larger than that for gels based purely on physical crosslinking.[77] Moreover, the DDD-AKAP interactions have been well studied by structural biologists, providing a strong molecular understanding of DDD-AD interactions and a library of association domains to engineer into adaptable hydrogels and other materials.[159–162]
3.2.4. Tetratricopeptide Repeat (TPR) Domains + Peptide
As another example of biorecognition, the tetratricopeptide Repeat (TPR) has been exploited to create a family of protein/polymer hydrogels (Figure 6D).[163, 164] TPR proteins are composed of tandem repeats of a basic structural unit that adopts a helix-turn-helix structure.[165] The properties of individual TPR units can be manipulated through sequence design, and the stability of arrays of TPRs can be predicted based on their constituent units.[165, 166] Additionally, TPR modules can be engineered to achieve highly specific binding with different peptide ligands, [167–169] which forms the basis for a physically crosslinked network. The resulting adaptable hydrogels are nontoxic and cytocompatible.[163]
3.2.5. Other Possible Biorecognition Mechanisms
In addition to the previously discussed biorecognition examples, there are many other potential physical associations that can be useful in adaptable hydrogel design. For example, calmodulin (CaM), which is important in the regulation of many Ca2+ sensitive pathways, can undergo a conformational change to an extended dumbbell-shaped protein upon binding to four calcium ions.[170] This conformation allows CaM to bind to several different calmodulin-binding domains (CBDs) in a reversible way.[171–174] Moreover, the diversity of natural and engineered CBD sequences and CaM variants with different binding affinity or ion sensitivity can be exploited to facilitate the design of an adaptable network.[175–179] As another example, the interactions between heparin and its binding partners may also be useful for creating adaptable hydrogels. Heparin is a type of linear polysaccharide with a high negative charge density that leads to essential interactions with many bioactive molecules.[1, 180, 181] Heparin-binding peptides (HBPs) have been shown to be effective in the non-covalent assembly of heparin-involved hydrogels. [182–186] In addition to HBPs, the heparin-binding domains of growth factors, such as vascular endothelial growth factor (VEGF)[187–189] and basic fibroblast growth factor (bFGF)[190, 191] can be used as crosslinking domain to form hydrogels. Furthermore, because biorecognition is ubiquitous in naturally evolved biological systems, a vast array of other potential binding partners could be explored for the design of adaptable hydrogels.
3.3. Hydrophobic Interaction
In the previous two sections, molecular recognition was used as a strategy to facilitate physical crosslinking within adaptable hydrogels. As an alternative strategy, hydrophobic interactions that promote self-assembly are also useful in creating hydrogels for cell encapsulation. The main driving force for this assembly is the net entropic increase that results from burying the hydrophobic faces away from the bulk aqueous environment, which releases surface-bound water molecules when two or more hydrophobic surfaces assemble together.
3.3.1. Coiled-coil peptides
Coiled-coil peptides are a type of structural motif widely found in fibrous proteins and as oligomerization domains in a variety of proteins.[192] Consisting of two or more α-helices, coiled-coils are built from heptad amino acid sequence repeats, which form right-handed α-helices that assemble to form helical bundles with left-handed supercoils.[193, 194] The primary structure of the coiled-coil motif is typically of the form (a-b-c-d-e-f-g)n, where positions ‘a’ and ‘d’ are usually occupied by hydrophobic amino acids, such as leucine and valine, and positions ‘e’ and ‘g’ are occupied by charged residues.[195] Upon folding into a helical structure, the ‘a’ and ‘d’ positions line up to form a hydrophobic face. Two or more of these aliphatic helices then associate with each other through their hydrophobic interfaces to form a super-helical structure.[193, 196]
Coiled-coil segments, such as leucine zipper domains, were one of the first recombinant peptide domains utilized in hydrogel design.[197–201] In a more recent example, a poly(ethylene glycol) (PEG) hydrogel formed via covalent Michael-addition also included coiled-coil physical associations within the gel (Figure 7A).[84] The coiled-coil domains were hypothesized to create constitutively open paths for cell movement through reversible dissociation and re-association. As a result, encapsulated epithelial cells could migrate and assemble to form hollow spherical cysts without requiring hydrogel degradation (Figure 7B). This design illustrated the promise of adaptable hydrogels to facilitate effective cell function while maintaining the overall stability of the hydrogel matrix. In a separate, modularly assembled hydrogel design, a leucine zipper domain that forms tetramers was chosen as the adaptable linkage.[174, 202] Through rational design and protein engineering, the coiled-coil motif was manipulated to alter the peptide stability, specificity, and hierarchical assembly.[203–206]
Figure 7.
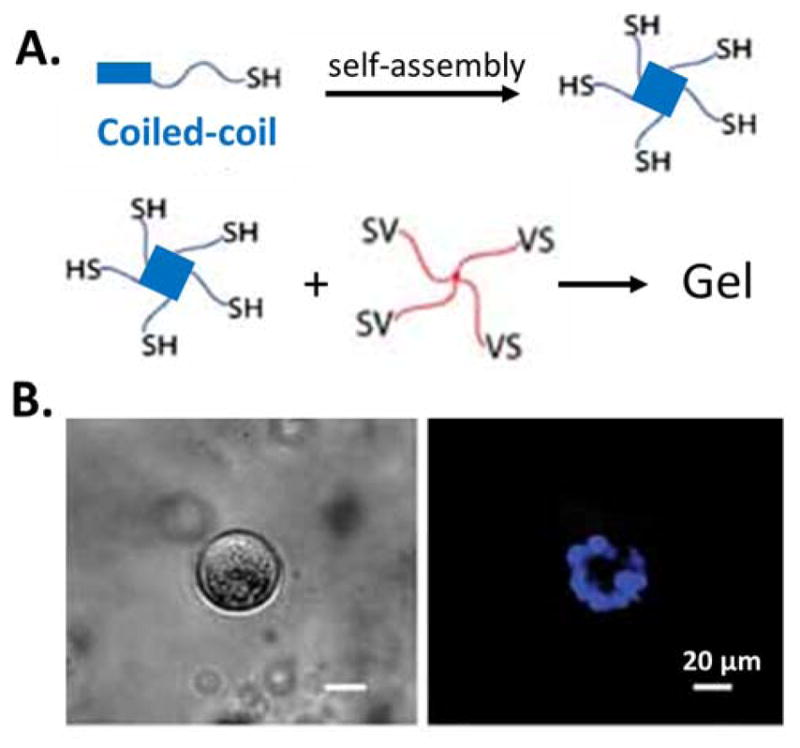
A) Schematic of a hydrogel combining physical crosslinking through the self-assembly of coiled-coil domains and chemical crosslinking between vinyl sulfone (VS) and thiol groups. B) Bright field (left) and fluorescent (right, nucleus staining in blue) of spherical epithelial cell aggregates formed within the hydrogel. Reproduced with permission.[84] Copyright 2011, John Wiley and Sons.
3.3.2. Amphiphilic Block Copolymers
Hydrophobic interactions are also widely exploited in the self-assembly of block copolymers to form a variety of interesting phases.[207, 208] When the constitutive amphiphilic molecules are designed appropriately, they are excellent candidates for the assembly of adaptable hydrogel networks. For biomaterials applications, successful designs have been demonstrated using amphiphilic block copolypeptides,[209–212] synthetic copolymers,[213–217] and hybrids including blocks of polypeptides and synthetic polymers [78, 218, 219].
The first example is an injectable and biocompatible amphiphilic, diblock copolypeptide hydrogel (DCH) (Figure 8A).[211] The diblock consisted of a charged, hydrophilic domain (poly lysine, K) and an α-helical hydrophobic domain (polyleucine, L), i.e. KmLn, where m and n represented the number of amino acid residues in each segment. Similar to the coiled-coil designs, the main driving force for assembly is to minimize exposure of the hydrophobic domains to the charged segments and the aqueous environment by maximizing helix-helix overlap.[220] DCHs with similar mechanical properties to brain tissue were injected into mouse forebrain and showed little toxicity in vivo. Because of its adaptable nature, K180L20 gel enabled time-dependent, in-growth of blood vessels and glial cells, indicating successful integration with brain tissue.
Figure 8.

A) Schematic of an amphiphilic diblock copolypeptide hydrogel composed of hydrophilic (blue) and hydrophobic (red) amino acids. Reproduced with permission.[211] Copyright 2009, Elsevier. B) Schematic of an injectable non-protease degradable poly(ethylene glycol)-poly(propylene sulfide) (PEG-PPS) hydrogel formed through hydrophobic interactions of a 4-arm, branched copolymer. Decoration with a cell-adhesive RGD domain enables long-term culture of induced pluripotent stem cell-derived neural progenitor cells (live-dead staining, green: live, red: dead). Reproduced with permission.[213] Copyright 2011, John Wiley and Sons. C) Schematic of a shear-thinning hydrogel formed by coiled-coil self-assembly and reinforced through temperature-responsive aggregation of PNIPAM. Reproduced with permission.[78] Copyright 2013, John Wiley and Sons.
As a second example, a synthetic, four-arm, branched copolymer was designed with a hydrophilic core and hydrophobic end blocks. The poly(ethylene glycol)-poly(propylene sulfide) (PEG-PPS) copolymers form adaptable hydrogels through physical association of the hydrophobic PPS segments (Figure 8B).[213] At the end of each assembling block, a pyridine dithione functional group was used to incorporate integrin-binding RGD peptides for cell adhesion via disulfide exchange reaction. Additionally, covalent bonds formed slowly via the same disulfide exchange reaction to progressively enhance the hydrogel’s mechanical properties, resulting in a ten-fold increase in the storage modulus after 14 days. Due to the dynamic nature of the physical association, the PEG-PPS hydrogel was shear-thinning and injectable. In this non-protease-degradable hydrogel, encapsulated mouse fibroblasts, MSCs, and human induced pluripotent stem cell derived-neural progenitor cells (iPS-NPC) spread and proliferated. This cell behavior was hypothesized to result from the cells’ ability to break the hydrophobic interactions in the hydrogel and rearrange the polymer blocks to generate enough space for cell spreading. In a comparison of encapsulated iPS-NPC injection into the mouse brain within PEG-PPS hydrogels or covalently crosslinked, protease-degradable HA hydrogels, transplanted cells could not be detected in the HA gel after 14 days, which completely degraded as a result of protease activity in the brain. In contrast, iPS-NPCs were still present within the PEG-PPS hydrogels. Therefore, adaptable hydrogels might be advantageous for cell implantation into tissues with high proteolytic activity.
Hydrophobic interactions can also be used to form hydrogels using hybrid molecules that include both polypeptides and synthetic polymer. As an example, a protein-polymer triblock copolymer was designed to form a hydrogel through assembly of two orthogonal association networks (Figure 8C).[78] First, a shear-thinning hydrogel was formed through assembly of the midblock, an engineered protein with coiled-coil domains. Next, a second, independent physical network was formed by hydrophobic interactions of the endblocks, a thermoresponsive, synthetic polymer poly(N-isopropylacrylamide) (PNIPAM), to reinforce the structure. PNIPAM has a lower critical solution temperature (LCST) of ~ 32 °C in water[221] and undergoes hydrophobic aggregation when the temperature is above the LCST.[222] PNIPAM has been used by many groups to adjust gelation temperature and to enhance the mechanical properties of injectable hydrogel systems.[74, 223–225] For example, the formation of a secondary PNIPAM network within a self-assembled hydrogel with peptide-based, physical crosslinks lead to an increase of G′ from ~13 Pa to ~100 Pa at physiological temperature. [74]
3.4. Hydrogen Bonding
Another useful binding mechanism, hydrogen bonding, is a type of non-covalent, secondary interaction that is relatively weak in isolation. However, when molecules are designed to facilitate the formation of multiple hydrogen bonds, the overall association constant is greatly increased,[71, 226, 227] making this a very useful interaction for the formation of adaptable hydrogels.
3.4.1. Nucleobase Pairing
One special type of assembly through hydrogen bonding is Watson-Crick base pairing of nucleobases: adenine (A) pairs with thymine (T), and guanine (G) pairs with cytosine (C) to form two and three hydrogen bond, respectively. In nature, double-stranded DNAs are assembled from complementary DNA strands through hydrogen bonding and further connected via covalent, enzyme-catalyzed assembly.[228] The binding pairs for DNA assembly have been utilized to engineer hydrogels for many different applications.[229–233] In a recent example, four-arm PEG was functionalized with either adenines or thymines (Figure 9A).[233] The two functionalized PEGs self-assembled into a hydrogel network after mixing due to specific hydrogen bonding between the nucleobase pairs. This hydrogel was injectable and demonstrated appropriate properties for potential applications in targeted growth factor delivery and cell encapsulation.
Figure 9.

A) Schematic of hydrogel self-assembly via intermolecular, hydrogen bonding of Watson-Crick base pairs between thymine (T) and adenine (A). Reproduced with permission.[233] Copyright 2012, Royal Society of Chemistry. B) Chemical structure of a self-assembling copolymer with a segmented, multiblock architecture including hydrophilic PEG (blue), hydrophobic chain-extenders (red), and UPy groups (green) that undergo self-complementary quadruple H-bonding to form a hydrogel. Reproduced with permission.[238] Copyright 2014, American Chemical Society.
3.4.2. Multiple Hydrogen Bonding within Synthetic Polymer Blocks
Similar to the Watson-Crick base pairing of nucleobases, other chemical moieties can undergo a type of molecular recognition to form assemblies through multiple hydrogen bonds. One particularly interesting example is ureidopyrimidinone (UPy), which assembles into strong dimers through quadruple hydrogen bonding.[234] Due to its facile synthesis and high equilibrium association constants (Keq = 6 × 107 M−1 in chloroform)[235], UPy has been explored in many non-cytocompatible organic systems.[71] Recently, however, a few studies have utilized UPy to create hydrogels in aqueous environments.[236–238] In these systems, UPy-UPy interactions are shielded within hydrophobic pockets to maintain the aqueous network. The self-complementary, hydrogen-bonding between the UPy units were hypothesized to form reversible and dynamic crosslinks that endowed the hydrogels with great flexibility and rapid self-healing capacity (Figure 9B). Similar to UPy, urea moieties have also been utilized in hydrogels with self-healing properties.[239, 240] Therefore, the design of synthetic block polymers that are decorated with moieties that can assemble through multiple hydrogen-bonding can be an effective strategy to build adaptable hydrogels for cell encapsulation.
In addition to the previously discussed examples, many other interesting biomaterials rely on hydrogen bonding for their self-assembly and are beyond the scope of our discussion. These include materials built from β-sheet assembly,[241–243] self-assembling β-hairpin peptides,[82, 244–246] and silk-like peptides.[247] Gelation in these systems is often a combined effect of hydrogen bonding and other interactions such as fiber entanglements, which may complicate their use in adaptable hydrogel designs.
3.5. Electrostatic (Ionic) Interactions
As mentioned above, electrostatic interactions can play a synergistic role with hydrogen bonding and hydrophobic interactions to form some types of adaptable linkages, such as coiled-coil assembly. In this section, we focus our discussion on hydrogels formed primarily through electrostatic crosslinking.
One particularly interesting and common example is alginate hydrogels, which are crosslinked with divalent ions, usually calcium. Owing to its low toxicity, relatively low cost, and biocompatibility, calcium-crosslinked alginate has been explored for a vast array of biomedical applications,[248–251] including injectable delivery of cell and growth factor.[252–255] In one recent example, blending of ionically crosslinked alginate with covalently crosslinked polyacrylamide was found to result in extremely stretchable and tough hydrogels.[256] In these interpenetrating networks, the covalent crosslinks preserved the memory of the initial state, while the adaptable electrostatic crosslinks were reversibly unzipped and re-zipped during loading and unloading, respectively, to recover the gel’s mechanical properties.
Adaptable, ionically crosslinked hydrogels have been used recently to probe the biophysics of cell-matrix interactions and its effects on cell behavior, some of which have shown similar results to studies using covalently crosslinked hydrogels.[66] MSCs encapsulated in adaptable alginate hydrogels were hypothesized to use their traction forces to reorganize and cluster cell-adhesion ligands within the gel, thereby altering differentiation.[63] Compared with covalently crosslinked alginate hydrogels, ionically crosslinked gels are viscoelastic and exhibit stress relaxation over time when exposed to a constant strain (Figure 10A).[62] Comparison of cell morphology on these two systems revealed that cell spreading was increased on substrates that can undergo stress relaxation (Figure 10B&C).[62] This observation suggests that the current view that cells use traction forces to sense matrix rigidity by gauging resistance cannot be directly applied to viscoelastic substrates. Because tissues in the body are viscoelastic, these data strongly encourage the development of adaptable hydrogels with tunable viscoelastic properties to more fully explore the mechanisms of cell-matrix mechanotransduction.
Figure 10.

A) Stress relaxation of covalently and ionically crosslinked alginate hydrogels. B) Percentage of cells with stress fibres on elastic and stress-relaxing alginate gels with initial moduli of 1.4_kPa. Data are shown as mean±s.d., and ***P<0.001 (Student’s t-test). C) Representative images of cell spreading on elastic and stress-relaxing alginate gels with initial moduli of 1.4_kPa (actin cytoskeleton in green, paxillin marker for focal adhesions in read). Reproduced with permission.[62] Copyright 2015, Nature Publishing Group.
In addition to alginate hydrogels, other systems based on electrostatic interactions have been developed recently as injectable hydrogels,[257–259] indicating that this strategy may have potential for further use in the design of adaptable hydrogels for cell encapsulation and delivery.
3.6. Dynamic Covalent Linkage
In addition to the physical crosslinking mechanisms discussed previously, crosslinks formed by dynamic covalent chemistry can also be used to build adaptable hydrogels. Dynamic covalent chemistry refers to chemical reactions where covalent bonds can be formed, broken, and reformed reversibly under equilibrium conditions.[44] According to our previously described requirements for building adaptable hydrogels, only dynamic covalent reactions with fast reaction kinetics under mild conditions are suitable for cell encapsulation. Other dynamic covalent reactions that rely on the use of catalysts or strong pH, such as reactions involving phenylboronate ester bonds,[41, 260] are not discussed here.
3.6.1. Schiff Base (Imine) Bonds
Schiff base reactions between amine and aldehyde groups have been used to create adaptable hydrogels where the dynamic uncoupling and recoupling of the reversible linkages impart self-healing capability without additional stimuli. For example, hydrogel with rapid gelation kinetics was created through dynamic Schiff base crosslinking between amine groups and benzaldehyde groups (Figure 11A).[261–263] Encapsulated HeLa cells maintained high viability during 3D hydrogel encapsulation and subsequent injection, demonstrating potential for use in injectable cell therapies.[261] Some studies have indicated that the aliphatic Schiff base crosslinking structure might be less stable than the reaction between benzaldehyde and amine groups.[264, 265] Nonetheless, some hydrogel systems based on Schiff base crosslinking between amine and aliphatic groups have been reported for cell encapsulation.[266–268] Interestingly, in all of the reported systems at least one component is a type of polysaccharide derivative, such as oxidized dextran, chitosan, or HA. One reason for this is the ease of controllable syntheses by oxidation of the sugar rings in polysaccharides, and another potential reason may be due to the large amount of crosslinking sites present on these natural biopolymer chains, which allows for a higher crosslinking density and greater hydrogel stability.
Figure 11.

A) A hydrogel was formed through the dynamic covalent Schiff base linkage between amine groups on chitosan (left structure) and benzaldehyde-modified PEG (right structure) under physiological conditions. Cell viability (live: green, dead: red) and spatial distribution of HeLa cells within the hydrogel after 24 hours is shown. Reproduced with permission.[261] Copyright 2012, Royal Society of Chemistry. B) Representatative images of multicellular behavior within two hydrogels formed through dynamic covalent hydrazone crosslinking. Gels with fast (left) or slow (right) stress relaxation behavior resulted in morphological changes in 3D cell structure after 10 days (F-actin in read and nuclei in blue). Reproduced with permission.[52] Copyright 2013, John Wiley and Sons.
3.6.2. Reversible Hydrazone Bonds
Hydrazone bonds, formed by reactions between aldehyde and hydrazine functionalities, are very close relatives to imines. Hydrazone reactions have been widely used in the fields of bioconjugation and materials science because of their stimuli-responsive nature and their rapid formation under physiological conditions.[269–272] Recently, in-situ forming hydrogels were developed for cell encapsulation based on hydrazone bonding.[52, 273–277] Studies have shown that the rate of formation and hydrolysis of the aliphatic aldehyde-derived hydrazone are much faster than a representative aryl aldehyde-derived hydrazone at neutral pH.[50, 272, 278] In one recent study, a cytocompatible, covalently adaptable PEG hydrogel was developed using a dynamic covalent hydrazone linkage (Figure 11B).[52] These PEG hydrogels have a reported Young’s modulus range of 1.8 – 27 kPa, higher than that of most physically crosslinked hydrogels. Hydrogels formed with 100% aliphatic crosslinks displayed rapid stress-relaxation kinetics. These gels allowed encapsulated C2C12 myoblasts to extend filopodia and lamellipodia and to fuse into multinucleated structures, which is a sign of the beginning stages of differentiation. In contrast, in the more slowly relaxing hydrogels formed with 100% aryl aldehyde crosslinks, most cells remained rounded after 10 days of culture with less than 30% of cells extending processes. They further used this adaptable hydrogel system to encapsulate embryonic stem cell-derived motor neurons to calculate the forces and energies required for neurite extension using fundamental physical relationships describing classical mechanics and viscoelastic materials.[35] In another recent study, an injectable hydrogel based on hydrazone linkages was developed for local, on-demand matrix metalloproteinase (MMP) inhibition.[279] A recombinant tissue inhibitor of MMP (rTIMP-3) was sequestered in the hydrogels through electrostatic interactions and released in response to MMP activity. This type of system may be helpful in translating MMP inhibitors to clinical application by overcoming their current dose-limiting side effects.
3.6.3. Oxime bonds
Another imine similar linkage, oxime bonds formed by the reaction between hydroxylamine and aldehyde or ketone, might be another good candidate to build adaptable hydrogels for cell encapsulation. Oxime bond formation has been shown to be biocompatible[280] and suitable for building injectable hydrogels[281]. Oximes exhibit higher hydrolytic stability than imines and hydrazones, with the equilibrium lying far toward the oxime.[282] This means that the oxime linkage may be harder to break with the same exerted force. Careful analysis of the kinetics of bond breaking and formation is needed before applying this reaction to make adaptable hydrogels that can be locally and reversibly remodeled by encapsulated cells.
3.6.4. Disulfide bonds
Disulfide bonds, formed through the thiol side chains of cysteine residues, play a crucial role in protein folding and assembly.[283–285] Although thiol groups are unreactive in the reduced state and the formation of disulfide-crosslinked hydrogels requires the presence of an oxidation agent, mild oxidative conditions including oxygen itself is found to be sufficient to drive cytocompatible crosslinking.[286–288] Disulfide crosslinking has been utilized in a variety of clever biomaterials design strategies,[289–291] and may provide an opportunity for building adaptable hydrogels. In particular, the thiol-disulfide exchange reaction was found to result in hydrogels with fast gelation kinetics and cytocompatibility with many different cell types.[292, 293] Potential limitations for this crosslinking mechanism include off-target reactions with encapsulated proteins which may interfere with their bioactivity. In addition, disulfide hydrogels may exhibit poor stability in the presence of reducing agents such as glutathione, a tripeptide that is found in high concentrations in some tissues.[289, 294, 295]
3.6.5. Reversible Diels-Alder Reaction
Reversible Diels-Alder (DA) reactions may also provide a promising route to adaptable hydrogel formation. DA reactions belong to the family of ‘click’ reactions, which are characterized by high selectivity and high yield with no appreciable side products.[296, 297] DA reactions can occur in aqueous media at physiologically compatible conditions and have been explored for biological applications in drug delivery[298, 299] and tissue engineering[299, 300]. The equilibrium of the DA reaction is thermally controlled; elevated temperatures induce the reformation of maleimide and furan moieties, whereas lower temperatures favor the adduct formation.[298] Previous studies suggest that very high temperatures (>100 °C) might be needed to trigger the reversible DA reaction, limiting its utility in biomedical applications.[42, 301] Nonetheless, recent studies have shown that it is possible to obtain self-healing hydrogels formed by reversible DA reactions under physiological conditions.[302] In one recent study, mixing of furyl and maleimide-modified PEGs resulted in hydrogel formation at 37 °C within 15 min.[303] Therefore, reversible DA reactions may be a useful component for future adaptable hydrogel designs.
4. Conclusions and Future Opportunities
In summary, a wide range of reversible linkages is being developed to generate adaptable hydrogels, which is an emerging new class of responsive materials for 3D cell encapsulation. This attractive platform presents many advantages compared with conventional, chemically crosslinked hydrogels. Adaptable hydrogels not only possess local adaptability to permit complex cell functions to occur, but also maintain their long-term overall stability due to the absence of irreversible bulk hydrogel degradation.
Although it shows great promise, the development of adaptable hydrogels for cell encapsulation is not without its challenges. As mentioned before, the time scales of cell action and hydrogel rearrangement need to be matched when developing adaptable hydrogels for 3D cell encapsulation. This requires study of both the dynamics of cell behavior and the kinetics of hydrogel rearrangement, including gelation and relaxation. For reference, several studies on the dynamics of cell spreading, migration, proliferation, and differentiation in synthetic 3D matrices have been performed.[304–307] Additionally, kinetic studies using representative small molecules have been proved to be helpful in characterizing hydrogel evolution and rearrangement. In one recent study, the rheological characteristics of a hydrazone-crosslinked PEG hydrogel was described by the kinetics of a small-molecule model system containing the reactive end groups.[50] The rates of gelation and gel relaxation were found to correspond closely with the rates of bond formation and hydrolysis, respectively. However, it should be noted that this method may not necessarily be sufficient to describe every hydrogel system, especially for synthetic polymers that are randomly copolymerized or pendant-modified and those consisting of naturally derived polymers with unevenly dispersed functional groups. In addition, the presence of cells may alter the kinetics and thermodynamics of crosslink formation and breakage. In particular, cells may interact with adaptable crosslinks through both physical and biochemical mechanisms. For example, as described in section 2, cells may exert local forces on crosslinks to modulate their coupling and uncoupling. Similarly, cells may secrete biomolecules that alter the local pH or redox environment resulting in modification of dynamic crosslink kinetics or thermodynamics. Therefore, individual systems still require careful analysis and modification. For a comprehensive review of the characterization techniques for dynamic structures, readers are referred to two excellent reviews published recently.[308, 309]
As researchers gain a better understanding of dynamic linkages and adaptable hydrogel design criteria, the application of adaptable hydrogels is likely to be expanded to many areas of bioengineering. For example, adaptable linkages can be applied in drug delivery systems to achieve control over drug release profiles.[110, 112, 127, 281, 310] Adaptable hydrogels can also be used as injectable systems for delivery of regenerative cell therapies due to their shear-thinning and self-healing properties. These same properties make adaptable hydrogels ideal for use in extrusion-based 3D bioprinting.[311] Furthermore, as a viscoelastic platform for in vitro cell culture, adaptable hydrogels could be useful for a variety of fundamental studies of cell biophysics, including quantification of cell-exerted forces over time and cell responses to stress-relaxation. Since native tissues are viscoelastic, but he majority of cell mechanotransduction studies to date have relied on use of elastic materials. Adaptable hydrogels may offer a more biomimetic scaffold for biophysical studies.
Acknowledgments
The authors gratefully acknowledge the suggestions and advice from Amy Proctor, Karen Dubbin and Dr. Lei Cai. The authors acknowledge funding provided by NSF DMR-0846363, NIH R21-AR062359, NIH DP2-OD-006477, and NIH R01-DK085720 (S.C.H.), and Kodak fellowship (H.W.).
Biographies
 Prof. Sarah C. Heilshorn is an Associate Professor of Materials Science and Engineering at Stanford University. She obtained a B.S. in Chemical Engineering from the Georgia Institute of Technology in 1998 and a Ph.D. in Chemical Engineering from the California Institute of Technology in 2004. She worked in the Department of Molecular and Cell Biology at the University of California, Berkeley as a postdoctoral scholar. The Heilshorn Biomaterials Group studies the design of materials and scaffolds that mimic the micro- and nano-scale order found in nature for applications in energy, tissue engineering, and regenerative medicine.
Prof. Sarah C. Heilshorn is an Associate Professor of Materials Science and Engineering at Stanford University. She obtained a B.S. in Chemical Engineering from the Georgia Institute of Technology in 1998 and a Ph.D. in Chemical Engineering from the California Institute of Technology in 2004. She worked in the Department of Molecular and Cell Biology at the University of California, Berkeley as a postdoctoral scholar. The Heilshorn Biomaterials Group studies the design of materials and scaffolds that mimic the micro- and nano-scale order found in nature for applications in energy, tissue engineering, and regenerative medicine.
 Huiyuan Wang obtained a B.S. in Polymer Science and Engineering from the University of Science and Technology of China in 2012. Now she is pursuing her Ph.D. in the Department of Materials Science and Engineering at Stanford University. Her research project involves the synthesis of hybrid hydrogels based on blends of engineered proteins and synthetic polymers and their applications as in vitro mimics of the extracellular matrix.
Huiyuan Wang obtained a B.S. in Polymer Science and Engineering from the University of Science and Technology of China in 2012. Now she is pursuing her Ph.D. in the Department of Materials Science and Engineering at Stanford University. Her research project involves the synthesis of hybrid hydrogels based on blends of engineered proteins and synthetic polymers and their applications as in vitro mimics of the extracellular matrix.
References
- 1.Kharkar PM, Kiick KL, Kloxin AM. Chem Soc Rev. 2013;42:7335. doi: 10.1039/c3cs60040h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan G, Mooney DJ. Trends Biotechnol. 2008;26:382. doi: 10.1016/j.tibtech.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 3.Cai L, Heilshorn SC. Acta Biomater. 2014;10:1751. doi: 10.1016/j.actbio.2013.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoffman BD, Grashoff C, Schwartz MA. Nature. 2011;475:316. doi: 10.1038/nature10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Engler AJ, Sen S, Sweeney HL, Discher DE. Cell. 2006;126:677. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 6.Pek YS, Wan ACA, Ying JY. Biomaterials. 2010;31:385. doi: 10.1016/j.biomaterials.2009.09.057. [DOI] [PubMed] [Google Scholar]
- 7.Park JS, Chu JS, Tsou AD, Diop R, Tang ZY, Wang AJ, Li S. Biomaterials. 2011;32:3921. doi: 10.1016/j.biomaterials.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhong CL, Chrzanowska-Wodnicka M, Brown J, Shaub A, Belkin AM, Burridge K. J Cell Biol. 1998;141:539. doi: 10.1083/jcb.141.2.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lutolf MP, Hubbell JA. Nature biotechnology. 2005;23:47. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 10.Nicodemus GD, Bryant SJ. Tissue Eng Pt B-Rev. 2008;14:149. doi: 10.1089/ten.teb.2007.0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tibbitt MW, Anseth KS. Biotechnol Bioeng. 2009;103:655. doi: 10.1002/bit.22361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drury JL, Mooney DJ. Biomaterials. 2003;24:4337. doi: 10.1016/s0142-9612(03)00340-5. [DOI] [PubMed] [Google Scholar]
- 13.Peppas NA, Hilt JZ, Khademhosseini A, Langer R. Adv Mater. 2006;18:1345. [Google Scholar]
- 14.Cruise GM, Scharp DS, Hubbell JA. Biomaterials. 1998;19:1287. doi: 10.1016/s0142-9612(98)00025-8. [DOI] [PubMed] [Google Scholar]
- 15.Lopergolo LC, Lugao AB, Catalani LH. Polymer. 2003;44:6217. [Google Scholar]
- 16.Liu Y, Chan-Park MB. Biomaterials. 2009;30:196. doi: 10.1016/j.biomaterials.2008.09.041. [DOI] [PubMed] [Google Scholar]
- 17.Benoit DS, Schwartz MP, Durney AR, Anseth KS. Nat Mater. 2008;7:816. doi: 10.1038/nmat2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khetan S, Guvendiren M, Legant WR, Cohen DM, Chen CS, Burdick JA. Nat Mater. 2013;12:458. doi: 10.1038/nmat3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kutty JK, Cho E, Lee JS, Vyavahare NR, Webb K. Biomaterials. 2007;28:4928. doi: 10.1016/j.biomaterials.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 20.Qiu YZ, Lim JJ, Scott L, Adams RC, Bui HT, Temenoff JS. Acta biomaterialia. 2011;7:959. doi: 10.1016/j.actbio.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 21.Burkoth AK, Anseth KS. Biomaterials. 2000;21:2395. doi: 10.1016/s0142-9612(00)00107-1. [DOI] [PubMed] [Google Scholar]
- 22.Bryant SJ, Anseth KS. J Biomed Mater Res A. 2003;64A:70. doi: 10.1002/jbm.a.10319. [DOI] [PubMed] [Google Scholar]
- 23.Sahoo S, Chung C, Khetan S, Burdick JA. Biomacromolecules. 2008;9:1088. doi: 10.1021/bm800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benoit DSW, Durney AR, Anseth KS. Tissue Eng. 2006;12:1663. doi: 10.1089/ten.2006.12.1663. [DOI] [PubMed] [Google Scholar]
- 25.Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. P Natl Acad Sci USA. 2003;100:5413. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Place ES, Evans ND, Stevens MM. Nat Mater. 2009;8:457. doi: 10.1038/nmat2441. [DOI] [PubMed] [Google Scholar]
- 27.Sabeh F, Shimizu-Hirota R, Weiss SJ. J Cell Biol. 2009;185:11. doi: 10.1083/jcb.200807195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kloxin AM, Kloxin CJ, Bowman CN, Anseth KS. Adv Mater. 2010;22:3484. doi: 10.1002/adma.200904179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeForest CA, Polizzotti BD, Anseth KS. Nat Mater. 2009;8:659. doi: 10.1038/nmat2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Potter SM, DeMarse TB. J Neurosci Meth. 2001;110:17. doi: 10.1016/s0165-0270(01)00412-5. [DOI] [PubMed] [Google Scholar]
- 31.Rodin S, Domogatskaya A, Strom S, Hansson EM, Chien KR, Inzunza J, Hovatta O, Tryggvason K. Nature biotechnology. 2010;28:611. doi: 10.1038/nbt.1620. [DOI] [PubMed] [Google Scholar]
- 32.Lu PF, Weaver VM, Werb Z. J Cell Biol. 2012;196:395. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu PF, Takai K, Weaver VM, Werb Z. Csh Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Romano NH, Sengupta D, Chung C, Heilshorn SC. Bba-Gen Subjects. 2011;1810:339. doi: 10.1016/j.bbagen.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKinnon DD, Domaille DW, Brown TE, Kyburz KA, Kiyotake E, Cha JN, Anseth KS. Soft Matter. 2014;10:9230. doi: 10.1039/c4sm01365d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fairbanks BD, Singh SP, Bowman CN, Anseth KS. Macromolecules. 2011;44:2444. doi: 10.1021/ma200202w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kloxin AM, Kasko AM, Salinas CN, Anseth KS. Science. 2009;324:59. doi: 10.1126/science.1169494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adzima BJ, Kloxin CJ, Bowman CN. Adv Mater. 2010;22:2784. doi: 10.1002/adma.200904138. [DOI] [PubMed] [Google Scholar]
- 39.Scott TF, Schneider AD, Cook WD, Bowman CN. Science. 2005;308:1615. doi: 10.1126/science.1110505. [DOI] [PubMed] [Google Scholar]
- 40.Gillette BM, Jensen JA, Wang MX, Tchao J, Sia SK. Adv Mater. 2010;22:686. doi: 10.1002/adma.200902265. [DOI] [PubMed] [Google Scholar]
- 41.Roberts MC, Hanson MC, Massey AP, Karren EA, Kiser PF. Adv Mater. 2007;19:2503. [Google Scholar]
- 42.Chen XX, Dam MA, Ono K, Mal A, Shen HB, Nutt SR, Sheran K, Wudl F. Science. 2002;295:1698. doi: 10.1126/science.1065879. [DOI] [PubMed] [Google Scholar]
- 43.Hoffman AS. Adv Drug Deliver Rev. 2012;64:18. [Google Scholar]
- 44.Rowan SJ, Cantrill SJ, Cousins GR, Sanders JK, Stoddart JF. Angewandte Chemie. 2002;41:898. doi: 10.1002/1521-3773(20020315)41:6<898::aid-anie898>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 45.Lehn JM. Chem-Eur J. 1999;5:2455. [Google Scholar]
- 46.Ghosh B, Urban MW. Science. 2009;323:1458. doi: 10.1126/science.1167391. [DOI] [PubMed] [Google Scholar]
- 47.Zheng PW, McCarthy TJ. J Am Chem Soc. 2012;134:2024. doi: 10.1021/ja2113257. [DOI] [PubMed] [Google Scholar]
- 48.Imato K, Nishihara M, Kanehara T, Amamoto Y, Takahara A, Otsuka H. Angew Chem Int Edit. 2012;51:1138. doi: 10.1002/anie.201104069. [DOI] [PubMed] [Google Scholar]
- 49.Ying HZ, Zhang YF, Cheng JJ. Nat Commun. 2014;5 doi: 10.1038/ncomms4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McKinnon DD, Domaille DW, Cha JN, Anseth KS. Chem Mater. 2014;26:2382. doi: 10.1002/adma.201303680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jin YH, Yu C, Denman RJ, Zhang W. Chem Soc Rev. 2013;42:6634. doi: 10.1039/c3cs60044k. [DOI] [PubMed] [Google Scholar]
- 52.McKinnon DD, Domaille DW, Cha JN, Anseth KS. Adv Mater. 2014;26:865. doi: 10.1002/adma.201303680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guvendiren M, Lu HD, Burdick JA. Soft Matter. 2012;8:260. [Google Scholar]
- 54.Kretlow JD, Klouda L, Mikos AG. Adv Drug Deliver Rev. 2007;59:263. doi: 10.1016/j.addr.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 55.Yu L, Ding JD. Chem Soc Rev. 2008;37:1473. doi: 10.1039/b713009k. [DOI] [PubMed] [Google Scholar]
- 56.Li YL, Rodrigues J, Tomas H. Chem Soc Rev. 2012;41:2193. doi: 10.1039/c1cs15203c. [DOI] [PubMed] [Google Scholar]
- 57.Wust S, Godla ME, Muller R, Hofmann S. Acta biomaterialia. 2014;10:630. doi: 10.1016/j.actbio.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 58.Kolesky DB, Truby RL, Gladman AS, Busbee TA, Homan KA, Lewis JA. Adv Mater. 2014;26:3124. doi: 10.1002/adma.201305506. [DOI] [PubMed] [Google Scholar]
- 59.Ferris CJ, Gilmore KJ, Beirne S, McCallum D, Wallace GG, Panhuis MIH. Biomater Sci-Uk. 2013;1:224. doi: 10.1039/c2bm00114d. [DOI] [PubMed] [Google Scholar]
- 60.Billiet T, Gevaert E, De Schryver T, Cornelissen M, Dubruel P. Biomaterials. 2014;35:49. doi: 10.1016/j.biomaterials.2013.09.078. [DOI] [PubMed] [Google Scholar]
- 61.Rodell CB, MacArthur JW, Dorsey SM, Wade RJ, Wang LL, Woo YJ, Burdick JA. Adv Funct Mater. 2015;25:636. doi: 10.1002/adfm.201403550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chaudhuri O, Gu L, Darnell M, Klumpers D, Bencherif SA, Weaver JC, Huebsch N, Mooney DJ. Nat Commun. 2015;6:6365. doi: 10.1038/ncomms7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huebsch N, Arany PR, Mao AS, Shvartsman D, Ali OA, Bencherif SA, Rivera-Feliciano J, Mooney DJ. Nat Mater. 2010;9:518. doi: 10.1038/nmat2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muller C, Muller A, Pompe T. Soft Matter. 2013;9:6207. [Google Scholar]
- 65.Murrell M, Kamm R, Matsudaira P. Biophys J. 2011;101:297. doi: 10.1016/j.bpj.2011.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cameron AR, Frith JE, Cooper-White JJ. Biomaterials. 2011;32:5979. doi: 10.1016/j.biomaterials.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 67.Cameron AR, Frith JE, Gomez GA, Yap AS, Cooper-White JJ. Biomaterials. 2014;35:1857. doi: 10.1016/j.biomaterials.2013.11.023. [DOI] [PubMed] [Google Scholar]
- 68.Brandl F, Sommer F, Goepferich A. Biomaterials. 2007;28:134. doi: 10.1016/j.biomaterials.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 69.Rubinstein M, Colby RH. Polymer physics. Oxford University Press; Oxford ; New York: 2003. [Google Scholar]
- 70.Gennes PGd. Scaling concepts in polymer physics. Cornell University Press; Ithaca, N.Y: 1979. [Google Scholar]
- 71.Appel EA, del Barrio J, Loh XJ, Scherman OA. Chem Soc Rev. 2012;41:6195. doi: 10.1039/c2cs35264h. [DOI] [PubMed] [Google Scholar]
- 72.Brunsveld L, Folmer BJB, Meijer EW, Sijbesma RP. Chem Rev. 2001;101:4071. doi: 10.1021/cr990125q. [DOI] [PubMed] [Google Scholar]
- 73.Seiffert S, Sprakel J. Chem Soc Rev. 2012;41:909. doi: 10.1039/c1cs15191f. [DOI] [PubMed] [Google Scholar]
- 74.Cai L, Dewi RE, Heilshorn SC. Advanced Functional Materials. 2015 doi: 10.1002/adfm.201403631. n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Foo CTSWP, Lee JS, Mulyasasmita W, Parisi-Amon A, Heilshorn SC. P Natl Acad Sci USA. 2009;106:22067. doi: 10.1073/pnas.0904851106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mulyasasmita W, Lee JS, Heilshorn SC. Biomacromolecules. 2011;12:3406. doi: 10.1021/bm200959e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lu HD, Soranno DE, Rodell CB, Kim IL, Burdick JA. Advanced healthcare materials. 2013;2:1028. doi: 10.1002/adhm.201200343. [DOI] [PubMed] [Google Scholar]
- 78.Glassman MJ, Chan J, Olsen BD. Adv Funct Mater. 2013;23:1182. doi: 10.1002/adfm.201202034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Erman B, Mark JE. Structures and properties of rubberlike networks. Oxford University Press; New York: 1997. [Google Scholar]
- 80.Treloar LRG. The physics of rubber elasticity/by L.R.G. Treloar. Clarendon Press ; Oxford University Press; Oxford New York: 2005. [Google Scholar]
- 81.Jay JI, Langheinrich K, Hanson MC, Mahalingam A, Kiser PF. Soft Matter. 2011;7:5826. [Google Scholar]
- 82.Haines-Butterick L, Rajagopal K, Branco M, Salick D, Rughani R, Pilarz M, Lamm MS, Pochan DJ, Schneider JP. P Natl Acad Sci USA. 2007;104:7791. doi: 10.1073/pnas.0701980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Spiller DG, Wood CD, Rand DA, White MRH. Nature. 2010;465:736. doi: 10.1038/nature09232. [DOI] [PubMed] [Google Scholar]
- 84.Liu Y, Liu B, Riesberg JJ, Shen W. Macromol Biosci. 2011;11:1325. doi: 10.1002/mabi.201100119. [DOI] [PubMed] [Google Scholar]
- 85.del Rio A, Perez-Jimenez R, Liu RC, Roca-Cusachs P, Fernandez JM, Sheetz MP. Science. 2009;323:638. doi: 10.1126/science.1162912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sawada Y, Tamada M, Dubin-Thaler BJ, Cherniavskaya O, Sakai R, Tanaka S, Sheetz MP. Cell. 2006;127:1015. doi: 10.1016/j.cell.2006.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Friedland JC, Lee MH, Boettiger D. Science. 2009;323:642. doi: 10.1126/science.1168441. [DOI] [PubMed] [Google Scholar]
- 88.Lv C, Gao X, Li W, Xue B, Qin M, Burtnick LD, Zhou H, Cao Y, Robinson RC, Wang W. Nat Commun. 2014;5:4623. doi: 10.1038/ncomms5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Marshall BT, Long M, Piper JW, Yago T, McEver RP, Zhu C. Nature. 2003;423:190. doi: 10.1038/nature01605. [DOI] [PubMed] [Google Scholar]
- 90.Thomas WE, Vogel V, Sokurenko E. Annual review of biophysics. 2008;37:399. doi: 10.1146/annurev.biophys.37.032807.125804. [DOI] [PubMed] [Google Scholar]
- 91.Thomas W, Forero M, Yakovenko O, Nilsson L, Vicini P, Sokurenko E, Vogel V. Biophys J. 2006;90:753. doi: 10.1529/biophysj.105.066548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Neuman KC, Nagy A. Nature methods. 2008;5:491. doi: 10.1038/nmeth.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stigler J, Ziegler F, Gieseke A, Gebhardt JC, Rief M. Science. 2011;334:512. doi: 10.1126/science.1207598. [DOI] [PubMed] [Google Scholar]
- 94.Brown AE, Litvinov RI, Discher DE, Weisel JW. Biophys J. 2007;92:L39. doi: 10.1529/biophysj.106.101261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Harada A, Takashima Y, Nakahata M. Accounts of chemical research. 2014;47:2128. doi: 10.1021/ar500109h. [DOI] [PubMed] [Google Scholar]
- 96.Schmidt BVKJ, Hetzer M, Ritter H, Barner-Kowollik C. Progress in polymer science. 2014;39:235. [Google Scholar]
- 97.Qu DH, Wang QC, Zhang QW, Ma X, Tian H. Chem Rev. 2015 doi: 10.1021/cr5006342. [DOI] [PubMed] [Google Scholar]
- 98.Bender ML, Komiyama M. Cyclodextrin chemistry. Springer-Verlag; Berlin ; New York: 1978. [Google Scholar]
- 99.Li J, Loh XJ. Adv Drug Deliver Rev. 2008;60:1000. doi: 10.1016/j.addr.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 100.Li J, Harada A, Kamachi M. Polym J. 1994;26:1019. [Google Scholar]
- 101.Harada A, Kawaguchi Y, Nishiyama T, Kamachi M. Macromol Rapid Comm. 1997;18:535. [Google Scholar]
- 102.Hernandez R, Rusa M, Rusa CC, Lopez D, Mijangos C, Tonelli AE. Macromolecules. 2004;37:9620. [Google Scholar]
- 103.Harada A, Li J, Kamachi M. Nature. 1992;356:325. [Google Scholar]
- 104.Jing B, Chen X, Wang XD, Zhao YR, Qiu HY. Chemphyschem. 2008;9:249. doi: 10.1002/cphc.200700625. [DOI] [PubMed] [Google Scholar]
- 105.Harada A, Okada M, Li J, Kamachi M. Macromolecules. 1995;28:8406. [Google Scholar]
- 106.Harada A, Li J, Kamachi M. Nature. 1994;370:126. [Google Scholar]
- 107.Kawabata R, Katoono R, Yamaguchi M, Yui N. Macromolecules. 2007;40:1011. [Google Scholar]
- 108.Koopmans C, Ritter H. Macromolecules. 2008;41:7418. [Google Scholar]
- 109.Wang J, Main DT, Guo XH, Li L, Lincoln SF, Luo ZF, Ke HL, Zheng L, Prud’homme RK. Ind Eng Chem Res. 2010;49:609. [Google Scholar]
- 110.Rodell CB, Kaminski AL, Burdick JA. Biomacromolecules. 2013;14:4125. doi: 10.1021/bm401280z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.van de Manakker F, van der Pot M, Vermonden T, van Nostrum CF, Hennink WE. Macromolecules. 2008;41:1766. [Google Scholar]
- 112.van de Manakker F, Braeckmans K, el Morabit N, De Smedt SC, van Nostrum CF, Hennink WE. Adv Funct Mater. 2009;19:2992. [Google Scholar]
- 113.Kim J, Jung IS, Kim SY, Lee E, Kang JK, Sakamoto S, Yamaguchi K, Kim K. J Am Chem Soc. 2000;122:540. [Google Scholar]
- 114.Appel EA, Loh XJ, Jones ST, Dreiss CA, Scherman OA. Biomaterials. 2012;33:4646. doi: 10.1016/j.biomaterials.2012.02.030. [DOI] [PubMed] [Google Scholar]
- 115.Uzunova VD, Cullinane C, Brix K, Nau WM, Day AI. Org Biomol Chem. 2010;8:2037. doi: 10.1039/b925555a. [DOI] [PubMed] [Google Scholar]
- 116.Hettiarachchi G, Nguyen D, Wu J, Lucas D, Ma D, Isaacs L, Briken V. Plos One. 2010:5. doi: 10.1371/journal.pone.0010514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Appel EA, Loh XJ, Jones ST, Biedermann F, Dreiss CA, Scherman OA. J Am Chem Soc. 2012;134:11767. doi: 10.1021/ja3044568. [DOI] [PubMed] [Google Scholar]
- 118.Rowland MJ, Appel EA, Coulston RJ, Scherman OA. J Mater Chem B. 2013;1:2904. doi: 10.1039/c3tb20180e. [DOI] [PubMed] [Google Scholar]
- 119.Park KM, Yang JA, Jung H, Yeom J, Park JS, Park KH, Hoffman AS, Hahn SK, Kim K. Acs Nano. 2012;6:2960. doi: 10.1021/nn204123p. [DOI] [PubMed] [Google Scholar]
- 120.Jung H, Park JS, Yeom J, Selvapalam N, Park KM, Oh K, Yang JA, Park KH, Hahn SK, Kim K. Biomacromolecules. 2014;15:707. doi: 10.1021/bm401123m. [DOI] [PubMed] [Google Scholar]
- 121.Yeom J, Kim SJ, Jung H, Namkoong H, Yang J, Hwang BW, Oh K, Kim K, Sung YC, Hahn SK. Advanced healthcare materials. 2014 [Google Scholar]
- 122.Perkins JR, Diboun I, Dessailly BH, Lees JG, Orengo C. Structure. 2010;18:1233. doi: 10.1016/j.str.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 123.Macias MJ, Gervais V, Civera C, Oschkinat H. Nat Struct Biol. 2000;7:375. doi: 10.1038/75144. [DOI] [PubMed] [Google Scholar]
- 124.Macias MJ, Wiesner S, Sudol M. Febs Lett. 2002;513:30. doi: 10.1016/s0014-5793(01)03290-2. [DOI] [PubMed] [Google Scholar]
- 125.Pires JR, Taha-Nejad F, Toepert F, Ast T, Hoffmuller U, Schneider-Mergener J, Kuhne R, Macias MJ, Oschkinat L. J Mol Biol. 2001;314:1147. doi: 10.1006/jmbi.2000.5199. [DOI] [PubMed] [Google Scholar]
- 126.Verdecia MA, Bowman ME, Lu KP, Hunter T, Noel JP. Nat Struct Biol. 2000;7:639. doi: 10.1038/77929. [DOI] [PubMed] [Google Scholar]
- 127.Mulyasasmita W, Cai L, Dewi RE, Jha A, Ullmann SD, Luong RH, Huang NF, Heilshorn SC. Journal of controlled release : official journal of the Controlled Release Society. 2014 doi: 10.1016/j.jconrel.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Parisi-Amon A, Mulyasasmita W, Chung C, Heilshorn SC. Advanced healthcare materials. 2013;2:428. doi: 10.1002/adhm.201200293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mulyasasmita W, Cai L, Hori Y, Heilshorn SC. Tissue engineering. Part A. 2014 doi: 10.1089/ten.tea.2013.0357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kanelis V, Rotin D, Forman-Kay JD. Nat Struct Biol. 2001;8:407. doi: 10.1038/87562. [DOI] [PubMed] [Google Scholar]
- 131.Russ WP, Lowery DM, Mishra P, Yaffe MB, Ranganathan R. Nature. 2005;437:579. doi: 10.1038/nature03990. [DOI] [PubMed] [Google Scholar]
- 132.Socolich M, Lockless SW, Russ WP, Lee H, Gardner KH, Ranganathan R. Nature. 2005;437:512. doi: 10.1038/nature03991. [DOI] [PubMed] [Google Scholar]
- 133.Wang JY, Zhang JX, Zhang XL, Zhou H. Plos One. 2013:8. [Google Scholar]
- 134.Wang HM, Han AT, Cai YB, Xie Y, Zhou H, Long JF, Yang ZM. Chem Commun. 2013;49:7448. doi: 10.1039/c3cc43711f. [DOI] [PubMed] [Google Scholar]
- 135.Zhang X, Zhou H, Xie Y, Ren C, Ding D, Long J, Yang Z. Advanced healthcare materials. 2014 doi: 10.1002/adhm.201300660. [DOI] [PubMed] [Google Scholar]
- 136.Zhang XL, Chu XL, Wang L, Wang HM, Liang GL, Zhang JX, Long JF, Yang ZM. Angew Chem Int Edit. 2012;51:4388. doi: 10.1002/anie.201108612. [DOI] [PubMed] [Google Scholar]
- 137.Guan DL, Ramirez M, Shao L, Jacobsen D, Barrera I, Lutkenhaus J, Chen ZL. Biomacromolecules. 2013;14:2909. doi: 10.1021/bm400814u. [DOI] [PubMed] [Google Scholar]
- 138.Ito F, Usui K, Kawahara D, Suenaga A, Maki T, Kidoaki S, Suzuki H, Taiji M, Itoh M, Hayashizaki Y, Matsuda T. Biomaterials. 2010;31:58. doi: 10.1016/j.biomaterials.2009.09.026. [DOI] [PubMed] [Google Scholar]
- 139.Rousset R, Fabre S, Desbois C, Bantignies F, Jalinot P. Oncogene. 1998;16:643. doi: 10.1038/sj.onc.1201567. [DOI] [PubMed] [Google Scholar]
- 140.Kanamori M, Sandy P, Marzinotto S, Benetti R, Kai C, Hayashizaki Y, Schneider C, Suzuki H. J Biol Chem. 2003;278:38758. doi: 10.1074/jbc.M306324200. [DOI] [PubMed] [Google Scholar]
- 141.Zhang JX, Yan XJ, Shi CW, Yang X, Guo Y, Tian CL, Long JF, Shen YQ. J Mol Biol. 2008;384:255. doi: 10.1016/j.jmb.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 142.Skelton NJ, Koehler MFT, Zobel K, Wong WL, Yeh S, Pisabarro MT, Yin JP, Lasky LA, Sidhu SS. J Biol Chem. 2003;278:7645. doi: 10.1074/jbc.M209751200. [DOI] [PubMed] [Google Scholar]
- 143.Yan XJ, Zhou H, Zhang JX, Shi CW, Xie XQ, Wu YN, Tian CL, Shen YQ, Long JF. J Mol Biol. 2009;392:967. doi: 10.1016/j.jmb.2009.07.060. [DOI] [PubMed] [Google Scholar]
- 144.Sheng M, Sala C. Annu Rev Neurosci. 2001;24:1. doi: 10.1146/annurev.neuro.24.1.1. [DOI] [PubMed] [Google Scholar]
- 145.Kim EJ, Sheng M. Nat Rev Neurosci. 2004;5:771. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- 146.Lamprecht G, Seidler U. Am J Physiol-Gastr L. 2006;291:G766. doi: 10.1152/ajpgi.00135.2006. [DOI] [PubMed] [Google Scholar]
- 147.Eo HS, Kim S, Koo H, Kim W. Molecules and cells. 2009;27:629. doi: 10.1007/s10059-009-0091-2. [DOI] [PubMed] [Google Scholar]
- 148.Chen JR, Chang BH, Allen JE, Stiffler MA, MacBeath G. Nature biotechnology. 2008;26:1041. doi: 10.1038/nbt.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Shao X, Tan CS, Voss C, Li SS, Deng N, Bader GD. Bioinformatics. 2011;27:383. doi: 10.1093/bioinformatics/btq657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Smith CA, Kortemme T. J Mol Biol. 2010;402:460. doi: 10.1016/j.jmb.2010.07.032. [DOI] [PubMed] [Google Scholar]
- 151.Kaufmann K, Shen N, Mizoue L, Meiler J. Journal of molecular modeling. 2011;17:315. doi: 10.1007/s00894-010-0725-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tanaka Y, Tsumoto K, Nakanishi T, Yasutake Y, Sakai N, Yao M, Tanaka I, Kumagai I. Febs Lett. 2004;556:167. doi: 10.1016/s0014-5793(03)01402-9. [DOI] [PubMed] [Google Scholar]
- 153.Ramirez M, Guan DL, Ugaz V, Chen ZL. J Am Chem Soc. 2013;135:5290. doi: 10.1021/ja401075s. [DOI] [PubMed] [Google Scholar]
- 154.Tanaka T, Sawano M, Ogasahara K, Sakaguchi Y, Bagautdinov B, Katoh E, Kuroishi C, Shinkai A, Yokoyama S, Yutani K. Febs Lett. 2006;580:4224. doi: 10.1016/j.febslet.2006.06.084. [DOI] [PubMed] [Google Scholar]
- 155.Shen W, Zhang KC, Kornfield JA, Tirrell DA. Nat Mater. 2006;5:153. doi: 10.1038/nmat1573. [DOI] [PubMed] [Google Scholar]
- 156.Lu HD, Charati MB, Kim IL, Burdick JA. Biomaterials. 2012;33:2145. doi: 10.1016/j.biomaterials.2011.11.076. [DOI] [PubMed] [Google Scholar]
- 157.Gold MG, Lygren B, Dokurno P, Hoshi N, McConnachie G, Tasken K, Carlson CR, Scott JD, Barford D. Mol Cell. 2006;24:383. doi: 10.1016/j.molcel.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 158.Newlon MG, Roy M, Morikis D, Hausken ZE, Coghlan V, Scott JD, Jennings PA. Nat Struct Biol. 1999;6:222. doi: 10.1038/6663. [DOI] [PubMed] [Google Scholar]
- 159.Alto NM, Soderling SH, Hoshi N, Langeberg LK, Fayos R, Jennings PA, Scott JD. P Natl Acad Sci USA. 2003;100:4445. doi: 10.1073/pnas.0330734100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Foss KB, Solberg R, Simard J, Myklebust F, Hansson V, Jahnsen T, Tasken K. Bba-Gene Struct Expr. 1997;1350:98. doi: 10.1016/s0167-4781(96)00152-2. [DOI] [PubMed] [Google Scholar]
- 161.Diviani D, Soderling J, Scott JD. J Biol Chem. 2001;276:44247. doi: 10.1074/jbc.M106629200. [DOI] [PubMed] [Google Scholar]
- 162.Chang CH, Rossi EA, Goldenberg DM. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13:5586s. doi: 10.1158/1078-0432.CCR-07-1217. [DOI] [PubMed] [Google Scholar]
- 163.Grove TZ, Forster J, Pimienta G, Dufresne E, Regan L. Biopolymers. 2012;97:508. doi: 10.1002/bip.22033. [DOI] [PubMed] [Google Scholar]
- 164.Grove TZ, Osuji CO, Forster JD, Dufresne ER, Regan L. J Am Chem Soc. 2010;132:14024. doi: 10.1021/ja106619w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kajander T, Cortajarena AL, Mochrie S, Regan L. Acta Crystallogr D. 2007;63:800. doi: 10.1107/S0907444907024353. [DOI] [PubMed] [Google Scholar]
- 166.Cortajarena AL, Lois G, Sherman E, O’Hern CS, Regan L, Haran G. J Mol Biol. 2008;382:203. doi: 10.1016/j.jmb.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Cortajarena AL, Liu TY, Hochstrasser M, Regan L. Acs Chem Biol. 2010;5:545. doi: 10.1021/cb9002464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Grove TZ, Hands M, Regan L. Protein Eng Des Sel. 2010;23:449. doi: 10.1093/protein/gzq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jackrei ME, Valverde R, Regan L. Protein Sci. 2009;18:762. doi: 10.1002/pro.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Chattopadhyaya R, Meador WE, Means AR, Quiocho FA. J Mol Biol. 1992;228:1177. doi: 10.1016/0022-2836(92)90324-d. [DOI] [PubMed] [Google Scholar]
- 171.Crivici A, Ikura M. Annu Rev Bioph Biom. 1995;24:85. doi: 10.1146/annurev.bb.24.060195.000505. [DOI] [PubMed] [Google Scholar]
- 172.Hultschig C, Hecht HJ, Frank R. J Mol Biol. 2004;343:559. doi: 10.1016/j.jmb.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 173.Vandonselaar M, Hickie RA, Quail JW, Delbaere LTJ. Nat Struct Biol. 1994;1:795. doi: 10.1038/nsb1194-795. [DOI] [PubMed] [Google Scholar]
- 174.Topp S, Prasad V, Cianci GC, Weeks ER, Gallivan JP. J Am Chem Soc. 2006;128:13994. doi: 10.1021/ja064546i. [DOI] [PubMed] [Google Scholar]
- 175.Venema RC, Sayegh HS, Kent JD, Harrison DG. J Biol Chem. 1996;271:6435. doi: 10.1074/jbc.271.11.6435. [DOI] [PubMed] [Google Scholar]
- 176.Matsubara M, Hayashi N, Titani K, Taniguchi H. J Biol Chem. 1997;272:23050. doi: 10.1074/jbc.272.37.23050. [DOI] [PubMed] [Google Scholar]
- 177.Yap KL, Yuan T, Mal TK, Vogel HJ, Ikura M. J Mol Biol. 2003;328:193. doi: 10.1016/s0022-2836(03)00271-7. [DOI] [PubMed] [Google Scholar]
- 178.Yuan T, Vogel HJ. J Biol Chem. 1998;273:30328. doi: 10.1074/jbc.273.46.30328. [DOI] [PubMed] [Google Scholar]
- 179.Montigiani S, Neri G, Neri P, Neri D. J Mol Biol. 1996;258:6. doi: 10.1006/jmbi.1996.0229. [DOI] [PubMed] [Google Scholar]
- 180.Liang Y, Kiick KL. Acta biomaterialia. 2014;10:1588. doi: 10.1016/j.actbio.2013.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Rabenstein DL. Natural product reports. 2002;19:312. doi: 10.1039/b100916h. [DOI] [PubMed] [Google Scholar]
- 182.Seal BL, Panitch A. Biomacromolecules. 2003;4:1572. doi: 10.1021/bm0342032. [DOI] [PubMed] [Google Scholar]
- 183.Seal BL, Panitch A. Macromolecules. 2006;39:2268. [Google Scholar]
- 184.Jeong KJ, Panitch A. Biomacromolecules. 2009;10:1090. doi: 10.1021/bm801270k. [DOI] [PubMed] [Google Scholar]
- 185.Yamaguchi N, Chae BS, Zhang L, Kiick KL, Furst EM. Biomacromolecules. 2005;6:1931. doi: 10.1021/bm0500042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Wieduwild R, Lin W, Boden A, Kretschmer K, Zhang Y. Biomacromolecules. 2014;15:2058. doi: 10.1021/bm500186a. [DOI] [PubMed] [Google Scholar]
- 187.Fairbrother WJ, Champe MA, Christinger HW, Keyt BA, Starovasnik MA. Structure. 1998;6:637. doi: 10.1016/s0969-2126(98)00065-3. [DOI] [PubMed] [Google Scholar]
- 188.Kim SH, Kiick KL. Macromol Rapid Commun. 2010;31:1231. doi: 10.1002/marc.201000130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Yamaguchi N, Zhang L, Chae BS, Palla CS, Furst EM, Kiick KL. J Am Chem Soc. 2007;129:3040. doi: 10.1021/ja0680358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Cai S, Liu Y, Zheng Shu X, Prestwich GD. Biomaterials. 2005;26:6054. doi: 10.1016/j.biomaterials.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 191.Nie T, Baldwin A, Yamaguchi N, Kiick KL. Journal of controlled release : official journal of the Controlled Release Society. 2007;122:287. doi: 10.1016/j.jconrel.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Lupas A. Trends Biochem Sci. 1996;21:375. [PubMed] [Google Scholar]
- 193.Woolfson DN. Advances in protein chemistry. 2005;70:79. doi: 10.1016/S0065-3233(05)70004-8. [DOI] [PubMed] [Google Scholar]
- 194.Kopecek J, Yang J. Angewandte Chemie. 2012;51:7396. doi: 10.1002/anie.201201040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Parry DAD, Fraser RDB, Squire JM. J Struct Biol. 2008;163:258. doi: 10.1016/j.jsb.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 196.Oakley MG, Hollenbeck JJ. Curr Opin Struc Biol. 2001;11:450. doi: 10.1016/s0959-440x(00)00232-3. [DOI] [PubMed] [Google Scholar]
- 197.DiMarco RL, Heilshorn SC. Adv Mater. 2012;24:3923. doi: 10.1002/adma.201200051. [DOI] [PubMed] [Google Scholar]
- 198.Petka WA, Harden JL, McGrath KP, Wirtz D, Tirrell DA. Science. 1998;281:389. doi: 10.1126/science.281.5375.389. [DOI] [PubMed] [Google Scholar]
- 199.Kennedy SB, deAzevedo ER, Petka WA, Russell TP, Tirrell DA, Hong M. Macromolecules. 2001;34:8675. [Google Scholar]
- 200.Olsen BD, Kornfield JA, Tirrell DA. Macromolecules. 2010;43:9094. doi: 10.1021/ma101434a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Shen W, Lammertink RGH, Sakata JK, Kornfield JA, Tirrell DA. Macromolecules. 2005;38:3909. [Google Scholar]
- 202.Liu B, Liu Y, Lewis AK, Shen W. Biomaterials. 2010;31:4918. doi: 10.1016/j.biomaterials.2010.02.069. [DOI] [PubMed] [Google Scholar]
- 203.Lumb KJ, Kim PS. Biochemistry-Us. 1995;34:8642. doi: 10.1021/bi00027a013. [DOI] [PubMed] [Google Scholar]
- 204.Lau SYM, Taneja AK, Hodges RS. J Biol Chem. 1984;259:3253. [PubMed] [Google Scholar]
- 205.Oshea EK, Rutkowski R, Stafford WF, Kim PS. Science. 1989;245:646. doi: 10.1126/science.2503872. [DOI] [PubMed] [Google Scholar]
- 206.Banwell EF, Abelardo ES, Adams DJ, Birchall MA, Corrigan A, Donald AM, Kirkland M, Serpell LC, Butler MF, Woolfson DN. Nat Mater. 2009;8:596. doi: 10.1038/nmat2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Rosler A, Vandermeulen GWM, Klok HA. Adv Drug Deliver Rev. 2012;64:270. doi: 10.1016/s0169-409x(01)00222-8. [DOI] [PubMed] [Google Scholar]
- 208.Grubbs RB, Sun Z. Chem Soc Rev. 2013;42:7436. doi: 10.1039/c3cs60079c. [DOI] [PubMed] [Google Scholar]
- 209.Nowak AP, Breedveld V, Pakstis L, Ozbas B, Pine DJ, Pochan D, Deming TJ. Nature. 2002;417:424. doi: 10.1038/417424a. [DOI] [PubMed] [Google Scholar]
- 210.Shroff K, Rexeisen EL, Arunagirinathan MA, Kokkoli E. Soft Matter. 2010;6:5064. [Google Scholar]
- 211.Yang CY, Song BB, Ao Y, Nowak AP, Abelowitz RB, Korsak RA, Havton LA, Deming TJ, Sofroniew MV. Biomaterials. 2009;30:2881. doi: 10.1016/j.biomaterials.2009.01.056. [DOI] [PubMed] [Google Scholar]
- 212.Fernandez-Colino A, Arias FJ, Alonso M, Rodriguez-Cabello JC. Biomacromolecules. 2014;15:3781. doi: 10.1021/bm501051t. [DOI] [PubMed] [Google Scholar]
- 213.Zhang JJ, Tokatlian T, Zhong J, Ng QKT, Patterson M, Lowry WE, Carmichael ST, Segura T. Adv Mater. 2011;23:5098. doi: 10.1002/adma.201103349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Li J, Li X, Ni XP, Wang X, Li HZ, Leong KW. Biomaterials. 2006;27:4132. doi: 10.1016/j.biomaterials.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 215.Gupta MK, Martin JR, Werfel TA, Shen T, Page JM, Duvall CL. J Am Chem Soc. 2014;136:14896. doi: 10.1021/ja507626y. [DOI] [PubMed] [Google Scholar]
- 216.Ahn HH, Kim KS, Lee JH, Lee JY, Kim BS, Lee IW, Chun HJ, Kim JH, Lee HB, Kim MS. Tissue engineering Part A. 2009;15:1821. doi: 10.1089/ten.tea.2008.0386. [DOI] [PubMed] [Google Scholar]
- 217.Kwon JS, Kim SW, Kwon DY, Park SH, Son AR, Kim JH, Kim MS. Biomaterials. 2014;35:5337. doi: 10.1016/j.biomaterials.2014.03.045. [DOI] [PubMed] [Google Scholar]
- 218.Glassman MJ, Olsen BD. Soft Matter. 2013;9:6814. doi: 10.1039/C3SM00102D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Fathi A, Mithieux SM, Wei H, Chrzanowski W, Valtchev P, Weiss AS, Dehghani F. Biomaterials. 2014;35:5425. doi: 10.1016/j.biomaterials.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Deming TJ. Soft Matter. 2005;1:28. doi: 10.1039/b500307e. [DOI] [PubMed] [Google Scholar]
- 221.Van Durme K, Van Assche G, Van Mele B. Macromolecules. 2004;37:9596. [Google Scholar]
- 222.Boutris C, Chatzi EG, Kiparissides C. Polymer. 1997;38:2567. [Google Scholar]
- 223.Cao YX, Zhang C, Shen WB, Cheng ZH, Yu LL, Ping QN. Journal of Controlled Release. 2007;120:186. doi: 10.1016/j.jconrel.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 224.Tan HP, Ramirez CM, Miljkovic N, Li H, Rubin JP, Marra KG. Biomaterials. 2009;30:6844. doi: 10.1016/j.biomaterials.2009.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Ibusuki S, Fujii Y, Iwamoto Y, Matsuda T. Tissue Eng. 2003;9:371. doi: 10.1089/107632703764664846. [DOI] [PubMed] [Google Scholar]
- 226.Keten S, Xu Z, Ihle B, Buehler MJ. Nat Mater. 2010;9:359. doi: 10.1038/nmat2704. [DOI] [PubMed] [Google Scholar]
- 227.Qin Z, Buehler MJ. Physical review E, Statistical, nonlinear, and soft matter physics. 2010;82:061906. doi: 10.1103/PhysRevE.82.061906. [DOI] [PubMed] [Google Scholar]
- 228.Roh YH, Ruiz RCH, Peng SM, Lee JB, Luo D. Chem Soc Rev. 2011;40:5730. doi: 10.1039/c1cs15162b. [DOI] [PubMed] [Google Scholar]
- 229.Um SH, Lee JB, Park N, Kwon SY, Umbach CC, Luo D. Nat Mater. 2006;5:797. doi: 10.1038/nmat1741. [DOI] [PubMed] [Google Scholar]
- 230.Cheng EJ, Xing YZ, Chen P, Yang Y, Sun YW, Zhou DJ, Xu LJ, Fan QH, Liu DS. Angew Chem Int Edit. 2009;48:7660. doi: 10.1002/anie.200902538. [DOI] [PubMed] [Google Scholar]
- 231.Park N, Um SH, Funabashi H, Xu JF, Luo D. Nat Mater. 2009;8:432. doi: 10.1038/nmat2419. [DOI] [PubMed] [Google Scholar]
- 232.Dave N, Chan MY, Huang PJJ, Smith BD, Liu JW. J Am Chem Soc. 2010;132:12668. doi: 10.1021/ja106098j. [DOI] [PubMed] [Google Scholar]
- 233.Tan HP, Xiao C, Sun JC, Xiong DS, Hu XH. Chem Commun. 2012;48:10289. doi: 10.1039/c2cc35449g. [DOI] [PubMed] [Google Scholar]
- 234.Beijer FH, Sijbesma RP, Kooijman H, Spek AL, Meijer EW. J Am Chem Soc. 1998;120:6761. [Google Scholar]
- 235.Sontjens SHM, Sijbesma RP, van Genderen MHP, Meijer EW. J Am Chem Soc. 2000;122:7487. [Google Scholar]
- 236.Cui JX, del Campo A. Chem Commun. 2012;48:9302. doi: 10.1039/c2cc34701f. [DOI] [PubMed] [Google Scholar]
- 237.Dankers PYW, Hermans TM, Baughman TW, Kamikawa Y, Kieltyka RE, Bastings MMC, Janssen HM, Sommerdijk NAJM, Larsen A, van Luyn MJA, Bosman AW, Popa ER, Fytas G, Meijer EW. Adv Mater. 2012;24:2703. doi: 10.1002/adma.201104072. [DOI] [PubMed] [Google Scholar]
- 238.Guo MY, Pitet LM, Wyss HM, Vos M, Dankers PYW, Meijer EW. J Am Chem Soc. 2014;136:6969. doi: 10.1021/ja500205v. [DOI] [PubMed] [Google Scholar]
- 239.Cui YL, Tan M, Zhu AD, Guo MY. J Mater Chem B. 2014;2:2978. doi: 10.1039/c4tb00315b. [DOI] [PubMed] [Google Scholar]
- 240.Pawar GM, Koenigs M, Fahimi Z, Cox M, Voets IK, Wyss HM, Sijbesma RP. Biomacromolecules. 2012;13:3966. doi: 10.1021/bm301242v. [DOI] [PubMed] [Google Scholar]
- 241.Dong H, Paramonov SE, Aulisa L, Bakota EL, Hartgerink JD. J Am Chem Soc. 2007;129:12468. doi: 10.1021/ja072536r. [DOI] [PubMed] [Google Scholar]
- 242.Aulisa L, Dong H, Hartgerink JD. Biomacromolecules. 2009;10:2694. doi: 10.1021/bm900634x. [DOI] [PubMed] [Google Scholar]
- 243.Galler KM, Aulisa L, Regan KR, D’Souza RN, Hartgerink JD. J Am Chem Soc. 2010;132:3217. doi: 10.1021/ja910481t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Schneider JP, Pochan DJ, Ozbas B, Rajagopal K, Pakstis L, Kretsinger J. J Am Chem Soc. 2002;124:15030. doi: 10.1021/ja027993g. [DOI] [PubMed] [Google Scholar]
- 245.Sathaye S, Zhang H, Sonmez C, Schneider JP, MacDermaid CM, Von Bargen CD, Saven JG, Pochan DJ. Biomacromolecules. 2014;15:3891. doi: 10.1021/bm500874t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Yan CQ, Mackay ME, Czymmek K, Nagarkar RP, Schneider JP, Pochan DJ. Langmuir. 2012;28:6076. doi: 10.1021/la2041746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Vepari C, Kaplan DL. Progress in polymer science. 2007;32:991. doi: 10.1016/j.progpolymsci.2007.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Rowley JA, Madlambayan G, Mooney DJ. Biomaterials. 1999;20:45. doi: 10.1016/s0142-9612(98)00107-0. [DOI] [PubMed] [Google Scholar]
- 249.Tan WH, Takeuchi S. Adv Mater. 2007;19:2696. [Google Scholar]
- 250.Novikova LN, Mosahebi A, Wiberg M, Terenghi G, Kellerth JO, Novikov LN. J Biomed Mater Res A. 2006;77A:242. doi: 10.1002/jbm.a.30603. [DOI] [PubMed] [Google Scholar]
- 251.Awad HA, Wickham MQ, Leddy HA, Gimble JM, Guilak F. Biomaterials. 2004;25:3211. doi: 10.1016/j.biomaterials.2003.10.045. [DOI] [PubMed] [Google Scholar]
- 252.Park H, Kang SW, Kim BS, Mooney DJ, Lee KY. Macromol Biosci. 2009;9:895. doi: 10.1002/mabi.200800376. [DOI] [PubMed] [Google Scholar]
- 253.Hori Y, Winans AM, Huang CC, Horrigan EM, Irvine DJ. Biomaterials. 2008;29:3671. doi: 10.1016/j.biomaterials.2008.05.033. [DOI] [PubMed] [Google Scholar]
- 254.Zhao LA, Weir MD, Xu HHK. Biomaterials. 2010;31:6502. doi: 10.1016/j.biomaterials.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Silva EA, Mooney DJ. J Thromb Haemost. 2007;5:590. doi: 10.1111/j.1538-7836.2007.02386.x. [DOI] [PubMed] [Google Scholar]
- 256.Sun JY, Zhao XH, Illeperuma WRK, Chaudhuri O, Oh KH, Mooney DJ, Vlassak JJ, Suo ZG. Nature. 2012;489:133. doi: 10.1038/nature11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Park H, Woo EK, Lee KY. Journal of Controlled Release. 2014;196:146. doi: 10.1016/j.jconrel.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 258.Cui H, Zhuang X, He C, Wei Y, Chen X. Acta biomaterialia. 2015;11:183. doi: 10.1016/j.actbio.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 259.Ji DY, Kuo TF, Wu HD, Yang JC, Lee SY. Carbohydrate polymers. 2012;89:1123. doi: 10.1016/j.carbpol.2012.03.083. [DOI] [PubMed] [Google Scholar]
- 260.He LH, Fullenkamp DE, Rivera JG, Messersmith PB. Chem Commun. 2011;47:7497. doi: 10.1039/c1cc11928a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Yang B, Zhang YL, Zhang XY, Tao L, Li SX, Wei Y. Polym Chem-Uk. 2012;3:3235. [Google Scholar]
- 262.Zhang YL, Yang B, Zhang XY, Xu LX, Tao L, Li SX, Wei Y. Chem Commun. 2012;48:9305. doi: 10.1039/c2cc34745h. [DOI] [PubMed] [Google Scholar]
- 263.Zhang YL, Tao L, Li SX, Wei Y. Biomacromolecules. 2011;12:2894. doi: 10.1021/bm200423f. [DOI] [PubMed] [Google Scholar]
- 264.Curtis NF. J Chem Soc. 1960:4409. [Google Scholar]
- 265.Engel AK, Yoden T, Sanui K, Ogata N. J Am Chem Soc. 1985;107:8308. [Google Scholar]
- 266.Tan HP, Chu CR, Payne KA, Marra KG. Biomaterials. 2009;30:2499. doi: 10.1016/j.biomaterials.2008.12.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Bhatia SK, Arthur SD, Chenault HK, Kodokian GK. Biotechnol Lett. 2007;29:1645. doi: 10.1007/s10529-007-9465-8. [DOI] [PubMed] [Google Scholar]
- 268.Weng L, Romanov A, Rooney J, Chen W. Biomaterials. 2008;29:3905. doi: 10.1016/j.biomaterials.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Dirksen A, Dirksen S, Hackeng TM, Dawson PE. J Am Chem Soc. 2006;128:15602. doi: 10.1021/ja067189k. [DOI] [PubMed] [Google Scholar]
- 270.Dirksen A, Dawson PE. Bioconjugate Chem. 2008;19:2543. doi: 10.1021/bc800310p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Dirksen A, Yegneswaran S, Dawson PE. Angew Chem Int Edit. 2010;49:2023. doi: 10.1002/anie.200906756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Kool ET, Park DH, Crisalli P. J Am Chem Soc. 2013;135:17663. doi: 10.1021/ja407407h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Dahlmann J, Krause A, Moller L, Kensah G, Mowes M, Diekmann A, Martin U, Kirschning A, Gruh I, Drager G. Biomaterials. 2013;34:940. doi: 10.1016/j.biomaterials.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 274.Smeets NMB, Bakaic E, Patenaude M, Hoare T. Chem Commun. 2014;50:3306. doi: 10.1039/c3cc48514e. [DOI] [PubMed] [Google Scholar]
- 275.Patenaude M, Hoare T. Biomacromolecules. 2012;13:369. doi: 10.1021/bm2013982. [DOI] [PubMed] [Google Scholar]
- 276.Oommen OP, Wang SJ, Kisiel M, Sloff M, Hilborn J, Varghese OP. Adv Funct Mater. 2013;23:1273. [Google Scholar]
- 277.Yan S, Wang T, Feng L, Zhu J, Zhang K, Chen X, Cui L, Yin J. Biomacromolecules. 2014;15:4495. doi: 10.1021/bm501313t. [DOI] [PubMed] [Google Scholar]
- 278.Nguyen R, Huc I. Chem Commun. 2003:942. doi: 10.1039/b211645f. [DOI] [PubMed] [Google Scholar]
- 279.Purcell BP, Lobb D, Charati MB, Dorsey SM, Wade RJ, Zellars KN, Doviak H, Pettaway S, Logdon CB, Shuman JA, Freels PD, Gorman JH, III, Gorman RC, Spinale FG, Burdick JA. Nat Mater. 2014;13:653. doi: 10.1038/nmat3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Grover GN, Lam J, Nguyen TH, Segura T, Maynard HD. Biomacromolecules. 2012;13:3013. doi: 10.1021/bm301346e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Grover GN, Braden RL, Christman KL. Advanced Materials. 2013;25:2937. doi: 10.1002/adma.201205234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Kalia J, Raines RT. Angew Chem Int Edit. 2008;47:7523. doi: 10.1002/anie.200802651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Gething MJ, Sambrook J. Nature. 1992;355:33. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- 284.Sitia R, Braakman I. Nature. 2003;426:891. doi: 10.1038/nature02262. [DOI] [PubMed] [Google Scholar]
- 285.Ellgaard L, Molinari M, Helenius A. Science. 1999;286:1882. doi: 10.1126/science.286.5446.1882. [DOI] [PubMed] [Google Scholar]
- 286.Patenaude M, Smeets NMB, Hoare T. Macromol Rapid Comm. 2014;35:598. doi: 10.1002/marc.201300818. [DOI] [PubMed] [Google Scholar]
- 287.Swindle-Reilly KE, Shah M, Hamilton PD, Eskin TA, Kaushal S, Ravi N. Invest Ophth Vis Sci. 2009;50:4840. doi: 10.1167/iovs.08-2891. [DOI] [PubMed] [Google Scholar]
- 288.Wu ZM, Zhang XG, Zheng C, Li CX, Zhang SM, Dong RN, Yu DM. Eur J Pharm Sci. 2009;37:198. doi: 10.1016/j.ejps.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 289.Klaikherd A, Nagamani C, Thayumanavan S. J Am Chem Soc. 2009;131:4830. doi: 10.1021/ja809475a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Ryu JH, Chacko RT, Jiwpanich S, Bickerton S, Babu RP, Thayumanavan S. J Am Chem Soc. 2010;132:17227. doi: 10.1021/ja1069932. [DOI] [PubMed] [Google Scholar]
- 291.Oh JK, Siegwart DJ, Lee HI, Sherwood G, Peteanu L, Hollinger JO, Kataoka K, Matyjaszewski K. J Am Chem Soc. 2007;129:5939. doi: 10.1021/ja069150l. [DOI] [PubMed] [Google Scholar]
- 292.Choh SY, Cross D, Wang C. Biomacromolecules. 2011;12:1126. doi: 10.1021/bm101451k. [DOI] [PubMed] [Google Scholar]
- 293.Wu DC, Loh XJ, Wu YL, Lay CL, Liu Y. J Am Chem Soc. 2010;132:15140. doi: 10.1021/ja106639c. [DOI] [PubMed] [Google Scholar]
- 294.Ostergaard H, Tachibana C, Winther JR. J Cell Biol. 2004;166:337. doi: 10.1083/jcb.200402120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Carelli S, Ceriotti A, Cabibbo A, Fassina G, Ruvo M, Sitia R. Science. 1997;277:1681. doi: 10.1126/science.277.5332.1681. [DOI] [PubMed] [Google Scholar]
- 296.Kolb HC, Finn MG, Sharpless KB. Angew Chem Int Edit. 2001;40:2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 297.Nandivada H, Jiang XW, Lahann J. Adv Mater. 2007;19:2197. [Google Scholar]
- 298.Koehler KC, Anseth KS, Bowman CN. Biomacromolecules. 2013;14:538. doi: 10.1021/bm301789d. [DOI] [PubMed] [Google Scholar]
- 299.Koehler KC, Alge DL, Anseth KS, Bowman CN. Biomaterials. 2013;34:4150. doi: 10.1016/j.biomaterials.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Nimmo CM, Owen SC, Shoichet MS. Biomacromolecules. 2011;12:824. doi: 10.1021/bm101446k. [DOI] [PubMed] [Google Scholar]
- 301.Chen XX, Wudl F, Mal AK, Shen HB, Nutt SR. Macromolecules. 2003;36:1802. [Google Scholar]
- 302.Wei Z, Yang JH, Du XJ, Xu F, Zrinyi M, Osada Y, Li F, Chen YM. Macromol Rapid Comm. 2013;34:1464. doi: 10.1002/marc.201300494. [DOI] [PubMed] [Google Scholar]
- 303.Kirchhof S, Brandl FP, Hammer N, Goepferich AM. J Mater Chem B. 2013;1:4855. doi: 10.1039/c3tb20831a. [DOI] [PubMed] [Google Scholar]
- 304.Anderson SB, Lin CC, Kuntzler DV, Anseth KS. Biomaterials. 2011;32:3564. doi: 10.1016/j.biomaterials.2011.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Kyburz KA, Anseth KS. Acta Biomaterialia. 2013;9:6381. doi: 10.1016/j.actbio.2013.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Kraehenbuehl TP, Zammaretti P, Van der Vlies AJ, Schoenmakers RG, Lutolf MP, Jaconi ME, Hubbell JA. Biomaterials. 2008;29:2757. doi: 10.1016/j.biomaterials.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 307.Shepard JA, Stevans AC, Holland S, Wang CE, Shikanov A, Shea LD. Biotechnology and Bioengineering. 2012;109:830. doi: 10.1002/bit.24355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Yu GC, Yan XZ, Han CY, Huang FH. Chem Soc Rev. 2013;42:6697. doi: 10.1039/c3cs60080g. [DOI] [PubMed] [Google Scholar]
- 309.Brandt J, Oehlenschlaeger KK, Schmidt FG, Barner-Kowollik C, Lederer A. Adv Mater. 2014 doi: 10.1002/adma.201400521. [DOI] [PubMed] [Google Scholar]
- 310.Appel EA, Forster RA, Rowland MJ, Scherman OA. Biomaterials. 2014;35:9897. doi: 10.1016/j.biomaterials.2014.08.001. [DOI] [PubMed] [Google Scholar]
- 311.Murphy SV, Atala A. Nature biotechnology. 2014;32:773. doi: 10.1038/nbt.2958. [DOI] [PubMed] [Google Scholar]