

Abstract
Chemotherapy-induced peripheral neuropathy (CIPN) accompanied by chronic neuropathic pain is the major dose-limiting toxicity of several anticancer agents including the taxane paclitaxel (Taxol®). A critical mechanism underlying paclitaxel-induced neuropathic pain is the increased production of peroxynitrite (PN) in spinal cord generated in response to activation of the superoxide-generating enzyme, NADPH oxidase. PN in turn contributes to the development of neuropathic pain by modulating several redox-dependent events in spinal cord. We recently reported that activation of the Gi/Gq-coupled A3 adenosine receptor (A3AR) with selective A3AR agonists (i.e., IB-MECA) blocked the development of chemotherapy induced-neuropathic pain evoked by distinct agents, including paclitaxel, without interfering with anticancer effects. The mechanism(s) of action underlying these beneficial effects has yet to be explored. We now demonstrate that IB-MECA attenuates the development of paclitaxel-induced neuropathic pain by inhibiting the activation of spinal NADPH oxidase and two downstream redox-dependent systems. The first relies on inhibition of the redox-sensitive transcription factor (NFκB) and mitogen activated protein kinases (ERK and p38) resulting in a decreased production of neuroexcitatory/pro-inflammatory cytokines (TNF-α, IL-1β) and increased formation of the neuroprotective/anti-inflammatory IL-10. The second involves inhibition of redox-mediated posttranslational tyrosine nitration and modification (inactivation) of glia-restricted proteins known to play key roles in regulating synaptic glutamate homeostasis: the glutamate transporter GLT-1 and glutamine synthetase. Our results unravel a mechanistic link into biomolecular signaling pathways employed by A3AR activation in neuropathic pain while providing the foundation to consider use of A3AR agonists as therapeutic agents in CIPN patients.
Keywords: adenosine, A3, neuroinflammation, chemotherapy-induced peripheral neuropathy, neuropathic pain, paclitaxel, spinal cord
Introduction
Chemotherapy-induced peripheral neuropathy (CIPN) accompanied by chronic neuropathic pain represents the most common dose-limiting complication associated with several first-line chemotherapeutics [12] including the taxane, paclitaxel (Taxol®) used for breast, ovarian, non-small cell lung carcinomas, and Kaposi’s sarcoma. This chronic neuropathy can persist for years after treatment [55] diminishing quality-of-life [12] and restricting optimal chemotherapeutic dosages. Clinical management becomes problematic as the causative mechanisms are poorly understood and current pain drugs are only marginally effective with unacceptable side effects [12]. Identification of novel therapeutics as adjuncts to chemotherapeutics to minimize side-effects and maximize anticancer effects is urgently needed.
We recently identified that highly-specific A3 adenosine receptor (A3AR) agonism is a novel and viable therapeutic strategy for CIPN [7]. Adenosine exerts its effects via four G protein-coupled receptor subtypes: A1AR and A3AR couple to Gi/Gq and A2AAR and A2BAR to Gs/olf/o [17]. Selective A3AR agonists, like IB-MECA or its 2-chloro analogue, Cl-IB-MECA, block neuropathic pain caused by diverse chemotherapeutics including paclitaxel, oxaliplatin, and bortezomib without interfering with anticancer effects [7]. Noteworthy, A3AR agonists have advanced to clinical trials for cancer and autoimmune conditions displaying promising beneficial effects and a good safety profile [17]. The beneficial mechanism(s) underlying A3AR agonism remain unexplored.
A3AR is expressed in endothelial cells, inflammatory cells, glial cells, and neurons within the periphery and central nervous system (CNS) [23]. Pathways proposed to mediate A3AR’s actions include inhibition of redox-sensitive NFκB, modulation of glycogen synthase kinase (GSK) 3β, attenuation of TNF-α/IL-1β, and increased formation of the anti-inflammatory IL-10 [21]. A3AR agonists are also neuroprotective [17,23]. Neuroprotection may occur by activating pro-survival RhoA-phospholipase D (PLD) signaling pathways. In cardiomyocytes, PLD activity is decreased in response to prolonged reactive oxygen species production during apoptosis [2]; an A3AR agonist can prevent this [30]. In breast cancer cells, increased PLD-mTOR activity and corresponding decreases in GSK3β generate pro-survival signaling [6]. Activating PLD can also increase production of choline, which activates α7 nicotinic acetylcholine receptors [29] known to be antinociceptive in chronic neuropathic pain [13]. A3AR agonists additionally stimulate glial-production of neuroprotective substances like CCL2 [58] and inhibit glial-derived pro-inflammatory cytokines [33]. A3AR activation protects against the neurotoxic P2X7-mediated [62] or the glutamate and NMDA-mediated rise in Ca2+ and thus neuronal excitability of neurons in vitro [61], suggesting that A3AR impacts glutamatergic signaling. While the underlying mechanisms of CIPN are multifactorial and include changes in the periphery [5], prominent neuropathological CNS changes have been implicated in the dysregulation of spinal neuro-glia communication brought about by neuroinflammatory processes [10,18,25]. For example, activation of NFκB and MAPKs (ERK, p38) [18,25] and overt production of pro-inflammatory cytokines (TNF-α, IL-1β) [10,18,25] have been reported.
In a first attempt to define potential mechanisms underlying A3AR’s protective actions in CIPN, we examined whether these effects are exerted via attenuation of spinal neuroinflammatory processes known to contribute to alterations in neuro-glia communication.
Methods
Experimental animals
Male Sprague Dawley rats (200–220 g starting weight) from Harlan Laboratories (Indianapolis, IN; Frederick, MD breeding colony) were housed 3–4 per cage in a controlled environment (12 h light/dark cycle) with food and water available ad libitum. All experiments were performed in accordance with the International Association for the Study of Pain and the National Institutes of Health guidelines on laboratory animal welfare and the recommendations by Saint Louis University Institutional Animal Care and Use Committee (IACUC). Animal use at the University of Messina likewise complied with Italian regulations for the protection of animals used for experimental and other scientific purpose (D.M. 116192) and with European Economic Community regulations. All experiments were conducted with the experimenters blinded to treatment conditions.
Test Compounds
In prophylactic experiments performed for biochemical analysis, all test compounds were given 15–20 min prior to chemotherapeutic (D0, D2, D4, and D6) and then daily thereafter up to D16. In the shortened prophylactic dosing regimen (concomitant), IB-MECA or its vehicle was given 15–20 minutes prior to paclitaxel only on the same days as paclitaxel (D0, 2, 4, 6). MRS1523 was given 15–20 minutes prior to IB-MECA. IB-MECA (1-deoxy-1-[6-[[(3-iodophenyl)methyl]amino]-9H-purin-9-yl]-N-methyl-β-D-ribofuranuronamide) was purchased from Tocris Bioscience (Bristol, United Kingdom). MRS1523 (3-propyl-6-ethyl-5-[(ethylthio)carbonyl]-2-phenyl-4-propyl-3-pyridine-carboxylate) was obtained from Sigma-Aldrich (St. Louis, MO).
Paclitaxel-induced neuropathic pain
Rats were treated with paclitaxel (Parenta Pharmaceuticals; Yardley, PA) or its vehicle as previously described [10,25]. In brief, doses of pharmaceutical-grade paclitaxel were prepared from original stock (6 mg/ml in Cremophor EL and 95% dehydrated ethanol in 1:1 ratio, Sigma) by diluting in sterile saline. Paclitaxel (2 mg/kg in 0.2 ml) was delivered by intraperitoneal (i.p.) injection alternating days for four days (D): D0, D2, D4 and D6 with a final cumulative dose of 8 mg/kg [45]. Control animals received an equivalent volume of the vehicle with proportional amounts of Cremophor EL and 95% dehydrated ethanol diluted in saline. This paclitaxel dosing regimen produces a time-dependent development of mechano-allodynia and mechano-hyperalgesia (mechano-hypersensitivity) evidenced by a decrease in paw withdrawal thresholds (PWTs (g)) by D12 (onset) that peaked by D16, and plateaued throughout our observation period (D25; times relative to first injection) [10,25]. We saw no evidence for the development of thermal hypersensitivity. The delay between the last exposure to paclitaxel and onset of mechano-hypersensitivity mimics the clinical “coasting” phenomenon described in patients [12]. In this study, all measurements were done at time of peak pain behaviors, D16 or D25.
Behavioral testing
Behavioral measurements were always taken prior to the administration of test substances. Mechano-allodynia was measured as previously described [10,25]. Briefly, rats were allowed to acclimate to behavioral chambers for 15 min prior to measuring the mechanical paw withdrawal thresholds, grams [PWT, (g)] by manual von Frey filaments (Stoelting, Wood Dale, IL, ranging from 3.61 [0.407 g] to 5.46 [26 g] bending force) [9] or electronic Von Frey test (dynamic plantar aesthesiometer, model 37450; Ugo Basile, Italy) with a cut off set at 50 g. Mechano-allodynia was defined by a significant (P<0.05) reduction in mechanical mean absolute PWT (g) at forces that failed to elicit withdrawal responses before chemotherapy treatment (D0). Mechano-hyperalgesia was measured by stimulating the dorsum of the rat’s hind paw as previously described [10,25] by the Randall and Sellitto paw pressure test [47] using a Ugo-Basile analgesiometer (Italy. model 37215). The nociceptive paw withdrawal threshold [PWT, (g)] was defined as the force (g) which caused the rat withdrew its paw (cut off set at 250 g). Since, chemotherapy-induced neuropathy results in bilateral allodynia and hyperalgesia with no differences between left and right hind PWT (g), the values from both paws were averaged. Animals receiving chemotherapeutic agents in the presence or absence of the experimental test substances tested did not display signs of any toxicities, as previously described [10,25].
Tissue Collection
At a time point of peak mechano-hypersensitivity (D16 or D25), animals were terminally anesthetized and decapitated for tissue collection. Immediately afterwards, the lumbar enlargement portion of the spinal cord (L5–L6) was harvested, flash frozen in liquid nitrogen, and stored at -80 °C until the appropriate biochemical assay could be performed.
Measurement of NADPH oxidase activity
Spinal cord tissues (L4–L6) were homogenized in HEPES buffer [10 mM, pH 7.5, containing 250 mM sucrose, 1 mM EGTA, 25 mM potassium chloride, 10 μg/ml soybean trypsin inhibitor, 2 μg/ml aprotinin and 10 μg/ml leupeptin] and centrifuged at 1,000 × g to obtain the nuclear-free supernatants. The NADPH cytochrome c reductase activity in these supernatants was measured using Cytochrome c Reductase (NADPH) Assay Kit as previously described [10].
Western blot analysis for transcription factors
The levels of IκBα, phospho-NFκB p65 (serine 536), phospho-ERK1/2, ERK1/2, and phospho-p38 MAP kinase were quantified in cytosolic fractions from spinal cord tissue, while NFκB p65 levels were quantified in nuclear fractions prepared as previously described [43]. All lysates were stored at -80°C until further analysis. Western blot analysis was performed as previously described [43] using specific antibodies to IκBα, NFκB p65, ERK2, phospho-ERK1/2 (1:1000; Santa Cruz Biotechnology; Dallas, TX), phospho-p38 MAP kinase (threonine180/tyrosine182), phospho-NFκB p65 (serine 536) (1:1000; Cell Signaling Technology; Beverly, MA), horseradish peroxidase-conjugated secondary antibodies (1:2000; Jackson ImmunoResearch Labratories; West Grove, PA) and enhanced chemiluminescence system (Amersham Biosciences; Amersham, UK). For loading controls, membranes were stripped and probed with antibodies against β-actin (1:10,000 Sigma-Aldrich) for whole lysates or cytosolic fractions or laminin-B1 protein (1:10,000, Sigma-Aldrich) for nuclear lysates. The relative expression of the protein bands were quantified by densitometric scanning of the X-ray films with GS-700 Imaging Densitometer (GS-700, Bio-Rad Laboratories; Milan, Italy) and Molecular Analyst software (IBM).
Cytokine Assay
The levels of cytokines in spinal cord (L4–6) cytosolic fraction isolated for Western Blot were assessed using by commercially available ELISA kits (R&D Systems; Milano, Italy) as previously described [10,25] or by custom-ordered magnetic multiplex cytokine kit (Bio-Rad Laboratories; Hercules, CA) according to manufacturer’s protocol.
Measurement of GLT-1 and GS
Cytosolic fractions and P2 membranes were obtained as previously described [38,39] and stored immediately at -80°C. Immunoprecipitation and Western blot analyses were performed as described previously [38,39]. Proteins were resolved with 7.5% (GLT-1), or 10% (GS) SDS-PAGE and electrophoretically transferred to PVDF membrane. The membranes were probed with mouse monoclonal anti-glutamine synthetase (1:2000) or polyclonal rabbit anti-GLT-1 (1:1000). Membranes were visualized with horseradish peroxidase-conjugated secondary antibodies (1 h, RT) and enhanced chemiluminescence. Rat brain lysate containing our proteins of interest was used as positive control. The blots were stripped and probed with a murine monoclonal anti-β-actin antibody (1:2000). The relative density of the protein bands of interest was determined from film using ImageQuant 5.2 software (Molecular Dynamics) and normalized to β-actin bands.
Statistical Analysis
Data are expressed as mean ± SD for n animals. Data from the time course studies were analyzed by two-way repeated measures ANOVA with Bonferroni comparisons. All other biochemical data collected were analyzed by one-way ANOVA with Dunnett’s-corrected comparisons. Significance was defined as P<0.05. All statistical analyses were performed using GraphPad Prism (v5.03, GraphPad Software, Inc.; San Diego, CA).
Results
Spinal activation of NADPH oxidase in response to paclitaxel is blocked by IB-MECA
We recently reported that the development of paclitaxel-induced neuropathic pain is dependent upon activation of NADPH oxidase which provides superoxide, the key precursor in the biosynthesis of peroxynitrite; blocking spinal NADPH oxidase blocked the formation of peroxynitrite and the development of paclitaxel-induced neuropathic pain [10]. We now demonstrate that on D16, a time of peak mechano-hypersensitivity (mechano-allodynia and –hyperalgesia; Fig. 1A,B; n=6), activation of NADPH oxidase and thus superoxide production in the spinal cord (Fig. 1C; n=6) was blocked by prophylactic treatment with IB-MECA (0.1 mg/kg/d; given i.p. from D0 to D15) (Fig. 1C; n=6); this was associated with significant attenuation of mechano-allodynia (Fig. 1A; n=6) and mechano-hyperalgesia (Fig. 1B; n=6). These beneficial events are dependent on selective A3AR-mediated mechanisms since pretreatment with 2 mg/kg/d (n=6) [7] of the selective A3AR antagonist, MRS1523, blocked the effects of IB-MECA on NADPH oxidase activity (Fig. 1C; n=6) and mechano-hypersensitivity (Fig. 1A,B; n=6). We also examined whether similar protection would be afforded by restricting IB-MECA treatment to when paclitaxel was given; a preferred adjunct regimen for treating patients only when they receive the chemotherapeutic agent. IB-MECA (0.1 mg/kg; n=6) given at the same time as paclitaxel (D0, 2, 4, and 6) blocked neuropathic pain through D25 (not shown); inhibitory effects at peak pain displayed a 98% and 98.7% inhibition of mechano-allodynia and mechano–hyperalgesia, respectively (n=6, P<0.001).
Fig. 1. IB-MECA prevents paclitaxel-induced mechano-hypersensitivity and spinal NADPH oxidase activity.
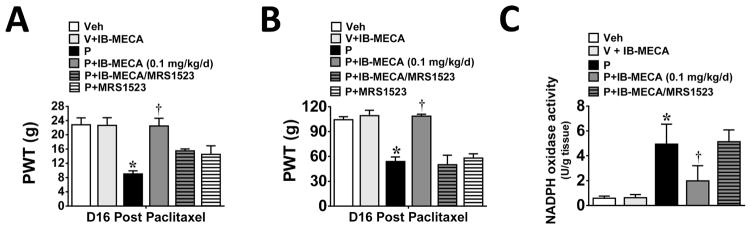
As compared to baseline on D16, administration of paclitaxel (P, black bars) but not vehicle (V, open bars) leads to the development of mechano-allodynia (A) and mechano–hyperalgesia (B). Spinal cords from paclitaxel-treated animals displayed higher levels of NADPH oxidase activity than their vehicle-treated counterparts (C). These paclitaxel-induced events were blocked by daily administration of the A3AR agonist IB-MECA (D0–15, 0.1 mg/kg/d; dark gray bars, A–C). The selective A3AR antagonist MRS1523 (2 mg/kg/d, D0–15, dark gray hatched bars) prevented IB-MECA’s beneficial actions (A–C). MRS1523 (white hatched bars) administered on its own to paclitaxel animals had no effect on PWTs (A,B). Results are expressed as mean ± SD for n=5–6 rats and analyzed by one-way ANOVA with Dunnett’s comparisons. *P<0.05 vs. Vehicle; †P<0.05 vs. Paclitaxel
IB-MECA prevents activation of NFκB and MAPK pathways and the production of TNF-α and IL-1β
The development of paclitaxel-induced neuropathic pain is dependent on increased production of peroxynitrite in the spinal cord that activates a series of redox-dependent signal transduction pathways, such as NFκB and MAPKs (ERK, p38) [25] and ultimately leading to the overt production of glia-dependent pro-inflammatory cytokines [10,25]. We now demonstrate that IB-MECA impinges upon these peroxynitrite-driven events. Consistent with our previous findings [25], administration of paclitaxel increased NFκB signaling in lumbar spinal cord indicated by IκBα degradation (Fig. 2A; n=5), p65 phosphorylation at serine 536 (Fig. 2B; n=5), and NFκB p65 nuclear translocation (Fig. 2C; n=5) as well as increased phosphorylation of MAPKs ERK1/2 (Fig. 2D; n=5) and p38 (Fig. 2E; n=5) and increased spinal formation of TNF-α and IL-1β (Fig. 3A; n=6). When compared to vehicle-treated rats, IB-MECA attenuated paclitaxel-induced activation of NFκB (Fig. 2A–C; n=5) and MAPKs (Fig. 2D,E; n=5) as well as the over-production of TNF-α and IL-1β (Fig. 3A; n=6) in the spinal cord measured by ELISA as previously reported [10,25]. Moreover, IB-MECA increased the level of the neuroprotective and anti-inflammatory cytokine, IL-10 (Fig. 3B; n=6). These events are A3AR-specific as MRS1523 (2 mg/kg/d, n=6) prevented IB-MECA-mediated reductions in TNF-α and IL-1β and enhancement of IL-10 formation (Fig. 3A,B). Similar results were obtained when IB-MECA treatment was restricted to coincide with paclitaxel dosing (Table 1; n=6).
Fig. 2. IB-MECA prevents activation of NFκB and MAPKs in the spinal cord during paclitaxel-induced neuropathic pain.

When compared to vehicle (V, open bars), administration of paclitaxel (P, black bars) increased IκBα degradation (A), cytosolic phosphorylation of NFκB p65 (B), nuclear translocation of NFκB p65 (C), and phosphorylation of ERK1/2 (D) and p38 (E). These paclitaxel-induced events were blocked by daily administration of IB-MECA (D0–15, 0.1 mg/kg/d; gray bars, A–E). Representative blots are shown. Results are expressed as mean ± SD for n=5–6 rats and analyzed by one-way ANOVA with Dunnett’s comparisons. *P<0.05 vs. Vehicle; †P<0.05 vs. Paclitaxel
Fig. 3. IB-MECA decreases paclitaxel-induced spinal formation of pro-inflammatory TNF-α and IL-1β and increases the anti-inflammatory IL-10.
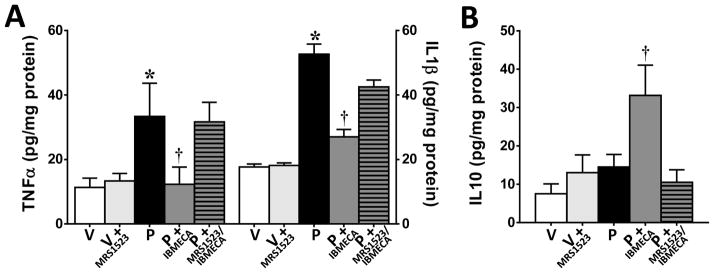
On D16, IB-MECA (0.1 mg/kg/d; D0–15; dark gray bars) attenuated paclitaxel-induced (P, black bars) elevations in TNF-α and IL-1β expression (A) and increased the levels of the anti-inflammatory cytokine, IL-10 (B) as compared to vehicle (V, open bars). The A3AR antagonist MRS1523 blocked IB-MECA’s effects (dark gray hatched bars) but had no effect on vehicle-treated (light gray bars) animals (A,B). Results are expressed as mean ± SD for n=6 and analyzed by one-way ANOVA with Dunnett’s post hoc comparisons. *P<0.05 vs. Vehicle; †P<0.05 vs. Paclitaxel
Table 1. Cytokine levels for animals treated concomitantly with IB-MECA.
Concurrent IB-MECA retains efficacy in modulating spinal pro-/anti-inflammatory cytokines with paclitaxel-treatment.
IB-MECA (0.1 mg/kg/d; D0–4) given 15–20 minutes prior to paclitaxel (D0, 2, 4, and 6) effectively prevented the increase in pro-inflammatory cytokines (TNF-α, IL-1β) and increased the anti-inflammatory cytokine IL-10 as measured on D25. Results are expressed as mean ± SD for n=6 and analyzed by one-way ANOVA with Dunnett’s post hoc comparisons.
| Treatment | TNF-α (pg/mg protein) | IL-1β (pg/mg protein) | IL-10 (pg/mg protein) | |||
|---|---|---|---|---|---|---|
| Mean ± SD | 95% CI | Mean ± SD | 95% CI | Mean ± SD | 95% CI | |
|
| ||||||
| Vehicle | 27.1 ± 5.2 | 21.6 – 32.5 | 40.0 ± 13.4 | 26.0 – 54.0 | 110.9 ± 19.5 | 90.4 – 131.3 |
|
| ||||||
| Paclitaxel | 40 ± 8.3* | 31.5 – 49.0 | 73.1 ± 13.8* | 58.6 – 87.6 | 94.6 ± 19.4 | 74.3 – 114.9 |
|
| ||||||
| P+IB-MECA | 26 ± 4.6** | 22.1 – 31.8 | 40.5 ± 12.6** | 27.3 – 53.7 | 146.9 ± 13.1 | 133.2 – 160.7** |
P<0.05 vs. Vehicle;
P<0.05 vs. Paclitaxel
IB-MECA prevents peroxynitrite-mediated post-translational tyrosine nitration of the spinal glutamate transporter, GLT-1, and glutamine synthetase
A key property of peroxynitrite lies in its ability to post-translationally nitrate tyrosine and consequently modify protein function [22]. Protein nitration is increasingly recognized as an important occurrence during cell signaling and regulation of protein activity (resulting in no effect, loss or gain of function) [22]. The loss of function is best demonstrated in the development of central sensitization [49], including paclitaxel-induced neuropathic pain [10], with the in vivo nitration of glial glutamate transporters and glutamine synthetase; proteins known to be essential in regulating synaptic concentrations of glutamate and glutamate neurotransmission. As can be seen in figure 4, when compared to vehicle-treated rats, the development of paclitaxel-induced mechano-hypersensitivity was associated with increased nitration of GLT-1 (Fig. 4A; n=5) and GS (Fig. 4B; n=5) in spinal cords harvested on D16 from paclitaxel-treated rats, and this nitration was significantly (P<0.01) attenuated by IB-MECA (0.1 mg/kg/d, Fig. 4; n=5).
Fig. 4. IB-MECA prevents post-translational nitration of GLT-1 & GS in the spinal cord during paclitaxel-induced neuropathic pain.
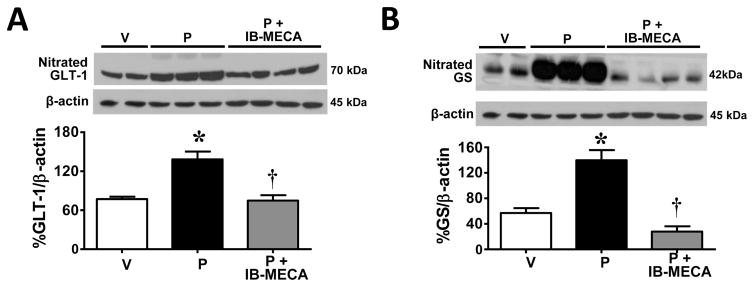
Compared with vehicle (V, open bars), paclitaxel (P, black bars) led to significant nitration of the glutamate transporter GLT-1 (A) and GS (B) in spinal cord tissues, events attenuated in rats treated with IB-MECA (0.1 mg/kg/d, D0–15; gray bars). Representative blots are shown. Results are expressed as mean ± SD for n=5 rats and analyzed by one-way ANOVA with Dunnett’s comparisons. *P<0.05 vs. Vehicle; †P<0.05 vs. Paclitaxel
Discussion
A3AR agonists are potent non-narcotic analgesics able to block and reverse neuropathic pain [7] by acting at peripheral, spinal, and supraspinal sites [32]. This study, the first to examine potential signaling pathways, identifies inhibition of NADPH oxidase with subsequent modulation of two well-described glia-restricted redox-dependent signaling pathways as an important spinal mechanism of action (Fig. 5).
Fig. 5. Proposed schematic representation of mechanisms underlying IB-MECA’s beneficial actions on CIPN.

Chemotherapy (paclitaxel)-induced neuropathic pain is associated with increased NADPH oxidase activity within the spinal cord contributing to enhanced peroxynitrite (PN) production. Due to its ability to post-translationally modify protein function, peroxynitrite could be a driver behind many of the spinal neuropathological changes underlying CIPN including 1) the nitration/inactivation of glutamate transporter 1 (GLT-1) and glutamine synthetase (GS) and 2) the activation of redox-dependent signaling pathways (NFκB and MAPK) leading to a surge in glial-associated pro-inflammatory cytokine production (TNF-α and IL-1β). Treatment with the selective A3AR agonist, IB-MECA, not only inhibits paclitaxel-induced pain and the associated spinal events, it also increases the formation of the neuroprotective/anti-inflammatory cytokine, IL-10.
Accumulating evidence implicates neuroinflammatory processes in the alteration of spinal glia-neuronal communication during paclitaxel-induced neuropathic pain. For example, the hyperactivation of glial cells (astrocytes [18,60] and microglia [44]), the activation of redox-sensitive NFκB and MAPKs (ERK, p38) [18,25], and overt production of glia-derived pro-inflammatory cytokines (TNF-α and IL-1β) [10,18,25] have been documented. We recently reported that increased production of spinal superoxide-derived peroxynitrite after nerve injury is the linchpin in setting into motion well-defined processes essential to the initiation, propagation, and maintenance of central sensitization associated with paclitaxel-induced neuropathic pain [10]. Noteworthy, these findings are not unique to CIPN but are also reported in inflammatory pain, neuropathic pain, and opioid-induced antinociceptive tolerance and hyperalgesia underscoring the key contribution of peroxynitrite to the development of central sensitization underlying pain of several etiologies (reviewed in [49]). In spinal cord, two important enzymatic sources have been identified in providing sustained elevated levels of peroxynitrite: activation of NADPH oxidase (expressed in neurons, astrocytes and microglia [4]) and inactivation of mitochondrial manganese superoxide dismutase (MnSOD) following post-translational nitration of Tyr-34 by peroxynitrite [36]. This disruption in enzymatic activity provides a “feed-forward” mechanism sustaining elevated peroxynitrite through elevated superoxide (reviewed in [49]). Our results demonstrate that IB-MECA attenuated spinal activation of NADPH oxidase with subsequent inhibition of NFκB and MAPKs resulting in decreased production of TNF-α and IL-1β and increased formation of IL-10. In addition to its well-recognized anti-inflammatory role, IL-10 is also a powerful neuroinhibitory cytokine; therapeutic manipulations aimed at increasing its presence in spinal cord (i.e., with plasmid DNA encoding IL-10) [28] or by indirectly increasing its production through the removal of peroxynitrite [10] blocked paclitaxel-induced neuropathic pain. Therefore, increased spinal formation of IL-10 may represent a major component of A3AR’s beneficial actions.
A recent study revealed that increased GSK3β activation in spinal cord contributes to paclitaxel-induced neuropathic pain by activating astrocytes and causing overt production of IL-1β; GSK3β inhibition with lithium was found to be beneficial [18]. Whether paclitaxel-induced activation of spinal GSK3β is also redox-modulated remains to be established, but is a clear possibility considering previous findings in non-pain related fields demonstrating a direct involvement of superoxide/peroxynitrite in Akt/GSK3β signaling [51] and since the pharmacological profile of lithium in the paclitaxel model [18] is identical to the one reported with peroxynitrite decomposition catalysts [10]. Once formed, nitroxidative species [40] and cytokines like IL-1β [59] contribute to excessive activation of synaptic glutamate receptors through several mechanisms including increasing the activities of AMPA and NMDA receptors in spinal dorsal horn neurons, and glutamate release from presynaptic terminals which has been reported to accompany paclitaxel-induced neuropathic pain [18]. It is currently unknown how A3AR inhibits NADPH oxidase activation; however, a recent report revealed that IB-MECA inhibits NADPH oxidase activation in prostate cancer cells by inhibiting a cyclic AMP/PKA pathway [24]. Additionally, IB-MECA treatment correlated with a reduction in the expression of the Rac1 and p47phox subunits of NADPH oxidase by inhibiting ERK1/2 activity [24]. Other mechanisms of A3AR-mediated inhibition of NADPH oxidase activation may stem from the observed A3AR-mediated shift from pro-inflammatory to anti-inflammatory environments. The provocation of NADPH oxidase activity by TNF-κ and toll-like receptors (TLRs) is well-established [4], and enhanced expression of endogenous IL-10 attenuates the production of pro-inflammatory cytokines and NADPH oxidase activity in LPS-stimulated cerebral microglia and prevents neuronal death [42]. The mitoprotective effects ascribed to A3AR agonists [11] may also be attributed to attenuation of NADPH oxidase activity. Excessive glutamatergic signaling [50] sparks mitochondrial uptake of Ca2+ leading to increased superoxide production [56]. Elevations in superoxide from mitochondria can then trigger NADPH oxidase activity to further exacerbate mitochondrial dysfunction [8]. Moreover, the addition of pro-inflammatory mediators promotes increased metabotropic glutamate receptor (mGluR) 3 and reduced mGluR5 expression in cultured glia [1]. Such mGluR expression profiles favor increased NADPH oxidase activity in microglia [37] as well as promote the development of their neurotoxic phenotype [53]. In neurons, A3AR agonists inhibit mGluR signaling [34]; thus, it is possible that A3AR’s effects on glial NADPH oxidase activity occur through similar inhibition of mGluR signaling.
Alterations in glutamatergic neurotransmission and increased neuronal excitability are also present in paclitaxel-induced neuropathic pain [10,60]. Synaptic levels of glutamate are tightly regulated by GTs whose appropriate function is critical in ensuring optimal glutamatergic signaling [19]. Three GT subtypes are found in spinal cord: GLAST and GLT-1 in glia [48] and the excitatory amino acid carrier-1 (EACC1) in neurons [26]. Glia-restricted GTs account for >90% of glutamate reuptake and thus control the termination of glutamatergic signaling [19]. Compromising the glutamate reuptake efficiencies of GTs by either downregulating their expression and/or inactivating their transport activity ensures excessive activation of AMPA and NMDA receptors in the spinal dorsal horn and failure to terminate excitatory signaling [19]. Downregulation of spinal GTs is reported to accompany paclitaxel-induced neuropathic pain [60], but the mechanism(s) involved are unclear. However, inactivation of GTs is the consequence of specific tyrosine nitration and post-translational modifications, a process carried out uniquely by peroxynitrite [54]. In contradistinction to GT-regulation of extracellular glutamate homeostasis, GS plays a pivotal role in its intracellular metabolic fate [52]. In CNS, GS is located mainly in astrocytes and protects neurons against excitotoxicity by converting excess ammonia and glutamate into non-toxic glutamine [52] and returning it to neurons as a precursor for glutamate and GABA; its inactivation maintains neuronal excitability [52]. Spinal astrocyte hyperactivation plays a central role in paclitaxel-induced neuroapthic pain [60]; therefore, compromising the enzymatic activity of GS is expected to maintain neuronal excitation [52]. GS is exquisitively sensitive to peroxynitrite with nitration on Tyr-160 leading to significant loss of enzymatic activity [20]. Results of our study revealed that a second consequence of A3AR activation is the inhibition of peroxynitrite-mediated posttranslational nitration and modification (inactivation) of GLT-1 and GS. It is therefore possible that A3AR agonists, by lowering the production of spinal peroxynitrite and preventing GT and GS nitration, “reset” optimal glutamatergic neurotransmission by reducing glutamatergic post-synaptic excitability.
The mechanistic connections between paclitaxel and activation of NADPH oxidase resulting in peroxynitrite formation in spinal cord and downstream effects remain unknown. A growing body of data recently emerged to implicate activation of TLR4 on glial cells in the development of neuropathic pain [57]. More recently activation of TLR4 expressed on spinal astrocytes has also been linked to paclitaxel-induced neuropathic pain [31]. It is well established that redox-signaling following activation of NADPH oxidase is essential to the downstream effects (i.e., NFκB activation) engaged by TLR4 [41]. Noteworthy, peroxynitrite can sustain the activation of NADPH oxidase by nitrating and increasing PKC activity [3]. PKC phosphorylates the p47phox subunit facilitating its translocation to the membrane and binding to the catalytic p67phox subunit forming the active holoenzyme [27]. Moreover, PKC also phosphorylates the membrane-associated gp91phox increasing its diaphorase activity and it’s binding of the Rac2, p67phox, and p47phox cytosolic subunits to form the active complex [46]. It is therefore tempting to speculate that TLR4 provides the link to peroxynitrite formation through NADPH oxidase activation; peroxynitrite then sustains a “nitroxidative” rich milieu in spinal cord by maintaining an activated NADPH oxidase and by inactivating mitochondrial MnSOD [35]. In this scenario, activation of A3AR disrupts this interplay therefore breaking the vicious circle. These reciprocal interactions are expected to prolong and exacerbate CIPN by instituting feedback loops that then sustain local neuroinflammatory processes in spinal cord. A similar process is envisioned in dorsal root ganglia where TLR4 activation has been implicated in paclitaxel-induced neuropathic pain [31].
Collectively, our study provides the first mechanistic insight into beneficial effects exerted by A3AR agonists. A3AR are highly expressed in a broad spectrum of malignant cells [14–16]. Furthermore, A3AR agonists possess direct anticancer effects in a variety of cancer cells; as shown in in vitro and in vivo studies [7,14–16], they do not interfere with the anticancer effects of widely used chemotherapeutics including paclitaxel [7] but can actually enhance their actions [7,14,15] and attenuate other undesireable side effects of chemotherapeutics including myelosuppression [14,16]. In addition, Cl-IB-MECA is already advancing in clinical trials as an anticancer agent, thus providing potential for fast-track proof-of-concept testing for CIPN. Noteworthy, in 2012 the FDA granted orphan drug status to Cl-IB-MECA for the treatment of hepatocellular carcinoma. Use of A3AR agonists may thus provide a distinct advantage/opportunity in this therapeutic area; direct anticancer effects and mitigation of a major dose limiting toxicity, CIPN. Molecules endowed with such complimentary and potentially synergistic properties have yet to be identified, so this is unprecedented. Further understanding of the adenosine-to-A3AR axis could provide the framework for clinical development of A3AR agonists in CIPN and perhaps other chronic neuropathic pain states such as diabetic neuropathy whose underlying pathology shares commonalities with CIPN [5].
Summary.
A3AR agonists prevent paclitaxel-induced neuropathic pain via modulating spinal redox-dependent signaling pathways (restoring glutamatergic homeostasis, attenuating pro-inflammatory pathways).
Acknowledgments
We would like to thank Leesa Bryant for her technical assistance in performing some of the animal experiments. This study was funded by grants from National Cancer Institute (RO1CA169519) and the Saint Louis University President Research Fund with additional support from the Saint Louis Cancer Center. KAJ and DKT acknowledge support from the NIDDK Intramural Research Program.
Footnotes
Conception and design: KJ, DS
Analysis and interpretation of data: KJ, EE, TD, SC, DTK, KAJ, DS
Drafting of manuscript: KJ, DS
Intellectual contribution of content: KJ, KAJ, DS
The authors claim no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aronica E, Gorter JA, Ijlst-Keizers H, Rozemuller AJ, Yankaya B, Leenstra S, Troost D. Expression and functional role of mGluR3 and mGluR5 in human astrocytes and glioma cells: opposite regulation of glutamate transporter proteins. The European journal of neuroscience. 2003;17(10):2106–2118. doi: 10.1046/j.1460-9568.2003.02657.x. [DOI] [PubMed] [Google Scholar]
- 2.Asemu G, Dent MR, Singal T, Dhalla NS, Tappia PS. Differential changes in phospholipase D and phosphatidate phosphohydrolase activities in ischemia-reperfusion of rat heart. Archives of biochemistry and biophysics. 2005;436(1):136–144. doi: 10.1016/j.abb.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 3.Balafanova Z, Bolli R, Zhang J, Zheng Y, Pass JM, Bhatnagar A, Tang XL, Wang O, Cardwell E, Ping P. Nitric oxide (NO) induces nitration of protein kinase Cepsilon (PKCepsilon), facilitating PKCepsilon translocation via enhanced PKCepsilon -RACK2 interactions: a novel mechanism of no-triggered activation of PKCepsilon. The Journal of biological chemistry. 2002;277(17):15021–15027. doi: 10.1074/jbc.M112451200. [DOI] [PubMed] [Google Scholar]
- 4.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological reviews. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 5.Bennett GJ, Doyle T, Salvemini D. Mitotoxicity in distal symmetrical sensory peripheral neuropathies. Nature reviews Neurology. 2014 doi: 10.1038/nrneurol.2014.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Y, Rodrik V, Foster DA. Alternative phospholipase D/mTOR survival signal in human breast cancer cells. Oncogene. 2005;24(4):672–679. doi: 10.1038/sj.onc.1208099. [DOI] [PubMed] [Google Scholar]
- 7.Chen Z, Janes K, Chen C, Doyle T, Bryant L, Tosh DK, Jacobson KA, Salvemini D. Controlling murine and rat chronic pain through A3 adenosine receptor activation. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2012;26(5):1855–1865. doi: 10.1096/fj.11-201541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free radical biology & medicine. 2011;51(7):1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- 10.Doyle T, Chen Z, Muscoli C, Bryant L, Esposito E, Cuzzocrea S, Dagostino C, Ryerse J, Rausaria S, Kamadulski A, Neumann WL, Salvemini D. Targeting the overproduction of peroxynitrite for the prevention and reversal of paclitaxel-induced neuropathic pain. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32(18):6149–6160. doi: 10.1523/JNEUROSCI.6343-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Emanuelov AK, Shainberg A, Chepurko Y, Kaplan D, Sagie A, Porat E, Arad M, Hochhauser E. Adenosine A3 receptor-mediated cardioprotection against doxorubicin-induced mitochondrial damage. Biochemical pharmacology. 2010;79(2):180–187. doi: 10.1016/j.bcp.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 12.Farquhar-Smith P. Chemotherapy-induced neuropathic pain. Curr Opin Support Palliat Care. 2011;5(1):1–7. doi: 10.1097/SPC.0b013e328342f9cc. [DOI] [PubMed] [Google Scholar]
- 13.Feuerbach D, Lingenhoehl K, Olpe HR, Vassout A, Gentsch C, Chaperon F, Nozulak J, Enz A, Bilbe G, McAllister K, Hoyer D. The selective nicotinic acetylcholine receptor alpha7 agonist JN403 is active in animal models of cognition, sensory gating, epilepsy and pain. Neuropharmacology. 2009;56(1):254–263. doi: 10.1016/j.neuropharm.2008.08.025. [DOI] [PubMed] [Google Scholar]
- 14.Fishman P, Bar-Yehuda S, Liang BT, Jacobson KA. Pharmacological and therapeutic effects of A3 adenosine receptor agonists. Drug discovery today. 2012;17(7–8):359–366. doi: 10.1016/j.drudis.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fishman P, Bar-Yehuda S, Madi L, Cohn I. A3 adenosine receptor as a target for cancer therapy. Anticancer Drugs. 2002;13(5):437–443. doi: 10.1097/00001813-200206000-00001. [DOI] [PubMed] [Google Scholar]
- 16.Fishman P, Bar-Yehuda S, Synowitz M, Powell JD, Klotz KN, Gessi S, Borea PA. Adenosine receptors and cancer. Handbook of experimental pharmacology. 2009;(193):399–441. doi: 10.1007/978-3-540-89615-9_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fredholm BB, AP IJ, Jacobson KA, Linden J, Muller CE. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors--an update. Pharmacol Rev. 2011;63(1):1–34. doi: 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao M, Yan X, Weng HR. Inhibition of glycogen synthase kinase 3beta activity with lithium prevents and attenuates paclitaxel-induced neuropathic pain. Neuroscience. 2013;254:301–311. doi: 10.1016/j.neuroscience.2013.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gegelashvili G, Robinson MB, Trotti D, Rauen T. Regulation of glutamate transporters in health and disease. Prog Brain Res. 2001;132:267–286. doi: 10.1016/S0079-6123(01)32082-4. [DOI] [PubMed] [Google Scholar]
- 20.Gorg B, Qvartskhava N, Voss P, Grune T, Haussinger D, Schliess F. Reversible inhibition of mammalian glutamine synthetase by tyrosine nitration. FEBS Lett. 2007;581(1):84–90. doi: 10.1016/j.febslet.2006.11.081. [DOI] [PubMed] [Google Scholar]
- 21.Hasko G, Szabo C, Nemeth ZH, Kvetan V, Pastores SM, Vizi ES. Adenosine receptor agonists differentially regulate IL-10, TNF-alpha, and nitric oxide production in RAW 264.7 macrophages and in endotoxemic mice. Journal of immunology. 1996;157(10):4634–4640. [PubMed] [Google Scholar]
- 22.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nature reviews. 2005;6(2):150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 23.Jacobson KA. Adenosine A3 receptors: novel ligands and paradoxical effects. Trends Pharmacol Sci. 1998;19(5):184–191. doi: 10.1016/s0165-6147(98)01203-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jajoo S, Mukherjea D, Watabe K, Ramkumar V. Adenosine A(3) receptor suppresses prostate cancer metastasis by inhibiting NADPH oxidase activity. Neoplasia. 2009;11(11):1132–1145. doi: 10.1593/neo.09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janes K, Little JW, Li C, Bryant L, Chen C, Chen Z, Kamocki K, Doyle T, Snider A, Esposito E, Cuzzocrea S, Bieberich E, Obeid L, Petrache I, Nicol G, Neumann WL, Salvemini D. The Development and Maintenance of Paclitaxel-Induced Neuropathic Pain Requires Activation of the Sphingosine 1-Phosphate Receptor Subtype 1. The Journal of biological chemistry. 2014 doi: 10.1074/jbc.M114.569574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanai Y, Hediger MA. Primary structure and functional characterization of a high-affinity glutamate transporter. Nature. 1992;360(6403):467–471. doi: 10.1038/360467a0. [DOI] [PubMed] [Google Scholar]
- 27.Kitada M, Koya D, Sugimoto T, Isono M, Araki S, Kashiwagi A, Haneda M. Translocation of glomerular p47phox and p67phox by protein kinase C-beta activation is required for oxidative stress in diabetic nephropathy. Diabetes. 2003;52(10):2603–2614. doi: 10.2337/diabetes.52.10.2603. [DOI] [PubMed] [Google Scholar]
- 28.Ledeboer A, Jekich BM, Sloane EM, Mahoney JH, Langer SJ, Milligan ED, Martin D, Maier SF, Johnson KW, Leinwand LA, Chavez RA, Watkins LR. Intrathecal interleukin-10 gene therapy attenuates paclitaxel-induced mechanical allodynia and proinflammatory cytokine expression in dorsal root ganglia in rats. Brain, behavior, and immunity. 2007;21(5):686–698. doi: 10.1016/j.bbi.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee HC, Fellenz-Maloney MP, Liscovitch M, Blusztajn JK. Phospholipase D-catalyzed hydrolysis of phosphatidylcholine provides the choline precursor for acetylcholine synthesis in a human neuronal cell line. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(21):10086–10090. doi: 10.1073/pnas.90.21.10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee JE, Bokoch G, Liang BT. A novel cardioprotective role of RhoA: new signaling mechanism for adenosine. FASEB J. 2001;15(11):1886–1894. doi: 10.1096/fj.01-0212com. [DOI] [PubMed] [Google Scholar]
- 31.Li Y, Zhang H, Kosturakis AK, Jawad AB, Dougherty PM. Toll-Like Receptor 4 Signaling Contributes to Paclitaxel-Induced Peripheral Neuropathy. J Pain. 2014 doi: 10.1016/j.jpain.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Little J, Chen Z, Ford A, Janes K, Doyle T, Tosh D, Jacobson K, Salvemini D. (297) Central adenosine A3 receptor (A3AR) activation reverses neuropathic pain. The journal of pain: official journal of the American Pain Society. 2014;15(4):S50. [Google Scholar]
- 33.Luo C, Yi B, Tao G, Li M, Chen Z, Tang W, Zhang JH, Feng H. Adenosine A3 receptor agonist reduces early brain injury in subarachnoid haemorrhage. Neuroreport. 2010;21(13):892–896. doi: 10.1097/WNR.0b013e32833dbd13. [DOI] [PubMed] [Google Scholar]
- 34.Macek TA, Schaffhauser H, Conn PJ. Protein kinase C and A3 adenosine receptor activation inhibit presynaptic metabotropic glutamate receptor (mGluR) function and uncouple mGluRs from GTP-binding proteins. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1998;18(16):6138–6146. doi: 10.1523/JNEUROSCI.18-16-06138.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Macmillan-Crow LA, Cruthirds DL. Invited review: manganese superoxide dismutase in disease. Free radical research. 2001;34(4):325–336. doi: 10.1080/10715760100300281. [DOI] [PubMed] [Google Scholar]
- 36.MacMillan-Crow LA, Thompson JA. Tyrosine modifications and inactivation of active site manganese superoxide dismutase mutant (Y34F) by peroxynitrite. Archives of biochemistry and biophysics. 1999;366(1):82–88. doi: 10.1006/abbi.1999.1202. [DOI] [PubMed] [Google Scholar]
- 37.Mead EL, Mosley A, Eaton S, Dobson L, Heales SJ, Pocock JM. Microglial neurotransmitter receptors trigger superoxide production in microglia; consequences for microglial-neuronal interactions. Journal of neurochemistry. 2012;121(2):287–301. doi: 10.1111/j.1471-4159.2012.07659.x. [DOI] [PubMed] [Google Scholar]
- 38.Muscoli C, Cuzzocrea S, Ndengele MM, Mollace V, Porreca F, Fabrizi F, Esposito E, Masini E, Matuschak GM, Salvemini D. Therapeutic manipulation of peroxynitrite attenuates the development of opiate-induced antinociceptive tolerance in mice. The Journal of clinical investigation. 2007;117(11):3530–3539. doi: 10.1172/JCI32420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muscoli C, Doyle T, Dagostino C, Bryant L, Chen Z, Watkins LR, Ryerse J, Bieberich E, Neumman W, Salvemini D. Counter-regulation of opioid analgesia by glial-derived bioactive sphingolipids. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30(46):15400–15408. doi: 10.1523/JNEUROSCI.2391-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishio N, Taniguchi W, Sugimura YK, Takiguchi N, Yamanaka M, Kiyoyuki Y, Yamada H, Miyazaki N, Yoshida M, Nakatsuka T. Reactive oxygen species enhance excitatory synaptic transmission in rat spinal dorsal horn neurons by activating TRPA1 and TRPV1 channels. Neuroscience. 2013;247:201–212. doi: 10.1016/j.neuroscience.2013.05.023. [DOI] [PubMed] [Google Scholar]
- 41.Park HS, Jung HY, Park EY, Kim J, Lee WJ, Bae YS. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. Journal of immunology. 2004;173(6):3589–3593. doi: 10.4049/jimmunol.173.6.3589. [DOI] [PubMed] [Google Scholar]
- 42.Park KW, Lee HG, Jin BK, Lee YB. Interleukin-10 endogenously expressed in microglia prevents lipopolysaccharide-induced neurodegeneration in the rat cerebral cortex in vivo. Experimental & molecular medicine. 2007;39(6):812–819. doi: 10.1038/emm.2007.88. [DOI] [PubMed] [Google Scholar]
- 43.Paterniti I, Mazzon E, Emanuela E, Paola RD, Galuppo M, Bramanti P, Cuzzocrea S. Modulation of inflammatory response after spinal cord trauma with deferoxamine, an iron chelator. Free radical research. 2010;44(6):694–709. doi: 10.3109/10715761003742993. [DOI] [PubMed] [Google Scholar]
- 44.Pevida M, Lastra A, Hidalgo A, Baamonde A, Menendez L. Spinal CCL2 and microglial activation are involved in paclitaxel-evoked cold hyperalgesia. Brain research bulletin. 2013;95:21–27. doi: 10.1016/j.brainresbull.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 45.Polomano RC, Mannes AJ, Clark US, Bennett GJ. A painful peripheral neuropathy in the rat produced by the chemotherapeutic drug, paclitaxel. Pain. 2001;94(3):293–304. doi: 10.1016/S0304-3959(01)00363-3. [DOI] [PubMed] [Google Scholar]
- 46.Raad H, Paclet MH, Boussetta T, Kroviarski Y, Morel F, Quinn MT, Gougerot-Pocidalo MA, Dang PM, El-Benna J. Regulation of the phagocyte NADPH oxidase activity: phosphorylation of gp91phox/NOX2 by protein kinase C enhances its diaphorase activity and binding to Rac2, p67phox, and p47phox. FASEB J. 2009;23(4):1011–1022. doi: 10.1096/fj.08-114553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Randall LO, Selitto JJ. A method for measurement of analgesic activity on inflamed tissue. Arch Int Pharmacodyn Ther. 1957;111(4):409–419. [PubMed] [Google Scholar]
- 48.Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13(3):713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 49.Salvemini D, Little JW, Doyle T, Neumann WL. Roles of reactive oxygen and nitrogen species in pain. Free radical biology & medicine. 2011;51(5):951–966. doi: 10.1016/j.freeradbiomed.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schinder AF, Olson EC, Spitzer NC, Montal M. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1996;16(19):6125–6133. doi: 10.1523/JNEUROSCI.16-19-06125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Son YO, Pratheeshkumar P, Wang L, Wang X, Fan J, Kim DH, Lee JY, Zhang Z, Lee JC, Shi X. Reactive oxygen species mediate Cr(VI)-induced carcinogenesis through PI3K/AKT-dependent activation of GSK-3beta/beta-catenin signaling. Toxicology and applied pharmacology. 2013;271(2):239–248. doi: 10.1016/j.taap.2013.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suarez I, Bodega G, Fernandez B. Glutamine synthetase in brain: effect of ammonia. Neurochem Int. 2002;41(2–3):123–142. doi: 10.1016/s0197-0186(02)00033-5. [DOI] [PubMed] [Google Scholar]
- 53.Taylor DL, Diemel LT, Cuzner ML, Pocock JM. Activation of group II metabotropic glutamate receptors underlies microglial reactivity and neurotoxicity following stimulation with chromogranin A, a peptide up-regulated in Alzheimer’s disease. Journal of neurochemistry. 2002;82(5):1179–1191. doi: 10.1046/j.1471-4159.2002.01062.x. [DOI] [PubMed] [Google Scholar]
- 54.Trotti D, Rossi D, Gjesdal O, Levy LM, Racagni G, Danbolt NC, Volterra A. Peroxynitrite inhibits glutamate transporter subtypes. The Journal of biological chemistry. 1996;271(11):5976–5979. doi: 10.1074/jbc.271.11.5976. [DOI] [PubMed] [Google Scholar]
- 55.Velasco R, Bruna J. Chemotherapy-induced peripheral neuropathy: an unresolved issue. Neurologia. 2010;25(2):116–131. [PubMed] [Google Scholar]
- 56.Wang JQ, Chen Q, Wang X, Wang QC, Wang Y, Cheng HP, Guo C, Sun Q, Chen Q, Tang TS. Dysregulation of mitochondrial calcium signaling and superoxide flashes cause mitochondrial genomic DNA damage in Huntington disease. The Journal of biological chemistry. 2013;288(5):3070–3084. doi: 10.1074/jbc.M112.407726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Watkins LR, Hutchinson MR, Rice KC, Maier SF. The “toll” of opioid-induced glial activation: improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol Sci. 2009;30(11):581–591. doi: 10.1016/j.tips.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wittendorp MC, Boddeke HW, Biber K. Adenosine A3 receptor-induced CCL2 synthesis in cultured mouse astrocytes. Glia. 2004;46(4):410–418. doi: 10.1002/glia.20016. [DOI] [PubMed] [Google Scholar]
- 59.Ye ZC, Sontheimer H. Cytokine modulation of glial glutamate uptake: a possible involvement of nitric oxide. Neuroreport. 1996;7(13):2181–2185. doi: 10.1097/00001756-199609020-00025. [DOI] [PubMed] [Google Scholar]
- 60.Zhang H, Yoon SY, Dougherty PM. Evidence That Spinal Astrocytes but Not Microglia Contribute to the Pathogenesis of Paclitaxel-Induced Painful Neuropathy. J Pain. 2012 doi: 10.1016/j.jpain.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang M, Hu H, Zhang X, Lu W, Lim J, Eysteinsson T, Jacobson KA, Laties AM, Mitchell CH. The A3 adenosine receptor attenuates the calcium rise triggered by NMDA receptors in retinal ganglion cells. Neurochem Int. 2010;56(1):35–41. doi: 10.1016/j.neuint.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang X, Zhang M, Laties AM, Mitchell CH. Balance of purines may determine life or death of retinal ganglion cells as A3 adenosine receptors prevent loss following P2X7 receptor stimulation. Journal of neurochemistry. 2006;98(2):566–575. doi: 10.1111/j.1471-4159.2006.03900.x. [DOI] [PubMed] [Google Scholar]