

Abstract
BACKGROUND
Personalized medicine is the provision of focused prevention, detection, prognostic, and therapeutic efforts according to an individual’s genetic composition. The actualization of personalized medicine will require combining a patient’s conventional clinical data with bioinformatics-based molecular-assessment profiles. This synergistic approach offers tangible benefits, such as heightened specificity in the molecular classification of cancer subtypes, improved prognostic accuracy, targeted development of new therapies, novel applications for old therapies, and tailored selection and delivery of chemotherapeutics.
CONTENT
Our ability to personalize cancer management is rapidly expanding through biotechnological advances in the postgenomic era. The platforms of genomics, proteomics, single-nucleotide polymorphism profiling and haplotype mapping, high-throughput genomic sequencing, and pharmacogenomics constitute the mechanisms for the molecular assessment of a patient’s tumor. The complementary data derived during these assessments is processed through bioinformatics analysis to offer unique insights for linking expression profiles to disease detection, tumor response to chemotherapy, and patient survival. Together, these approaches permit improved physician capacity to assess risk, target therapies, and tailor a chemotherapeutic treatment course.
SUMMARY
Personalized medicine is poised for rapid growth as the insights provided by new bioinformatics models are integrated with current procedures for assessing and treating cancer patients. Integration of these biological platforms will require refinement of tissue-processing and analysis techniques, particularly in clinical pathology, to overcome obstacles in customizing our ability to treat cancer.
Throughout the present “era of postgenomics,” technological breakthroughs and bioinformatics-based analysis have briskly expanded our ability to unravel the molecular composition and function of disease. These advancements in molecular assessment have heightened the anticipation for their application to improving patient care through personalized medicine (1). Cancer, despite sharing common aberrant physiological alterations, is a diverse constellation of disease processes (2). Various manifestations of cancer are immensely heterogeneous with respect to metastatic potential and resistance to treatment (3). Both of these factors contribute to the failure of modern cancer therapies to durably repress recurrence in patients, as evinced by the stagnant mortality rates over the past 3 decades (4). Thus, the heterogeneous nature of cancer and the shortcomings of currently available therapeutics suggest the potential for a central role for a personalized approach to cancer management. Indeed, the paradigm-shifting concept of targeting a dysregulated kinase in chronic myelogenous leukemia has focused therapeutic development on the comprehension of molecular mechanisms (5).
Current standards of cancer management are ripe for personalization, because opportunities for molecularly assessing mutational dysregulation exist throughout the clinical course of disease progression (Fig. 1). Even before the occurrence of malignant transformation, the interplay between genetic composition and environmental factors shapes an individual’s predisposition for cancer. Screening for these genetic factors can provide clinicians with the insight necessary to recommend modifications to behavior, lifestyle, and diet, while monitoring for disease onset. Upon malignant transformation, the management approach transitions from modifying risk to preventing progression. The bioinformatics-based analyses discussed in this review aim to integrate data from a patient’s clinical presentation and the molecular-expression profile of the patient’s tumor with molecular information from external databases. This multisource integrative approach to cancer management promises to provide unparalleled ability to assess risk, target therapies, and tailor treatments throughout the disease course.
Fig. 1. Pathways in personalized cancer management.
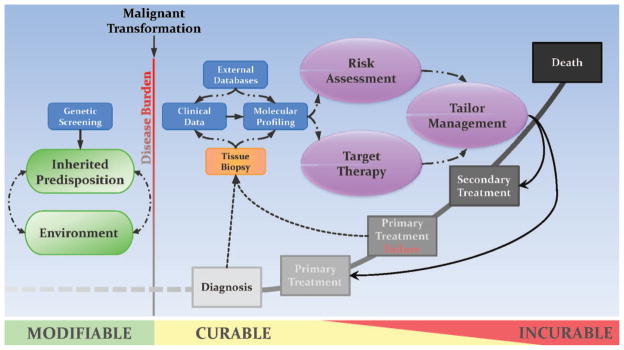
Cancer management is personalized by integrating synergistic molecular-assessment methods with conventional methods of clinical practice. In a prediagnostic setting, combining current knowledge of environmental factors with available screens for heritable cancer-inducing genetic mutations allows physicians an opportunity to modify preventive and monitoring guidelines. The onset of malignant transformation signals a transition from passive to active treatment in order to curtail an increase in disease burden. After diagnosis, the patient’s clinical data are combined with molecular-expression profile data for the tumor, and the data are analyzed with bioinformatics methods that draw upon archival expression data from external databases. This collaborative effort tailors cancer treatment by means of increased accuracy of risk assessment and improved methods so that therapies targeted to specific molecular dysregulations can be selected. Most importantly, the iterative nature of this approach to therapeutic assessment offers a continuous opportunity for physicians to reanalyze risk and select additional therapies to adapt to a patient’s changing molecular profile.
We describe existing molecular-profiling platforms and review applications that may prove useful for current or future contributions to personalized cancer management. We consider the complementary nature of data generated through the various modalities, including transcriptomics, proteomics, single-nucleotide polymorphism (SNP)3 profiling and haplotype mapping, high-throughput genomic sequencing, and pharmacogenomics. We subsequently detail the role of bioinformatics in combining complex, multivariate molecular data to refine networks of interconnectivity among classifications of morphologically distinct neoplasms, identify and validate biomarkers, and aid in the characterization of patients’ risk, prognosis, and therapeutic response. Finally, we consider the broadened role for clinical pathology in mediating the delivery of personalized cancer management.
Molecular Avenues toward Personalized Medicine
Genomic-assessment methods spawned by the sequencing of the human genome have catalyzed an increasingly molecular approach to studying the foundations of human disease. Indeed, profiling platforms that provide insight into the role of genetic sequences, RNA and protein concentrations, and activities of metabolic enzymes in the development of cancer are becoming prominent fixtures in the clinical management of cancer. These molecular-profiling techniques promise to complement current practices of clinical evaluation to permit a more comprehensive staging and assessment of tumor progression. Continuing on this path of complementarity should pave the way toward more personalized diagnoses, prognoses, and predictions of the response to treatment.
Current platforms for molecular assessment include transcriptomics, proteomics, SNP profiling and haplotype mapping, high-throughput genetic sequencing, and pharmacoproteomics. Each of these approaches relies on different formats of biological input; accordingly, each has unique advantages and limitations with respect to personalized medicine. By developing a comprehensive understanding of the strengths and weaknesses of these routes, we can better leverage the information they provide in a collaborative fashion (Table 1).
Table 1.
Molecular platforms for personalized medicine.
| Technology | Strengths | Weaknesses | Applications |
|---|---|---|---|
| Transcriptomics (genomic expression) |
|
|
|
| Proteomics (proteomic expression) |
|
|
|
| SNP/HapMap |
|
|
|
| High-throughput gene sequencing |
|
|
|
| Pharmacogenomics |
|
|
CGAP, Cancer Genome Anatomy Project; HCGP, Human Cancer Genome Project.
TRANSCRIPTOMICS: GENOMIC-EXPRESSION PROFILING
Given that many neoplastic processes are caused by mutations in the genetic code and subsequent errors in transcription, there have been concerted efforts to identify alterations in levels of gene expression that are associated with tumorigenesis. These transcriptomic studies rely on DNA-microarray technology to provide information on aberrant gene expression in human cancer. The 2 forms of microarray methods—those that detect cDNA and oligonucleotide methods—are provided in a variety of commercial and noncommercial platforms. The principle detection method, however, remains the same—mRNAs isolated from a patient’s tumor biopsy or from surgically resected tissue samples are used to create fluorescently labeled cDNA. These cDNA probes hybridize with complementary sequences bound to microarrays to produce a fluorescent signal. The observed intensity of each gene’s signal is proportional to the concentration of the original transcript in the tissue sample. The mechanics of microarrays and their utility in analyzing gene expression in cancer are thoroughly reviewed elsewhere (6).
Growing from a seminal study that classified acute leukemias on the basis of genomic expression (7), the widespread integration of microarray technology into biomedical and clinical oncologic research has led to the identification of the transcriptional status of many different tumors. Additional studies have subsequently identified gene expression signatures that serve as biomarkers, aid in prognostic prediction, and guide chemotherapeutic selection. Minna et al. have comprehensively summarized recent studies that have used expression profiling to derive biomarkers that predict a tumor’s response to chemotherapy (8). Additionally, the large quantities of data produced by these studies have prompted the development of Internet-based resources, such as the Oncomine Research Platform (http://www.oncomine.org/), to catalogue transcriptome profiles from published gene expression analyses into a rapidly accessible database.
Two other elements of transcriptomic assessment in cancer that deserve specific mention are the emerging field of microRNA (miRNA) and the study of epigenetic regulation. Genomic miRNAs are short non-coding sequences of 20–22 nucleotides that exert their regulatory effects by binding to a complementary 3′ untranslated region of mRNA sequences. Studies continue to identify miRNA mutations that can alter the homeostatic regulation of gene expression by acting either as oncogenes (MIR155,4 microRNA 155) or as tumor suppressors (MIR15A, microRNA 15a; MIR16–1, microRNA 16–1) (9). Moreover, microarray expression-profiling methods used for mRNAs have been developed for analysis of noncoding miRNAs (10). Preliminary analyses with this technology have screened both healthy and tumor tissues to identify signatures capable of stratifying patients into predictive prognostic cohorts and treatment subgroups (11, 12). Recent work has suggested an additional route of miRNA regulation, in which reductions in miRNA concentrations in tumors are frequently due to epigenetic modifications, particularly hypermethylation (13).
Dysfunctional epigenetic regulation is conventionally thought to have a broader role in tumor development, most commonly through alterations in DNA methylation (14). The classification of these aberrant methylation patterns and the identification of hypermethylation markers in cancer promise to improve clinical identification and management of individual cancers, as well as affect the personalized application of epigenetics-based treatment regimens, such as 5-azacytidine (Vidaza) and 5-aza-2′-deoxycytidine (decitabine) (14). Although inappropriate methylation remains the most comprehensively studied epi-genetic modification associated with tumorigenesis, histone alterations, such as lysine acetylation and serine phosphorylation, may ultimately become therapeutic targets for reprogramming a malignant cell’s epigenetic code (14).
The capability for multistudy, cross-institutional corroboration and the reproducibility of genomic-expression data currently make transcriptomics one of the most broad-scale, inclusive, and accurate means for personalizing oncologic care. These strengths account for the increasing reliance of expression profiling in assessing chemotherapeutic decisions, despite the often-cited potential disconnect between transcript concentrations and protein translational and functional status (15). Another limitation of transcriptomics, as well as of other molecular-assessment platforms, is the lack of access to patient biosamples, which can lead to the use of samples of heterogeneous quality and type. The lack of a unified, multi-institutional “tumor tissue library” with well-annotated clinical information continues to dampen the impact of transcriptomics on the individualization of cancer management. Finally, proper transcriptomic analysis requires the pathologic discrimination of relevant tumor cells from a heterogeneous background. This hurdle, however, is often overcome with such techniques as laser-capture microdissection and fluorescence-activated cell sorting that help isolate cells pertinent to a tumor’s etiology.
PROTEOMICS
Proteomics, although in an early stage of exponential growth, currently offers extensive capabilities for performing system-wide analyses targeted at elucidating functional protein interactions and discovering novel biomarkers for cancer therapeutics. Given that activated protein-signaling cascades represent the final stage of genomic expression, most modern therapies have been designed to target and disrupt dysfunctional cellular signaling. Thus, the insight that proteomics can provide into the functional status of known neoplastic signaling networks will prove to be of great benefit for tumor classification and subsequent selection of treatment based on pharmacoproteomics (15).
The protein microarray, a technology comparable to the widely used DNA microarray, stands as the cornerstone of proteomics research because of its efficiency in analyzing multiple proteins and their interactions with nucleic acids, lipids, and other small molecules (16). This style of targeted proteomics aims at characterizing the abundance, modification, activity, localization, and interaction of protein-signaling cascades that are widely dysregulated in cancer (16). The format of proteomic microarrays is tailored to the intended focus of the proteomics investigation, e.g., forward-phase arrays with antibodies immobilized to a surface or reversed-phase arrays with immobilized analytes (or each patient’s protein samples). Details of protein microarray formats can be found elsewhere (16, 17).
The potential of proteomic analysis has yet to be fully realized because of technological boundaries and limitations in applicability. Because the exact number of polypeptides produced in humans is uncertain, we are left with estimates that range from hundreds of thousands to millions, if one also counts splice variants and posttranslational modifications that can occur for a given protein species (18). The absence of currently unidentified proteins on modern protein arrays may lead to results that are inconclusive, underpowered, or have numerous false negatives. Further complicating array analysis is the fact that the relative abundances of polypeptides vary by orders of magnitude (18). Some studies have suggested that approximately 90% of proteins exist at moderate to low concentrations (19), potentially below thresholds for separation and detection. Thus, correlating plypeptide quantities obtained with current proteomics techniques to actual protein concentrations while accounting for the interactions of unknown protein species is a biological and computational hurdle. Additional insights into the existence and interactions of polypeptides are sorely needed to decrease the complexity of the nearly intractable algorithms required to account for these shortcomings.
SNP PROFILING AND THE HapMap PROJECT
SNP mapping arose out of a necessity to perform large-scale genomewide detection of genetic variants among patients. Coinciding with this need for efficient genomic analysis have been substantial technological advances that enable the discovery of disease associations among 10 × 106 SNPs in the entire human genome. For example, several commercial SNP arrays already can accurately survey >500 000 SNPs in the human genome (20). Combinations of specific alleles proximate to SNPs within a single chromosome are compiled into haplotypes. Screening for such haplotypes can then be used as efficient methods for ascertaining patterns of DNA sequence variation potentially linked with disease (21).
The task of constructing haplotype maps of human disease has been undertaken by the International HapMap Consortium, which aspires to provide linkage analysis of SNP variants to the public (21). Selective profiling of genes through haplotype mapping remains relatively inexpensive, a substantial benefit given the cost of genomewide sequencing. The ability to apply SNP profiling as a rapid and accurate means to survey populations for biomarkers is yet another beneficial characteristic.
Nevertheless, SNP profiling and haplotype mapping suffer from several limitations. Although understanding genetic variations helps to anticipate tumorigenic predisposition, a sole reliance on this approach generally does not provide information on functional mechanisms of disease or any associated therapeutic targets. Additionally, inferring an association from the binary nature of SNP genotype information often requires a large number of patients, e.g., a few thousand, to detect any genetic effects of relevant SNP biomarkers. SNP profiling also cannot account for any potential effects of posttranscriptional modifications or epigenetic factors (22). Such modifications can alter a protein’s isotype, its function in downstream signaling, interactions with other intracellular molecules, and localization within a cell. These variable alterations are not predictable from analyses of genomic sequences alone, and other approaches must be used to gather a more complete view of a cell’s neoplastic potential.
HIGH-THROUGHPUT GENE SEQUENCING
Breakthroughs in high-throughput sequencing methods have rejuvenated interest in scouring the entire genomes of patients for mutations associated with a predisposition to neoplastic processes. Future applications of the improved techniques can be envisioned for both direct sequencing and heteroduplex-detection methods (22). Such efforts as the Cancer Genome Anatomy Project, the Human Cancer Genome Project, the Cancer Genome Project, and the Cancer Genome Atlas promise to showcase the benefits of genomewide sequencing by uncovering novel disease-associated mutations (23). More tangibly, the results of a pilot study that used high-throughput, sequence-based mutational profiling in primary human acute myelogenous leukemia cells have demonstrate the potential of this technology to detect mutational errors that lead to tumorigenesis (24). The validity of these investigators’ high-throughput screen was supported by their identification of 6 previously described sequences and 7 novel sequences associated with acute myelogenous leukemia tumorigenesis (24). By debunking fears that chance mutations would cloud the identification of pathologically relevant mutations, this study establishes a foundation for future genomewide screens of primary tumors.
The genomic-sequencing approach suffers from limitations similar to those of the SNP-profiling technique, however. The lack of insight into various epigenetic and gene-product modifications supports the argument that genomewide screening techniques should be paired with functional assessments of a specific neoplastic process. High cost has persisted as a principal hurdle to broad-scale application of high-throughput sequencing. Although next-generation sequencing platforms will decrease expenses substantially (25), the sheer quantity of information delivered by a fully sequenced genome offers minimal guidance on how best to approach the task of analysis and interpretation. Distinguishing benign polymorphisms from tumorigenic mutations is still a formidable task, as is the ability to predict which detected mutations are actually transcribed and therefore biologically relevant (22).
PHARMACOGENOMICS
Pharmacogenomics is based on assessing a patient’s genetic profile for known polymorphisms in specific networks of essential genes that encode either metabolic pathway effectors (enzymes and transporters) or drug targets (receptors) that affect the pharmacokinetics of drug metabolism and distribution (26).
One such genetic screen designed for metabolic analysis is the AmpliChip (Roche Molecular Systems), which has been approved by the US Food and Drug Administration (FDA). This screening system identifies a patient’s cytochrome P450 genotype encoded by the CYP2D6 (cytochrome P450, family 2, subfamily D, polypeptide 6) and CYP2C19 (cytochrome P450, family 2, subfamily C, polypeptide 19) genes. This development demonstrates the utility of pharmacogenomics by fully using the genomic knowledge of polymorphism-based differences in isoenzymes that affect the metabolism of a host of drugs, including the common chemotherapeutics tamoxifen and cyclophosphamide (27). Pharmacogenomics thus offers a superior method for optimizing the dosage profiles of existing therapeutics. This approach, however, is limited by its narrowness of scope. Predicting the pharmacokinetics of a given therapeutic agent with our incomplete knowledge of polymorphisms in metabolic pathways is unlikely to encompass the full complexity associated with drug metabolism. Nevertheless, an increasing capacity to perform high-throughput genomewide scans for polymorphisms in genes involved in metabolism will lead to a coincident increase in the utility of pharmacogenomics.
Current Genomic Applications in Personalized Cancer Management
The genomics revolution has laid a foundation for deriving genetic signatures for prediagnostic genetic screening, tumor classification, evaluation of patient prognosis, determining the risk of recurrence, and therapeutic response (Table 2). Moreover, recent developments in the use of in vitro drug-sensitivity data in methods of bioinformatics extrapolation promise to provide in silico prediction of the therapeutic response.
Table 2.
Expression signatures and commercial products for personalizing cancer management.
| Studies/products | Platform | Sample | Cancer type |
|---|---|---|---|
| • Classification (human patient–based modeling) | |||
| Golub et al. (7 ) | Affymetrix HU600 microarray – 50-gene predictor signature | Fresh | AML/ALL |
| Lymphochip [Alizadeh et al. (35 )] | cDNA array – 3186 genes | Fresh/frozen tissue | DLBCL |
| [Perou et al. (36 )] | cDNA array – 8102 genes | Fresh/frozen tissue | Breast |
| Breast Bioclassifier™ [University Genomics (57 )] | 55-Gene RT-PCR | Fresh/FFPE tissue | Breast |
| MapQuant Dx™ [Ipsogen (58 )] | Affymetrix GCS3000Dx2 microarray | Fresh | Breast |
| Pulmotype™ [Applied Genomics (59 )] | IHC | FFPE | NSCLC |
| CancerTYPE ID® [bioTheranostics (60 )] | 92-Gene RT-PCR | FFPE | 39 Tumor types |
| Pathwork® Tissue of Origin Test [Dumur et al. (37 )] | cDNA array – 1550 genes | Frozen | Associates tumor with 1 of 15 tissues of origin |
| • Prognosis (human patient–based modeling) | |||
| Yeoh et al. (61 ) | Affymetrix U95Av2 microarray | BM aspirate | Pediatric ALL (classification and PVAD failure) |
| Rosenwald et al. (62 ) | 17-Gene signature cDNA microarray | FFPE | DLBCL |
| van ‘t Veer et al. (38 ) | Oligonucleotide microarray | Frozen tissue | Breast |
| Paik et al. (63 ) | 21-Gene RT-PCR | FFPE | Breast (recurrence after tamoxifen therapy) |
| Rotterdam Signature [Ross et al. (64 )] | 76-Gene signature from Affymetrix U133a microarray | Fresh/frozen | Breast |
| MammaPrint™ (based on van ‘t Veer study) [Agendia (65 )] | 70-Gene oligonucleotide microarray | Fresh/RNARetain® tissue | Breast |
| eXagenBC™ [eXagen (66 )] | FISH | FFPE | Breast |
| Mammostrat® [Applied Genomics (67 )] | IHC | FFPE | Breast |
| PathVysion® [Abbott (68 )] | FISH | FFPE | Breast (for HER-2 status) |
| HerScan™ [Combimatrix Molecular Diagnostics (69 )] | DNA microarray | Fresh/frozen DNA | Breast (for HER-2 status) |
| Pulmostrat™ [Applied Genomics (70 )] | IHC | FFPE | NSCLC |
| Prostate Px [Aureon Laboratories (71 )] | IHC and automated pattern analysis | FFPE | Prostate |
| • Response to therapy (human patient–based modeling) | |||
| Cario et al. (72 ) | 54-Gene signature cDNA microarray | BM aspirate | Pediatric ALL (multi-drug chemotherapy) |
| Okutsu et al. (73 ) | 28-Gene signature cDNA microarray | Mononuclear cells | AML (multi-drug chemotherapy) |
| Takata et al. (74 ) | 14-Gene signature from cDNA microarray for 27648 genes | Frozen biopsy | Bladder (M-VAC response) |
| Frank et al. (75 ) | 128-Gene signature of Affymetrix U133A microarray | BM aspirate | CML (imatinib mesylate resistance) |
| Dressman et al. (76 ) | 38-Gene signature of Affymetrix U1332 Plus 2.0 microarray | Frozen core biopsy | Breast (doxorubicin/paclitaxel |
| Dressman et al. (40 ) | 1727-Gene signature of Affymetrix U1332 Plus 2.0 microarray | Fresh/frozen tissue | Ovarian (platinum resistance) |
| Oncotype DX® [Genomic Health (77 )] | RT-PCR | FFPE | Breast (early stage, ER+) |
| NuvoSelect™ [Ayers et al. (78 )] | 30-Gene signature cDNA microarray | Fresh/frozen | Breast (TFAC chemotherapy efficacy) |
| HERmark™ assay [Monogram Biosciences (79 )] | Protein expression quantification | FFPE | Breast (for HER-2 status) |
| PharmacoDiagnostic® tests (EGFR, HER-2, cKit) [Dako (80 )] | IHC, FISH | FFPE | Colorectal (cetuximab and panitumumab efficacy); breast (trastuzumab efficacy); GIST (imatinib mesylate efficacy) |
| TheraScreen K-Ras [Diagnostic Innovations (81 )] | RT-PCR | Fresh/frozen/FFPE tissue | Colorectal (cetuximab and panitumumab efficacy) |
| Leumeta™ tests [Quest Diagnostics (82 )] | DNA, RNA, protein analysis | Blood (plasma) | Leukemias (CLL, CML, ALL) |
| PGxPredict™ [PGx Health (83 )] | SNP analysis in FCGR3A | Blood | NHL (rituximab efficacy) |
| AmpliChip® CYP450 Test [Roche Diagnostics (84 )] | Microarray for allelic variations in CYP2D6 and CYP2C19 | Blood | Predict phenotype for chemotherapy metabolism |
| • Response to therapy (in vitro panel–based modeling) | |||
| Potti et al. (41 ) | In vitro drug sensitivity + Affymetrix microarray | In vitro cell lines | Cancers with common cytotoxic agent treatments |
| Lee et al. (42 ) | In vitro drug sensitivity + Affymetrix microarray | In vitro cell lines | All cancers |
| • Targeted therapy (human patient–based modeling) | |||
| Imatinib mesylate (Gleevec®) | Small molecule tyrosine kinase inhibitor | CML | |
| Lapatinib ditosylate (Tykerb®) | Small molecule tyrosine kinase inhibitor for HER-2/neu and EGFR | Breast cancer and lung cancers | |
| Gefitinib (Iressa®) | Small molecule EGFR inhibitor | NSCLC | |
| Erlotinib (Tarceva®) | Small molecule EGFR inhibitor | NSCLC | |
| Trastuzumab (Herceptin®) | Humanized monoclonal antibody | HER-2 overexpressing breast cancer | |
| Cetuximab (Erbitux®) | Chimeric monoclonal antibody | Metastastic colorectal cancer | |
| Panitumumab (Vectibix®) | Humanized monoclonal antibody | Metastastic colorectal cancer | |
| Bevacizumab (Avastin®) | Humanized monoclonal antibody | Colorectal, lung, and breast cancer | |
AML, acute myelogenous leukemia; ALL, acute lymphoblastic leukemia; DLBCL, diffuse large B-cell lymphoma; RT-PCR, real-time PCR; IHC, immunohistochemistry; NSCLC, non–small cell lung carcinoma; BM, bone marrow; PVAD, prednisone, vincristine, asparaginase, and daunorubicin; FISH, fluorescence in situ hybridization; M-VAC, methotrexate, vinblastine, doxorubicin (Adriamycin™), and cisplatin; CML, chronic myelogenous leukemia; ER, estrogen receptor; TFAC, paclitaxel (Taxol®), 5-fluorouracil, doxorubicin (Adriamycin), and cyclophosphamide; EGFR, epidermal growth factor receptor; GIST, gastrointestinal stromal tumor; HER-2, ERBB2 gene; CLL, chronic lymphocytic leukemia; NHL, non-Hodgkin lymphoma.
Human genes: FCGR3A, Fc fragment of IgG, low affinity IIIa, receptor (CD16a).
PREDIAGNOSTIC GENETIC SCREENING
Even before malignant transformation, the interplay between genetic composition and environmental factors shape an individual’s predisposition for cancer. Knowledge of this genetic predisposition would facilitate the design of a course of preventive management for modifying the risk of neoplastic progression. Such knowledge is available through genetic screening, which is most applicable for families with a history of oncogenic gene mutations, such as in APC (adenomatous polyposis coli), BRCA1 (breast cancer 1, early onset), and BRCA2 (breast cancer 2, early onset), in which mutational status will guide a clinical decision (28). The creation of databases with comprehensive lists of genetic mutations known to promote tumorigenesis in humans, such as the Catalogue of Somatic Mutations in Cancer (COSMIC) (http://www.sanger.ac.uk/genetics/CGP/cosmic/), will undoubtedly alter the utility of genetic screening in the prediagnosis of cancer (29).
EARLY DETECTION
Genetic instability, abnormal gene transcription or translation, and altered protein production and modification lead to early transformational events that produce signaling changes in a cell’s molecular phenotype (30). These alterations are the foundation for genomic and proteomic analyses that can identify them. Following validation, these deviations are incorporated into biomarker measurements for detecting cancer in its nascent stages. Prostate-specific antigen has become a mainstay for detecting and monitoring prostate cancer, and the Early Detection Research Network (http://edrn.nci.nih.gov/) has accelerated biomarker discovery by creating cross-institutional collaborative alliances for identifying and verifying potential clinical biomarkers that are “. . . easy to detect, measurable across populations, and amenable to use in one or more of the following settings: detection at an early stage; identification of high-risk individuals; early detection of recurrence; or as intermediate endpoints in chemoprevention” (31). With the promising leads produced by proteomic (32), miRNA (33), and epigenetic (34) platforms, the holistic approach to biomarker identification is a model for the integrative initiative necessary for successful personalization of cancer management.
TUMOR CLASSIFICATION
The use of the microarray assessment of tumors for class prediction and discovery originated with the previously mentioned study of acute leukemias (7). With a newly developed method of “neighborhood analysis,” the investigators grouped genes highly expressed in one class and expressed at low levels in the comparative class into “idealized patterns of expression” (7). Through further bioinformatics processing, they derived a 50-gene predictor set for correctly assigning independent leukemia samples into acute myelogenous leukemia, acute lymphoblastic leukemia, or uncertain groups (7). Moreover, this study demonstrated the potential of assessing differential gene expression in cancers for identifying novel subclasses previously overlooked by conventional classification methods (7). Other investigators have used similar microarray platforms to apply selective assessment of gene expression to a variety of cancers, including diffuse large B-cell lymphoma (35), breast cancer (36), cancers with an unknown tissue of origin (37), and others (block 1 in Table 2).
PROGNOSIS AND PREDICTION OF PATIENT RESPONSE
Gene expression–based risk assessment for prognosis and prediction of disease recurrence is currently available for breast tumors through Genomic Health’s On-cotype Dx® test and Agendia’s MammaPrint™ assay. By screening breast tumor biopsies against the Oncotype Dx real-time PCR–based expression panel of 21 gene biomarkers, physicians can obtain a patient’s prognostic score to guide their clinical decisions (1). More than a dozen recent studies and commercially available products have demonstrated the utility of genomic biomarkers to predict a patient’s prognosis and risk of recurrence (blocks 2 and 3 in Table 2).
In an early study in this field, van ‘t Veer et al. used expression-microarray analysis to evaluate biopsies of primary tumors from patients with no signs of lymph node or organ metastases. The aim was to develop a gene expression signature predictive of early metastasis and therefore a poor prognosis (38). An unsupervised clustering of tumors according to their expression of approximately 5000 substantially regulated genes led to the identification of “good prognosis” and “bad prognosis” tumors (38). The number of “informative genes” was subsequently pared down to a 70-gene signature capable of correctly predicting disease outcome in 65 (85%) of 78 patients (38). Similar approaches for outcome prediction were used in trials involving non–small cell lung cancer and ovarian cancer (39, 40). Foresight into a patient’s likely outcome will aid in the delivery of adjuvant therapies to patients who require aggressive care, reduce unnecessary therapeutic toxicities and side effects in nonprogressing groups, and alleviate the financial toll of excessive treatments on the healthcare system.
IN SILICO PREDICTION OF THERAPEUTIC RESPONSE
Two innovative drug-sensitivity studies have called into question the necessity of in vivo models to predict chemotherapeutic response. Both studies developed bioinformatics-centric prediction models that use gene expression patterns of common tumors, in vitro activities of therapeutic compounds on these tumors, and retrospective data from matched clinical trials to predict and validate chemosensitivity for a variety of cancers (block 4 in Table 2) (41, 42). Potti et al. developed a genomic “predictor signature” consisting of genes whose expression correlated with single-drug sensitivity and resistance in the NCI-60 set of cancer cell lines (41). By applying this signature to previously published clinical-response data sets, these investigators demonstrated that patients’ responses to both single-drug therapies and combination therapies could be predicted by comparing the expression of genes in patient tumors with expression-based signatures for in vitro chemosensitivity (41).
Concurrently, Lee et al. applied their COXEN (COeXpression ExtrapolatioN) algorithm to integrate drug-sensitivity data and gene expression data from the NCI-60 panel with gene expression data from patients’ tumors in order to identify a COXEN biomarker panel (42). This biomarker panel consists of genes with strongly positive or negative correlations to in vitro chemosensitivity. Comparison of the gene expression levels for this panel with gene expression profiles of patient tumors will facilitate the projection of therapeutic sensitivity into the clinic (42). The COXEN algorithm also demonstrates applicability for predicting chemosensitivity in cancer subtypes not included in the NCI-60 panel, a particular strength because many tumor types are not represented in the NCI-60 panel (42). In addition to its therapeutic-prediction capabilities, COXEN can facilitate computational drug screening, thus expanding our capacity to identify agents that are likely to be effective in patients. A trial of such screening methods identified a new potential agent for treating bladder cancer, NSC637993 {6H-imidazo[4,5,1-de]acridin-6-one, 5-[2-(diethylamino) ethylamino]-8-methoxy-1-methyl-, dihydrochloride}, which exhibited similar predicted and actual chemo-sensitivities (42).
The major advantage of in vitro–derived genomic predictions (41, 42) is the ability to include prospective drugs and drug combinations in chemosensitivity predictions. This capability is particularly important when one considers the more than 2400 doublet combinations possible among the approximately 70 FDA-approved antineoplastic agents. The FDA has proposed guidelines for clinical-trial validation of these combinatorial therapies and future multidrug regimens that use combinations of current and novel therapeutics (43). This document acknowledges that in vitro drug validations would expedite current practices of drug development through the use of archival tissue samples and relevant clinical information from patients (43).
The potential of these in silico analyses to directly bridge in vitro chemosensitivity with predictions of clinical efficacy is highly attractive in its efficient use of resources. Deriving an approach to clinical treatment from in vitro results promises to shorten the time and decrease the resources necessary to match patients with a large number of treatment options. Eliminating the need for animal intermediates has the potential to abbreviate the time devoted to the research and development of novel therapeutics. Moreover, currently approved pharmaceuticals will find new application through cross-comparisons of in vitro screening chemosensitivities and tumor expression profiles.
TARGETED THERAPY
The concept of targeting therapies speaks to both identifying distinct molecular mechanisms for therapeutic recourse and selecting patient populations for which a particular treatment will be most efficacious. The success of molecularly targeted inhibitors of tumorigenic pathways is readily apparent in the treatment of hematogenous cancers. In these cancers, such as chronic myelogenous leukemia, the discovery of independent molecular drivers of neoplasia have aided in the development of targeted therapies (44). In the case of chronic myelogenous leukemia, linking the genetic lesion, a t(9;22) translocation, with the dysregulation of an ABL kinase led to the development of a selective kinase inhibitor, imatinib mesylate (Gleevec®; Novartis). Successful identification of rogue molecular-signaling pathways is ongoing for other cancers, and novel small-molecule inhibitors and monoclonal antibodies continue to see moderate clinical use. From the use of gefitinib (Iressa®; AstraZeneca) and erlotinib (Tarceva®; Genentech) as therapies for lung cancer to the use of trastuzumab (Herceptin®; Genentech) and lapatinib (Tykerb®; GlaxoSmithKline) as therapies for breast cancer, the potential for targeting individual signaling disruptions as a method for chemotherapeutic treatment is evident (block 5 in Table 2).
The integration of screening results provided by biomarker panels and pharmacodiagnostic tests will substantiate treatment decisions based on data from clinical trials (45). In the application of trastuzumab for breast cancer patients, determining whether the HER-2 protein is overproduced or whether ERBB2 [v-erb-b2 erythroblastic leukemia viral oncogene homolog 2, neuro/glioblastoma derived oncogene homolog (avian)] (HER-2) amplification can predict the efficacy of treatment (45). Screening tests such as Her-ceptest™ (Dako) for assessing HER-2 overproduction in tumor biopsies and fluorescence in situ hybridization analysis for ERBB2 amplification increase the likelihood for success in treatment groups. Moreover, screening patients for pertinent molecular dysregulations will bolster our knowledge of mutational frequency in a given pathway and thus expand our understanding of its absolute relevancy within disease populations.
Bioinformatics: Techniques and Challenges
Bioinformatics translates raw molecular data extracted from patient samples into interpretable, accessible, and distributable information. This step in the personalization process is necessary for interpreting most high-throughput microarray-based biomedical applications and typically consists of the following steps: (a) data preprocessing, (b) normalization, (c) biomarker discovery, (d) statistical modeling and validation of prediction, and (e) follow-up clinical confirmation. Some details of implementation of these procedures may vary slightly between different molecular platforms, such as cDNA arrays, oligonucleotide arrays, or mass spectrometry (46). Nevertheless, these approaches have been used across most platforms and therefore provide a systematic method for transcending the challenges encountered during the analysis of most high-throughput biotechnology data.
DATA PREPROCESSING
High-throughput molecular data possess rich digital information for each molecular target. For example, a single scan of a microarray chip produces hundreds of image pixels for each of >20 000 transcript probes. The initial preprocessing analysis of such high-density data is often tightly combined and encrypted within the manufacturing steps of biotechnology instrumentation. This step, however, is one of the most critical in the analysis for optimizing the molecular information obtainable from such massive amounts of biological data. In fact, preprocessing algorithms generated by third parties sometimes have demonstrated substantially higher reproducibility and sensitivity with certain commercial microarray platforms than those produced by the platform manufacturer (47). Additionally, identifying and quantifying specific protein species from a background of fragmented-peptide mass-spectrometry spectra are recognized as a challenging statistical and computational problem. The direct involvement of computational researchers in the development of these initial data-preprocessing steps will prove greatly beneficial.
NORMALIZATION
Normalization is required to standardize multiple data sets produced in independent experiments before they can be combined for analysis. This step is essential for obtaining data with universal applicability despite their having differing institutional origins. Before normalization, data are often log-transformed to make their distributions more appropriate for subsequent analyses. Data are then normalized via correction often with a simple constant factor. More sophisticated normalization methods such as nonparametric regression can be used if different data sets have nonlinear relationships (47).
BIOMARKER DISCOVERY AND PREDICTION MODELING
The identification and selection of biomarkers that are clinically relevant are a fundamental step that determines the success of downstream applications that advance personalized medicine. This kind of discovery can be performed in various ways: (a) simple comparison of contrasting groups (e.g., disease-free survivors vs relapsed patients, responders vs nonresponders to a therapeutic compound), (b) analysis of a statistical association between patients’ outcomes and phenotypes, and (c) analysis of correlations with continuous drug-sensitivity data, such as for GI50 (concentration inhibiting growth by 50%) in a cell line panel. An important key to biomarker discovery is the subsequent validation (46).
With a set of preidentified biomarkers, many different statistical approaches have been used in prediction modeling. Such approaches include gene voting, discriminant analysis, Bayesian regression or classification, random forest, Cox regression, and support vector machines, technical details of which are beyond the scope of this review and can be found elsewhere (7, 41, 42). These different prediction-modeling techniques have often been found to provide similar predictive powers if each is finely tuned. An integral aspect of modeling is to maintain extremely tight control of error and bias due to overtraining and multiple comparisons.
VALIDATION AND FOLLOW-UP CLINICAL CONFIRMATION
To avoid “selection bias” one must validate a trained prediction model with patient data sets that are completely independent of the original discovery and training data set (48). The follow-up preferably uses data from different places and clinical settings, because a patient cohort from a particular location and clinical setting may produce specific molecular results that may not occur in other patient populations. This step is particularly important to ensure the performance and accuracy of the molecular assay in clinical practice.
Considerations for Clinical Pathology
BIOSAMPLE QUALITY
The privileged access of the clinical pathology laboratory to patient biopsies and samples places it in a unique position to ensure sample integrity through the development and implementation of standardized guidelines for procuring and storing biosamples. The coordination of such efforts is currently facilitated by the US National Cancer Institute’s Office of Biorepositories and Biospecimen Research (http://biospecimens.cancer.gov/index.asp) (49). The establishment of this department in 2005 is a testament to the priority placed on the necessity to preserve reliable biosamples for expediting the development of molecular-based diagnostics and therapeutics.
The Biospecimen Research Network, the research branch of the Office of Biorepositories and Biospecimen Research, seeks to conduct and collaborate on projects to help establish best practices for sample storage and tracking, to identify high-quality samples in existing repositories, and to determine the impact of specific variables during sample handling (49). Whereas the latter 2 aims will provide a short-term solution for the immediate use and interpretation of results produced from currently preserved samples, the implementation of a best-practices policy for bio-sample handling will have a long-standing impact on protocols for the procurement and storage of pathology samples. Variables such as time from sample excision to fixation, optimal fixation solutions and conditions (e.g., ethanol vs formalin fixation), and sample storage, cataloging, and retrieval each require specific attention (49).
EXTRACTION OF MOLECULAR INFORMATION
Sustaining the recent momentum in the field of molecular cancer research requires a coalescence of well-annotated clinical information and molecular data derived from patient biosamples. An elegant and abundant cache of patient molecular material is the formalin-fixed, paraffin-embedded (FFPE) samples that have been produced in myriad clinical trials, in which the patient information has been well characterized and the outcomes are known. Retrieval of the full molecular information in archived FFPE samples, however, is hindered by changes in molecular structures that occur during the fixation process.
The challenge of extracting useful molecular information despite formalin-induced protein cross-linkages and the addition of monomethylol groups to nucleosides has been partially overcome with the emergence of commercially available kits, such as the RNeasy FFPE Kit (Qiagen), the Paraffin Block Isolation and the RecoverAll Total Nucleic Acid Isolation Kits (Ambion), and the Paradise Plus Reagent System (Arcturus). These kits predominantly rely on proteinase K digestion to facilitate mRNA release from the bonds to cross-linked proteins (50). Although successful extraction of intact mRNA is tightly tied to the quality of sample fixation, isolation of contiguous miRNA is less dependent on fixation methods, as evinced by the lower cross-sample variation found in a recent study (51). The superior potential for extracting miRNA from such samples is primarily attributable to the short lengths of miRNAs (51). Such distinctions underscore the necessity for skilled clinical pathologists to determine which biological platform to implement for accurate tumor assessment given the quality of the available samples.
QUANTITATIVE REAL-TIME PCR
The power of quantitative real-time PCR (qPCR) to obtain molecular-expression profiles from patient tumor biopsies must be harnessed. Although microarray technology remains the preeminent mode for biomarker discovery, qPCR analysis serves as a useful adjunct for validating microarray results, in addition to functioning as a targeted method for analyzing specific, previously verified biomarker concentrations. As an assessment platform, qPCR offers rapid, single-step amplification and quantification of molecular targets that are useful for clarifying diagnosis, predicting recurrence, and guiding treatment (52). For example, a qPCR-based ratio of HOXB13 (homeobox B13) expression to IL17RB (interleukin 17 receptor B) expression has been used to predict tumor recurrence in the setting of adjuvant tamoxifen monotherapy (53).
Although conventional qPCR methods are effective for assessing expression signatures for small batches of molecular targets, the advancement of multiplex qPCR permits concentrations of multiple targets to be analyzed in a single reaction (52). The consolidation of resources and time offered by a multiplexed qPCR model may provide the speed and cost efficiency necessary for individualized molecular-assessment practices to gain widespread clinical integration (25).
FOCUS ON EDUCATION
Efforts to emphasize the therapeutic potential of integrating molecular profiling and clinical assessments are necessary to accelerate the actualization of personalized care. The clinical pathology laboratory is well equipped to provide physicians with the knowledge to promote the regular incorporation of molecular tumor analysis into standard workups; however, the overall success of such efforts will reside in striking a cautionary balance of promoting personalized medicine without overselling a concept that lacks proper verification and implementation standards. This sentiment resonates with groups such as the Coriell Personalized Medicine Collaborative (54). A Coriell study hopes to clarify the impact of genomic assessment on clinical outcomes while shaping the social and legal ramifications associated with openly accessible genomic profiling (54 ).
Conclusions
Advancements in molecular-profiling techniques have provided unprecedented insight into the genetic etiologies and basic molecular dysfunctions that lead to tumorigenesis. Moreover, bioinformatics analysis of gene expression data has produced expression signatures that are useful for personalizing many aspects of cancer management, including genetic screening, early detection, tumor classification, and prediction of prognosis and therapeutic response. Successful incorporation of bioinformatics-based assessments alongside current methods of tissue and clinical evaluation will leverage the complementary natures of these biological platforms. Orchestrating this collaboration is one of the steepest challenges facing clinical pathologists in their quest to integrate bioinformatics with frontline clinical care. The likely payoff for such efforts appears enormous. Through mediation of conventional and molecular-assessment methods, the personalization of cancer management stands poised for success.
Footnotes
Nonstandard abbreviations: SNP, single-nucleotide polymorphism; miRNA, microRNA; FDA, Food and Drug Administration; COSMIC, Catalogue of Somatic Mutations in Cancer; FFPE, formalin-fixed, paraffin-embedded; qPCR, quantitative real-time PCR.
Human genes: MIR155, microRNA 155; MIR15A, microRNA 15a; MIR16-1, microRNA 16-1; CYP2D6, cytochrome P450, family 2, subfamily D, polypeptide 6; CYP2C19, cytochrome P450, family 2, subfamily C, polypeptide 19; APC, adenomatous polyposis coli; BRCA1, breast cancer 1, early onset; BRCA2, breast cancer 2, early onset; ERBB2 (HER-2), v-erb-b2 erythroblastic leukemia viral oncogene homolog 2, neuro/glioblastoma derived oncogene homolog (avian); HOXB13, homeobox B13; IL17RB, interleukin 17 receptor B.
Author Contributions: All authors confirmed they have contributed to the intellectual content of this paper and have met the following 3 requirements: (a) significant contributions to the conception and design, acquisition of data, or analysis and interpretation of data; (b) drafting or revising the article for intellectual content; and (c) final approval of the published article.
Authors’ Disclosures of Potential Conflicts of Interest: Upon manuscript submission, all authors completed the Disclosures of Potential Conflict of Interest form. Potential conflicts of interest: Employment or Leadership: J.K. Lee and D. Theodorescu are co-founders of Key Genomics.<br>Consultant or Advisory Role: None declared.
Stock Ownership: D. Theodorescu, Key Genomics; J.K. Lee, Key Genomics.
Honoraria: None declared.
Research Funding: J.B. Overdevest, NIH Cancer Research Training in Molecular Biology (T32A009109); D. Theodorescu, AstraZeneca and NIH grant R01CA075115; J.K. Lee, NIH grant R01HL081690.
Expert Testimony: None declared.
Role of Sponsor: The funding organizations played no role in the design of study, choice of enrolled patients, review and interpretation of data, or preparation or approval of manuscript.
References
- 1.Allison M. Is personalized medicine finally arriving? Nat Biotechnol. 2008;26:509–17. doi: 10.1038/nbt0508-509. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 3.Fidler IJ. Tumor heterogeneity and the biology of cancer invasion and metastasis. Cancer Res. 1978;38:2651–60. [PubMed] [Google Scholar]
- 4.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 5.Sherbenou DW, Druker BJ. Applying the discovery of the Philadelphia chromosome. J Clin Invest. 2007;117:2067–74. doi: 10.1172/JCI31988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramaswamy S, Golub TR. DNA microarrays in clinical oncology. J Clin Oncol. 2002;20:1932–41. doi: 10.1200/JCO.2002.20.7.1932. [DOI] [PubMed] [Google Scholar]
- 7.Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–7. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 8.Minna JD, Girard L, Xie Y. Tumor mRNA expression profiles predict responses to chemotherapy. J Clin Oncol. 2007;25:4329–36. doi: 10.1200/JCO.2007.12.3968. [DOI] [PubMed] [Google Scholar]
- 9.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–66. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 10.Liu CG, Calin GA, Meloon B, Gamliel N, Sevignani C, Ferracin M, et al. An oligonucleotide microchip for genome-wide microRNA profiling in human and mouse tissues. Proc Natl Acad Sci U S A. 2004;101:9740–4. doi: 10.1073/pnas.0403293101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu SL, Chen HY, Chang GC, Chen CY, Chen HW, Singh S, et al. MicroRNA signature predicts survival and relapse in lung cancer. Cancer Cell. 2008;13:48–57. doi: 10.1016/j.ccr.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 12.Merritt WM, Lin YG, Han LY, Kamat AA, Spannuth WA, Schmandt R, et al. Dicer, Drosha, and outcomes in patients with ovarian cancer. N Engl J Med. 2008;359:2641–50. doi: 10.1056/NEJMoa0803785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lujambio A, Ropero S, Ballestar E, Fraga MF, Cerrato C, Setien F, et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007;67:1424–9. doi: 10.1158/0008-5472.CAN-06-4218. [DOI] [PubMed] [Google Scholar]
- 14.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–59. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 15.Wulfkuhle J, Edmiston K, Liotta L, Petricoin E. Technology insight: pharmacoproteomics for cancer—promises of patient-tailored medicine using protein microarrays. Nat Clin Pract Oncol. 2006;3:256–68. doi: 10.1038/ncponc0485. [DOI] [PubMed] [Google Scholar]
- 16.MacBeath G. Protein microarrays and proteomics. Nat Genet. 2002;32:526–32. doi: 10.1038/ng1037. [DOI] [PubMed] [Google Scholar]
- 17.Liotta LA, Espina V, Mehta AI, Calvert V, Rosenblatt K, Geho D, et al. Protein microarrays: meeting analytical challenges for clinical applications. Cancer Cell. 2003;3:317–25. doi: 10.1016/s1535-6108(03)00086-2. [DOI] [PubMed] [Google Scholar]
- 18.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 19.Marko-Varga G, Ogiwara A, Nishimura T, Kawamura T, Fujii K, Kawakami T, et al. Personalized medicine and proteomics: lessons from non-small cell lung cancer. J Proteome Res. 2007;6:2925–35. doi: 10.1021/pr070046s. [DOI] [PubMed] [Google Scholar]
- 20.Shi MM. Enabling large-scale pharmacogenetic studies by high-throughput mutation detection and genotyping technologies. Clin Chem. 2001;47:164–72. [PubMed] [Google Scholar]
- 21.The International HapMap Consortium. The International HapMap Project. Nature. 2003;426:789–96. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 22.Weber BL. Cancer genomics. Cancer Cell. 2002;1:37–47. doi: 10.1016/s1535-6108(02)00026-0. [DOI] [PubMed] [Google Scholar]
- 23.Strausberg RL, Simpson AJG, Wooster R. Sequence-based cancer genomics: progress, lessons and opportunities. Nat Rev Genet. 2003;4:409–18. doi: 10.1038/nrg1085. [DOI] [PubMed] [Google Scholar]
- 24.Ley TJ, Minx PJ, Walter MJ, Ries RE, Sun H, McLellan M, et al. A pilot study of high-throughput, sequence-based mutational profiling of primary human acute myeloid leukemia cell genomes. Proc Natl Acad Sci U S A. 2003;100:14275–80. doi: 10.1073/pnas.2335924100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–45. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 26.Evans WE, Relling MV. Pharmacogenomics: translating functional genomics into rational therapeutics. Science. 1999;286:487–91. doi: 10.1126/science.286.5439.487. [DOI] [PubMed] [Google Scholar]
- 27.van Schaik RHN. CYP450 pharmacogenetics for personalizing cancer therapy. Drug Resist Updat. 2008;11:77–98. doi: 10.1016/j.drup.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 28.Ponder B. Genetic testing for cancer risk. Science. 1997;278:1050–4. doi: 10.1126/science.278.5340.1050. [DOI] [PubMed] [Google Scholar]
- 29.Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer. 2004;91:355–8. doi: 10.1038/sj.bjc.6601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sidransky D. Emerging molecular markers of cancer. Nat Rev Cancer. 2002;2:210–9. doi: 10.1038/nrc755. [DOI] [PubMed] [Google Scholar]
- 31.Srinivas PR, Kramer BS, Srivastava S. Trends in biomarker research for cancer detection. Lancet Oncol. 2001;2:698–704. doi: 10.1016/S1470-2045(01)00560-5. [DOI] [PubMed] [Google Scholar]
- 32.Wulfkuhle JD, Liotta LA, Petricoin EF. Proteomic applications for the early detection of cancer. Nat Rev Cancer. 2003;3:267–75. doi: 10.1038/nrc1043. [DOI] [PubMed] [Google Scholar]
- 33.Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105:10513–8. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verma M, Srivastava S. Epigenetics in cancer: implications for early detection and prevention. Lancet Oncol. 2002;3:755–63. doi: 10.1016/s1470-2045(02)00932-4. [DOI] [PubMed] [Google Scholar]
- 35.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–11. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 36.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–52. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 37.Dumur CI, Lyons-Weiler M, Sciulli C, Garrett CT, Schrijver I, Holley TK, et al. Interlaboratory performance of a microarray-based gene expression test to determine tissue of origin in poorly differentiated and undifferentiated cancers. J Mol Diagn. 2008;10:67–77. doi: 10.2353/jmoldx.2008.070099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van ’t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AAM, Mao M, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–6. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 39.Potti A, Mukherjee S, Petersen R, Dressman HK, Bild A, Koontz J, et al. A genomic strategy to refine prognosis in early-stage non-small-cell lung cancer. N Engl J Med. 2006;355:570–80. doi: 10.1056/NEJMoa060467. [DOI] [PubMed] [Google Scholar]
- 40.Dressman HK, Berchuck A, Chan G, Zhai J, Bild A, Sayer R, et al. An integrated genomic-based approach to individualized treatment of patients with advanced-stage ovarian cancer. J Clin Oncol. 2007;25:517–25. doi: 10.1200/JCO.2006.06.3743. [DOI] [PubMed] [Google Scholar]
- 41.Potti A, Dressman HK, Bild A, Riedel RF, Chan G, Sayer R, et al. Genomic signatures to guide the use of chemotherapeutics. Nat Med. 2006;12:1294–300. doi: 10.1038/nm1491. [DOI] [PubMed] [Google Scholar]
- 42.Lee JK, Havaleshko DM, Cho H, Weinstein JN, Kaldjian EP, Karpovich J, et al. A strategy for predicting the chemosensitivity of human cancers and its application to drug discovery. Proc Natl Acad Sci U S A. 2007;104:13086–91. doi: 10.1073/pnas.0610292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.US Food and Drug Administration. [Accessed October 2008];Draft guidance – in vitro diagnostic multivariate index assays. http://www.fda.gov/cdrh/oivd/guidance/1610.html.
- 44.Green MR. Targeting targeted therapy. N Engl J Med. 2004;350:2191–3. doi: 10.1056/NEJMe048101. [DOI] [PubMed] [Google Scholar]
- 45.Jorgensen JT, Nielsen KV, Ejlertsen B. Pharmacodi-agnostics and targeted therapies—a rational approach for individualizing medical anticancer therapy in breast cancer. Oncologist. 2007;12:397–405. doi: 10.1634/theoncologist.12-4-397. [DOI] [PubMed] [Google Scholar]
- 46.Macgregor PF, Squire JA. Application of microarrays to the analysis of gene expression in cancer. Clin Chem. 2002;48:1170–7. [PubMed] [Google Scholar]
- 47.Simon R, Radmacher MD, Dobbin K, McShane LM. Pitfalls in the use of DNA microarray data for diagnostic and prognostic classification. J Natl Cancer Inst. 2003;95:14–8. doi: 10.1093/jnci/95.1.14. [DOI] [PubMed] [Google Scholar]
- 48.Ambroise C, McLachlan GJ. Selection bias in gene extraction on the basis of microarray gene-expression data. Proc Natl Acad Sci U S A. 2002;99:6562–6. doi: 10.1073/pnas.102102699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Office of Biorepositories and Biospecimen Research. [Accessed December 2008]; http://biospecimens.cancer.gov/index.asp.
- 50.Lewis F, Maughan N, Smith V, Hillan K, Quirke P. Unlocking the archive— gene expression in paraffin-embedded tissue. J Pathol. 2001;195:66–71. doi: 10.1002/1096-9896(200109)195:1<66::AID-PATH921>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 51.Doleshal M, Magotra AA, Choudhury B, Cannon BD, Labourier E, Szafranska AE. Evaluation and validation of total RNA extraction methods for microRNA expression analyses in formalin-fixed, paraffin-embedded tissues. J Mol Diagn. 2008;10:203–11. doi: 10.2353/jmoldx.2008.070153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bernard PS, Wittwer CT. Real-time PCR technology for cancer diagnostics. Clin Chem. 2002;48:1178–85. [PubMed] [Google Scholar]
- 53.Ma XJ, Wang Z, Ryan PD, Isakoff SJ, Barmettler A, Fuller A, et al. A two-gene expression ratio predicts clinical outcome in breast cancer patients treated with tamoxifen. Cancer Cell. 2004;5:607–16. doi: 10.1016/j.ccr.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 54.Coriell Personalized Medicine Collaborative. [Accessed December 2008]; http://www.coriell.org/index.php/content/view/92/167/
- 55.Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 56.Araujo RP, Liotta LA, Petricoin EF. Proteins, drug targets and the mechanisms they control: the simple truth about complex networks. Nat Rev Drug Discov. 2007;6:871–80. doi: 10.1038/nrd2381. [DOI] [PubMed] [Google Scholar]
- 57.University Genomics, Inc. [Accessed October 2008];Breast Bioclassifier. http://www.bioclassifier.com/
- 58.Ipsogen. [Accessed October 2008];MapQuant DX. http://www.ipsogen.com/index.php?id=64.
- 59.Applied Genomics, Inc. [Accessed October 2008];PulmoType. http://www.applied-genomics.com/pulmotype.html.
- 60.bioTheranostics. [Accessed October 2008];Theros CancerTYPE ID. http://www.aviaradx.com/cTYPE/cType_overview.html.
- 61.Yeoh EJ, Ross ME, Shurtleff SA, Williams WK, Patel D, Mahfouz R, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 2002;1:133–43. doi: 10.1016/s1535-6108(02)00032-6. [DOI] [PubMed] [Google Scholar]
- 62.Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:1937–47. doi: 10.1056/NEJMoa012914. [DOI] [PubMed] [Google Scholar]
- 63.Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351:2817–26. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 64.Ross JS, Hatzis C, Symmans WF, Pusztai L, Hortobagyi GN. Commercialized multigene predictors of clinical outcome for breast cancer. Oncologist. 2008;13:477–93. doi: 10.1634/theoncologist.2007-0248. [DOI] [PubMed] [Google Scholar]
- 65.Agendia. [Accessed October 2008];MammaPrint. http://usa.agendia.com/en/mammaprint.html.
- 66.eXagen. [Accessed October 2008];eXagenBC. http://www.exagen.com/ourproducts/breastcancer.aspx.
- 67.Applied Genomics, Inc. [Accessed October 2008];MammoStrat. http://www.applied-genomics.com/mammostrat.html.
- 68.Abbott. [Accessed October 2008];PathVysion. http://www.pathvysion.com/
- 69.Combimatrix Molecular Diagnostics. [Accessed January 2009];HerScan. http://www.cmdiagnostics.com/testmenu.htm.
- 70.Applied Genomics, Inc. [Accessed October 2008];PulmoStrat. http://www.applied-genomics.com/pulmostrat.html.
- 71.Aureon Laboratories. [Accessed October 2008];Prostate Px. http://www.aureon.com/prognostic-tests-prostate-px.htm.
- 72.Cario G, Stanulla M, Fine BM, Teuffel O, Neuhoff NV, Schrauder A, et al. Distinct gene expression profiles determine molecular treatment response in childhood acute lymphoblastic leukemia. Blood. 2005;105:821–6. doi: 10.1182/blood-2004-04-1552. [DOI] [PubMed] [Google Scholar]
- 73.Okutsu J, Tsunoda T, Kaneta Y, Katagiri T, Kitahara O, Zembutsu H, et al. Prediction of chemosensitivity for patients with acute myeloid leukemia, according to expression levels of 28 genes selected by genome-wide complementary DNA microarray analysis. Mol Cancer Ther. 2002;1:1035–42. [PubMed] [Google Scholar]
- 74.Takata R, Katagiri T, Kanehira M, Tsunoda T, Shuin T, Miki T, et al. Predicting response to methotrexate, vinblastine, doxorubicin, and cis-platin neoadjuvant chemotherapy for bladder cancers through genome-wide gene expression profiling. Clin Cancer Res. 2005;11:2625–36. doi: 10.1158/1078-0432.CCR-04-1988. [DOI] [PubMed] [Google Scholar]
- 75.Frank O, Brors B, Fabarius A, Li L, Haak M, Merk S, et al. Gene expression signature of primary imatinib-resistant chronic myeloid leukemia patients. Leukemia. 2006;20:1400–7. doi: 10.1038/sj.leu.2404270. [DOI] [PubMed] [Google Scholar]
- 76.Dressman HK, Hans C, Bild A, Olson JA, Rosen E, Marcom PK, et al. Gene expression profiles of multiple breast cancer phenotypes and response to neoadjuvant chemotherapy. Clin Cancer Res. 2006;12:819–26. doi: 10.1158/1078-0432.CCR-05-1447. [DOI] [PubMed] [Google Scholar]
- 77.Genomic Health. [Accessed October 2008];Oncotype DX. http:/www.genomichealth.com/OncotypeDX/Index.aspx?Sid=33.
- 78.Ayers M, Symmans WF, Stec J, Damokosh AI, Clark E, Hess K, et al. Gene expression profiles predict complete pathologic response to neoadjuvant paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide chemotherapy in breast cancer. J Clin Oncol. 2004;22:2284–93. doi: 10.1200/JCO.2004.05.166. [DOI] [PubMed] [Google Scholar]
- 79.Monogram Biosciences, Inc. [Accessed October 2008];HERmark breast cancer assay. http://www.hermarkassay.com/
- 80.Dako. [Accessed January 2009];pharmacoDiagnostic Solution tests. http://www.dakousa.com/index/prod_search/prod_groups.htm?productareaid=39.
- 81.Diagnostic Innovations. [Accessed October 2008];TheraScreen. http://www.dxsdiagnostics.com/Content/TheraScreenKRAS.aspx.
- 82.Quest Diagnostics. [Accessed October 2008];Leumeta. http://www.questdiagnostics.com/hcp/topics/hem_onc/leumeta.html.
- 83.PGx Health. [Accessed January 2009];PGxPredict:Rituximab. http://www.pgxhealth.com/genetictests/rituximab/
- 84.Roche Diagnostics. [Accessed October 2008];AmpliChip CYP450 test. http://www.amplichip.us/