

Abstract
Objective
Endothelial cell (EC) migration is essential for healing of arterial injuries caused by angioplasty, but a high cholesterol diet inhibits endothelial repair. In vivo studies suggest that apolipoprotein A-I (apoA-I), the major protein constituent of HDL, is essential for normal healing of arterial injuries. ApoA-I mimetics, including 4F, have been designed to mimic the amphipathic portion of the apoA-I molecule. This study was undertaken to determine if 4F improves endothelial migration and healing.
Methods
A razor scrape assay was used to analyze the effect of 4F on EC migration in vitro. Endothelial healing in vivo was assessed following electrical injury of carotid arteries in mice. Markers of oxidative stress were also examined.
Results
Lipid oxidation products inhibited EC migration in vitro, but preincubation with L-4F preserved EC migration. Endothelial healing of carotid arterial injuries in mice on a high cholesterol diet was delayed compared with mice on a chow diet with 27.8% vs. 48.2% healing, respectively, at 5 days. Administration of D-4F improved endothelial healing in mice on a high cholesterol diet to 43.4%. D-4F administration had no effect on lipid levels but decreased markers of oxidation. In vivo, there was a significant inverse correlation between endothelial healing and plasma markers of oxidative stress.
Conclusion
These studies suggested that an apoA-I mimetic can improve endothelial healing of arterial injuries by decreasing oxidative stress.
Keywords: Endothelium, hypercholesterolemia, apolipoprotein A-I mimetic, D-4F, L-4F, endothelial migration, arterial healing
INTRODUCTION
Cardiovascular disease has a major impact on the length and quality of life. The population is aging and between the years 2010 and 2030, the number of people over the age of 65 is expected to increase by 73% in the United States [1], and the number of cardiovascular interventions (angioplasties, stents, and vascular grafts) performed in 2030 is expected to be 1.73 times the number performed in 2010. The long-term results of vascular interventions are compromised by intimal hyperplasia and continued thrombogenicity of the incompletely endothelialized surface. Until the endothelial cell (EC) surface continuity is restored, the angioplasty site remains thrombogenic relative to the adjacent normal artery. Additionally, the development of intimal hyperplasia is more severe if restoration of the endothelial monolayer is delayed [2, 3]. Many factors that inhibit EC migration in vitro or delay arterial healing in vivo have been identified. Oxidized LDL and lysophosphatidylcholine (lysoPC), the major lysophospholipid in oxidized LDL, block EC migration in vitro [4, 5], and a high cholesterol diet retards endothelial healing of arterial injuries in a mouse model [6]. Also, healing of carotid injuries is delayed in mice deficient in apolipoprotein A-I (apoA-I), the major protein component of HDL, and reconstitution of apoA-I allows normal healing [7]. These findings suggest the importance of apoA-I in EC migration.
ApoA-I has anti-oxidant and anti-inflammatory properties as well as cardiovascular protection and reverse cholesterol transport functions [8–11]. ApoA-I can inhibit LDL oxidation, remove lipid hydroperoxides, decrease monocyte chemotaxis, and protect EC from apoptosis [9, 12–15]; all properties that would aid in endothelial healing of arterial injuries. In fact, apoA-I Milano administration decreases intimal thickening and macrophage content after balloon injury of arteries in cholesterol-fed rabbits [16].
ApoA-I mimetics have been developed to replicate the anti-atherogenic functions of apoA-I. An 18 amino acid peptide, 4F (Ac-DWFKAFYDKVAEKFKEAF-NH3) [17], reproduces the helical and amphipathic portion of apoA-I which is key to its function [18]. Phenylalanine residues on the non-polar face increase the hydrophobicity and lipid binding ability [19]. D-4F and L-4F, composed of the D- and L-isomers of the amino acids, respectively, have comparable profiles of action [20], and function similarly to apoA-I. D-4F is stable when administered orally, but L-4F must be delivered parenterally [17]. ApoA-I mimetics can improve reverse cholesterol transport, increase levels of pre-beta HDL (the fraction most important in reverse cholesterol transport), decrease atherosclerotic lesion formation, prevent oxidation of LDL, decrease LDL-induced monocyte chemotactic activity, and increase the anti-inflammatory properties of HDL [12, 13, 17, 21–23].
This study was undertaken to determine if an apoA-I mimetic can promote EC migration. Using a razor scrape assay, the effect of L-4F on endothelial migration in vitro was assessed. The ability of D-4F to promote endothelial healing of a carotid injury in chow-fed mice and reverse the detrimental effect of a high cholesterol diet was also studied.
MATERIALS AND METHODS
Bovine aortic EC culture and migration study
Bovine aortic EC (BAEC) were isolated from adult bovine aortas, cultured, and used between passage 4 and 10, as previously described [24]. To assess early EC migration during a time period not influenced by cell proliferation, a razor scrape migration assay was used. In the appropriate study groups, cells were pretreated for 3 hours with medium, 0.43 μmol/L of L-4F [17], or 0.43 μmol/L of a scrambled peptide containing the same amino acids as in L-4F (Ac-DWFAKDYFKKAFVEEFAK-NH3) (GenScript, Piscataway, NJ). At the end of 3 hours, EC in one region were removed with a razor blade, as previously described [24]. LysoPC (1-palmitol-2-hydroxy-sn-glycerol-3-phosphocholine; 12.5 μmol/L, Avanti Polar Lipids, Alabaster, AL) was added to appropriate wells. After 24 hours, EC were fixed and an observer, blinded to experimental conditions, counted the number of migrating EC using NIH Image 1.63 software (http://rsb.info.nih.gov/ij/).
Measurement of reactive oxygen species (ROS)
Intracellular ROS were monitored by oxidation of dichloro-dihydro-fluorescein diacetate (DCF-DA) assay, as previously described [25]. EC were grown to 70% confluence and loaded with DCF-DA (10 μmol/L, Life Technologies, NY) for 20 min. EC were washed, incubated for 3 h with L-4F (0.43 μmol/L) or scrambled peptide (0.43 μmol/L), washed, then lysoPC was added for 15 min, as indicated. DCF fluorescence was detected at 480 nm with a Leica DM IRB inverted fluorescence microscope (Heidelberg, Germany) using an FITC filter. A minimum of 3 randomly selected locations were captured in each well for analysis.
Immunoblot analysis for NAD(P)H oxidase activation and externalization
NAD(P)H oxidase activation was assessed by evaluating p47phox phosphorylation. Confluent BAEC were incubated in serum-free DMEM containing 0.1% gelatin. Cells were incubated for 3 hours with either medium or L-4F (0.43 μmol/L). Following pretreatment, the cells were washed, and lysoPC (12.5 μmol/L) was added as appropriate. After a 15 minute incubation period, cells were lysed, proteins (50 μg/lane) resolved by SDS-PAGE and detected by immunoblot analysis using an antibody specific for phospho-p47phox (Ser345) (Assay Biotech, Sunnyvale, CA). The membranes were reprobed for actin (Chemicon, Temecula, CA) to confirm equal loading of the lanes.
Translocation of NAD(P)H oxidase subunits, p47phox and p67phox, to the cell membrane was evaluated. EC were incubated with L-4F (0.43 μmol/L) or the scrambled peptide (0.43 μmol/L) for 3 h, washed, then lysoPC (12.5 μmol/L) was added for 15 min as indicated. EC were lysed and cell membranes disrupted by passage through a 20- to 25-gauge needle. The cytosolic fraction was separated from the membrane fraction by centrifuged at 15,000 g for 20 min at 4°C. The supernatant containing the cytosolic fraction was removed for analysis. To disrupt the nuclear membranes, the pellet was resuspended in buffer (50 mM HEPES, 150 mM NaCl, 1 mM EDTA, 200 μM Na3VO4, 100 mM NaF, 1% Triton X-100, pH 7.4) containing protease inhibitors (Complete, Roche, Indianapolis, IN) and centrifuged at 15,000 g for 20 min at 4°C. The supernatant containing the nuclear proteins was discarded. The pellet containing the membrane fraction was resuspended for analysis. The presence of p47phox and p67phox in the cytosolic and membrane fractions was assessed by Western blot using anti-p47phox antibody (1:1000, Cell Signaling Technology) and anti-p67phox antibody (1:1000, Cell Signaling Technology, MA).
Mouse model
Six-week-old male C57Bl/6 mice were randomly divided into four study groups: 1) chow diet, 2) high cholesterol (HC) diet (Harlan Teklad, Madison, WI) containing 21% milk fat and 0.2% cholesterol by weight, 3) chow diet plus D-4F, and 4) HC diet plus D-4F. D-4F (21.7 μmol/L, GenScript) was administered in the drinking water starting two weeks prior to carotid injury and continued until the end of study. The Institutional Animal Care and Use Committee approved the study protocol. The animal protocols and care complied with the American Association of Laboratory Care Guidelines and the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, 1996).
At eight weeks of age, a perivascular electrical injury of the right common carotid artery was performed as previously described [6]. Electrocautery was applied to the artery using bipolar forceps with 4 mm wide tips. The injury was produced by applying 2 watts of power to the artery for 3 seconds.
Mouse carotid artery harvest and analysis for EC migration
At 120 hours after carotid artery injury, when the injury is approximately 50% healed in mice on a chow diet, the mice were anesthetized. Blood was drawn from the inferior vena cava and Evans Blue (5% in PBS, 100 μl) was injected intravenously to stain arteries not having an intact EC surface [6]. Arteries were perfusion-fixed, removed, pinned flat, imaged, and analyzed as previously described [6].
Plasma assays
Plasma levels of total cholesterol, HDL, LDL/VLDL, lysoPC, and thiobarbituric acid reactive substances (TBARS) were assessed at the conclusion of the study. Plasma from blood collected at the time of carotid removal, with added butylated hydroxytoluene (BHT), was stored under N2 at −80o C until analyzed for total cholesterol, HDL cholesterol, and LDL/VLDL cholesterol by a cholesterol oxidase method (Infinity Cholesterol Reagent; Thermo Fisher Scientific, Inc., Waltham, MA), as previously described [26]. Plasma lysoPC concentration was determined by an enzymatic method (Azwell LPC Assay Kit; Cosmo Bio USA Inc., Carlsbad, CA), and lipid peroxidation was measured as TBARS by a fluorometric assay, as previously described [6, 26].
Urine assays
Urine was collected during the first 24 hours after injury and for the 24 hours prior to carotid artery removal using a metabolic cage (Fisher Scientific, Pittsburgh, PA), and 8-isoprostanes measured as an additional marker of systemic oxidative stress, as previously described [27]. Urine 8-isoprostane levels were normalized to creatinine to correct for variation in urine concentration.
Tissue lipid analysis
The injured right and uninjured left common carotid arteries were isolated from surrounding tissue and immersed in a cold solution of 0.15 mol/L NaCl, 0.1 mmol/L BHT, and 1 mmol/L EDTA. Samples were stored under N2 at −80º C until analyzed. After combining carotid tissue from four mice to achieve a measurable level of cholesterol, tissue was minced, lipids extracted, and cholesterol was measured by HPLC, as previously described [26].
Immunostaining for oxidized LDL
Oxidized LDL in the carotid arteries, as a marker of oxidative stress, was assessed by immunohistochemistry as previously described [6]. OxLDL was identified by use of an antibody to hypochlorite-oxidized LDL (1:2000, Calbiochem, Gibbstown, NJ). Tissue stained for oxLDL was scored on a scale of 0 to 4 by three observers blinded to the treatment group, giving a qualitative measure of oxLDL content.
Statistical analysis
GraphPad Prism 5.0 (GraphPad Software, Inc., San Diego, CA) was utilized for statistical analysis. The results were represented as the mean±standard deviation (SD) or mean±standard error (SE) of the mean, as indicated. Student t-test and ANOVA followed by Tukey’s posthoc multiple comparison test were used for the analysis. P<0.05 was considered statistically significant.
RESULTS
LysoPC-induced inhibition of EC migration is attenuated by pretreatment with L-4F
The ability of an apoA-I mimetic peptide, L-4F, to alter EC migration in vitro was assessed in BAEC. Treatment with L-4F led to an insignificant increase in EC migration relative to control (n=3, P=NS, Fig. 1A & 1B)). Treatment with the scrambled peptide did not alter EC migration relative to control (n=3, P=NS, Fig. 1A & 1B). Next, the ability of L-4F to preserve migration of EC exposed to lysoPC was assessed. EC were pretreated with PBS, L-4F, or scrambled peptide for 3 hours, washed, then incubated with lysoPC. LysoPC inhibited EC migration by 72% (n=3, P<0.001; Fig. 1A & 1B). After pretreatment with L-4F, lysoPC only inhibited migration to 32% of control, a significant improvement over cells incubated with lysoPC without L-4F pretreatment (n=3, P<0.001 compared to lysoPC, Fig. 1A). Pretreatment with scrambled peptide did not change the inhibitory effect of lysoPC on EC migration. These results suggested that 4F has a specific action in countering the anti-migratory effect of lysoPC.
Fig. 1.
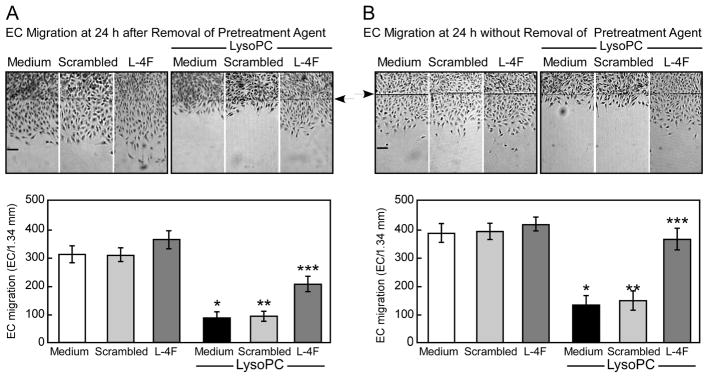
The anti-migratory effect of lysophosphatidylcholine (lysoPC) is attenuated in endothelial cells (EC) pretreated with the apoplipoprotein A-I mimetic peptide L-4F. Confluent bovine aortic EC (BAEC) were incubated with medium, L-4F (0.43 μmol/L), or scrambled peptide (0.43 μmol/L) for 3 hours prior to initiation of the migration assay. Migration was initiated, and lysoPC (12.5 μmol/L) was added as indicated during the migration assay. Migration was quantified after 24 hours. Arrow indicates starting line of migration. Original magnification, ×40. Bar, 100 μm. The bottom panel represents migration results by mean ± SD (n=3, * P<0.001 compared with medium control, ** P<0.001 compared with medium control and scrambled peptide control, *** P<0.01 compared with medium+lysoPC and with L-4F control). (A) EC were washed 3 times with PBS prior to the addition of lysoPC. (B) Pretreatment agents were not removed during incubation with lysoPC.
L-4F binds lipids, especially oxidized lipids [28], but in the studies above, L-4F was removed and cells washed prior to the addition of lysoPC. No L-4F was present in the medium to bind the lysoPC. To assess whether L-4F could further improve EC migration by directly binding lysoPC, the experiment was repeated but without removal of L-4F or washing of EC. Under these conditions, L-4F improved EC migration in the presence of lysoPC even further, with only a 4% decrease in migration from the medium alone (n=3, P<0.001 compared to lysoPC, Fig. 1B). Pretreatment with the scrambled peptide prior to incubation with lysoPC did not improve EC migration compared with cells incubated with lysoPC alone. The results suggested that L-4F partially preserved EC migration by a direct effect on EC in the presence of lysoPC, and also binds lysoPC to further preserve migration.
L-4F reduces intracellular ROS
Intracellular ROS was assessed by monitoring oxidation of DCF. LysoPC increased ROS in EC (Supplemental Fig. 1A). When EC were pretreated with L-4F, but not the scrambled peptide, lysoPC did not increase ROS in EC (Supplemental Fig. 1A).
L-4F does not inhibit lysoPC induced activation and externalization of NADP(H) oxidase
To explore the ability of L-4F to block lysoPC-induced NAD(P)H oxidase activation, p47phox phosphorylation was assessed. Incubation of BAEC with lysoPC resulted in phosphorylation of p47phox (n=3, Supplemental Fig. 1B). Pretreatment with L-4F did not alter the lysoPC-induced p47phox phosphorylation. (n=3, Supplemental Fig. 1B).
Externalization of NADH(P)H oxidase subunits p47phox and p67phox was assessed. The p47phox and p67phox subunits translocated from the cytoplasm to the membrane following BAEC incubation with lysoPC (n=3, Supplemental Fig. 1C). Pretreatment with L-4F did not alter the lysoPC-induced translocation of p47phox and p67phox. L-4F did not block the lysoPC-induced inhibition of EC migration by preventing the activation and externalization of NAD(P)H oxidase subunits.
Mouse study groups
Ninety-seven 8-week old C57Bl/6 mice were used to assess the effect of an apoA-I mimetic on endothelial healing of an arterial injury. Two weeks prior to carotid injury, the mice were assigned to a diet and treatment group and maintained on the assigned diet and medication until 120 hours after the injury. Of the mice that underwent the carotid injury, no mice died during the procedure or thrombosed the injured artery. Table 1 details the demographics for the mice that underwent carotid injury. Mice fed the chow and HC diets were not significantly different with respect to age, but the hypercholesterolemic mice weighed more at the time of injury as compared with the mice on a chow diet. Mice treated with D-4F were not significantly different with respect to age or weight at the time of injury as compared with mice on the corresponding diet not receiving D-4F.
Table 1.
Mouse demographics and plasma cholesterol data, presented as mean ± standard error
| Group | N | Age (days) | Weight (g) | Total Cholesterol (mmol/L) | HDL (mmol/L) | LDL/VLDL (mmol/L) |
|---|---|---|---|---|---|---|
| Chow | 24 | 56±0.4 | 23.7±0.3a | 2.54±0.03b | 1.61±0.04b | 0.67±0.03b |
| Chow+D-4F | 24 | 57±0.3 | 24.3±0.3 | 2.66±0.03b | 1.60±0.02b | 0.67±0.03b |
| High Cholesterol | 24 | 57±0.3 | 25.8±0.4 | 3.89±0.07 | 2.15±0.05 | 1.30±0.05 |
| HC+D-4F | 25 | 58±0.4 | 25.7±0.6 | 3.84±0.04 | 2.27±0.04 | 1.23±0.04 |
HC=High cholesterol
Significant differences (P≤0.05) are designated as:
Between Chow and High Cholesterol
Between all chow groups and all high cholesterol groups
Plasma cholesterol
Plasma cholesterol levels were determined from blood samples obtained 120 hours after carotid injury. Mice receiving a HC diet had significantly higher plasma total cholesterol levels than chow-fed mice (P<0.001, Table 1). Similarly, a HC diet was associated with a significant increase in plasma HDL and LDL/VLDL levels (P<0.001, Table 1). Treatment with D-4F did not significantly alter plasma total cholesterol, HDL, or LDL/VLDL levels compared with mice on the corresponding diet not receiving D-4F treatment (Table 1).
Reendothelialization after arterial injury
After the electrical injury, chow-fed mice reendothelialized 48.2±1.9% (Fig. 2) of the injured area by 120 hours. Reendothelialization was significantly decreased in the high cholesterol group with coverage of only 27.8±1.4% at 120 hours (P<0.0001, Fig. 2). Treatment with D-4F improved endothelial healing to 56.6±4.3% in chow-fed mice (Fig. 2), but the results did not reach statistical significance (P=0.1). Treatment with D-4F significantly increased reendothelialization from 27.8±1.4% in HC-fed mice to 43.4±3.4% in HC-fed mice treated with D-4F (P=0.002, Fig. 2). Thus, D-4F significantly improved endothelial healing in the hypercholesterolemic mice.
Fig. 2.
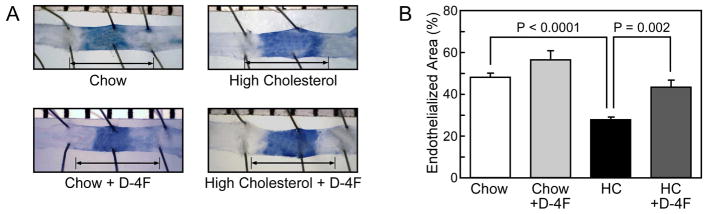
Endothelial cell migration is preserved in hypercholesterolemic C57Bl/6 mice treated with D-4F, an apoplipoprotein A-I mimetic peptide. (A) Representative carotid arteries 120 hours after injury for mice in the chow, chow plus D-4F, high cholesterol (HC), and the HC plus D-4F groups are shown. The area stained by Evans Blue is the area that remained deendothelialized. The arrow identifies the length of the original injury. (B) Reendothelialization reported as a percentage of the original injury. Results are expressed as the mean ± standard error for mice in each group: chow (n=6), chow plus D-4F (n=6), HC (n=6), and HC plus D-4F (n=6).
Tissue cholesterol and oxLDL
Tissue cholesterol levels were higher in the injured right carotid artery than the uninjured left carotid artery. They either approached or reached statistical significance when comparing the injured artery with the corresponding uninjured artery within the same treatment group (Table 2). D-4F treatment did not significantly change the tissue cholesterol levels compared to mice in the same dietary group without D-4F treatment (Table 2). In fact, neither uninjured arterial cholesterol levels nor injured carotid tissue cholesterol levels were significantly different across the treatment groups.
Table 2.
Mouse tissue cholesterol and oxidized LDL, presented as mean ± standard error
| Group | Tissue Cholesterol (μg/mg) | Tissue oxLDL | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| N | RCCA | LCCA | N | RCCA | LCCA | |
| Chow | 3 | 0.22±0.02 | 0.15±0.01a | 6 | 1.76±0.3b | 1.39±0.9 |
| Chow+D-4F | 3 | 0.25±0.03 | 0.17±0.02a | 5 | 2.87±0.3 | 2.20±0.2 |
| High Cholesterol | 3 | 0.23±0.03 | 0.17±0.02a | 6 | 3.15±0.2 | 2.34±0.1a |
| HC+D-4F | 3 | 0.37±0.08 | 0.21±0.03 | 7 | 2.97±0.2 | 2.32±0.1a |
RCCA=Right common carotid artery
LCCA=Left common carotid artery
HC=High cholesterol
Between injured RCCA and LCCA within same study group (P<0.05)
Between injured RCCA in the Chow and Chow+D-4F study groups (P<0.05)
Tissue oxLDL scores were a marker of local oxidative stress. OxLDL score was not significantly different between the injured and uninjured carotid artery in chow-fed mice or chow-fed mice treated with D-4F (Table 2). Mice on the HC and the HC+D-4F diet had a significantly higher tissue oxLDL score in the injured artery compared with the uninjured artery, but D-4F did not lower oxLDL scores compared with mice not treated with D-4F (Table 2).
Markers of systemic oxidative stress
Plasma TBARS and lysoPC levels were determined from blood samples obtained 120 hours after injury as markers of systemic oxidative stress. Mice on the HC diet had significantly higher TBARS levels compared with chow-fed mice (P<0.001, Table 3). Treatment with D-4F did not affect the TBARS levels in normocholesterolemic mice but significantly reduced TBARS levels in hypercholesterolemic mice (P<0.001, Table 3). Plasma lysoPC levels were also significantly higher in hypercholesterolemic mice compared to normocholesterolemic mice (P<0.001, Table 3). D-4F treatment significantly reduced plasma lysoPC levels in chow-fed (P<0.001, Table 3) and in HC mice (P<0.001, Table 3).
Table 3.
Mouse plasma and urine markers of oxidative stress, presented as mean ± standard error
| Group | LysoPC (μmol/L) (n=24) | TBARS (μmol MDA/L) (n=12) | 8-Iso/Cr - Day 1 (pg/mg) (n=5) | 8-Iso/Cr - Day 5 (pg/mg) (n=5) |
|---|---|---|---|---|
| Chow | 599.2±13.9a,b | 0.367±0.01a | 1359.2±59.4a | 946.0±30.5a,b |
| Chow+D-4F | 488.9±12.8 | 0.369±0.01 | 1126.9±54.7 | 514.7±51.9 |
| High Cholesterol | 827.0±9.9c | 0.623±0.01c | 2546.0±190.3c | 1151.3±57.6c |
| HC+D-4F | 722.9±7.7 | 0.477±0.01 | 1267.3±182.7 | 593.8±42.0 |
HC=High cholesterol
LysoPC=Lysophosphatidylcholine
TBARS=Thiobarbituric acid reactive substances
8-Iso=8-Isoprostane
Cr=Creatinine
Significant differences (P≤0.05) are designated as:
Between Chow and High Cholesterol
Between Chow and Chow+D-4F
Between High Cholesterol and HC+D-4F
To assess the immediate and longer impact of the injury and diet on systemic oxidative stress, urine was collected for 8-isoprostane determination for the first 24 hours after injury (Day 1) and for the 24 hours prior to sacrifice (Day 5). On Day 1 and 5, hypercholesterolemic mice had significantly higher levels of 8-isoprostane compared to chow-fed mice (P<0.001, Table 3). Treatment with D-4F did not affect the 8-isoprostane levels of chow-fed mice on Day 1, but D-4F was associated with a significant decrease in 8-isoprostane levels in hypercholesterolemic mice on Day 1 and in both dietary groups on Day 5 (P<0.001, Table 3). Overall, administration of D-4F to mice on a HC diet resulted in a decrease in markers of systemic oxidative stress. Considering plasma TBARS and lysoPC levels, and urinary 8-isoprostane levels as measures of systemic oxidative stress, mice on a HC diet had increased oxidative stress compared with those on a chow diet. D-4F reduced the level of oxidative stress. Interestingly, for all study groups, there was a significant inverse correlation between endothelial healing and plasma TBARS (r=−0.695, P<0.001), plasma lysoPC (r=−0.597, P=0.002), urine 8-isoprostane on Day 1 (r=−0.582, P<0.01), and urine 8-isoprostane levels on Day 5 (r=−0.590, P<0.01).
DISCUSSION
Our studies demonstrate the ability of 4F to ameliorate the inhibitory effects of lysoPC on EC migration in vitro and improve endothelial healing of arterial injuries in hypercholesterolemic mice. L-4F does not significantly stimulate EC migration under control conditions, but the inhibitory effect of lysoPC is significantly decreased when EC are pretreated with L-4F. This effect is seen when the L-4F is removed prior to the addition of lysoPC suggesting that L-4F has an effect directly on EC, perhaps by altering membrane mechanical properties that are important in migration [29, 30]. When L-4F is also present during incubation with lysoPC, inhibition of migration is completely prevented suggesting that L-4F can also bind lysoPC and prevent its inhibitory effect.
One of the mechanisms by which oxidized LDL and lysoPC inhibit EC migration is through an increase in EC production of superoxide by NAD(P)H oxidase [25]. L-4F is reported to decrease NAD(P)H oxidase activity and p47phox subunits in aortic tissue of rats with chronic kidney disease [31]. In our studies, L-4F decreased ROS, but L-4F did not acutely alter NAD(P)H oxidase activity as evidenced by no change in p47phox phosphorylation and p47phox and p67phox externalization. Thus, the efficacy of L-4F in preserving EC migration in vitro does not appear to be related to acute inhibition of NAD(P)H oxidase activity, but L-4F might be acting as a scavenger of ROS or increasing the activity of superoxide dismutase[32].
This study shows that a high cholesterol diet impairs endothelial healing of an arterial injury in mice, and that administration of an apoA-I mimetic improves healing. In mice on a chow diet, the improved healing seen with D-4F administration approaches but does not reach statistical significance (P=0.1). In hypercholesterolemic mice, D-4F blocks the inhibition and increases endothelial healing to a similar level as seen in chow-fed mice. The mechanism by which D-4F augments endothelial healing is unclear, but does not appear to be due to changes in the cholesterol or HDL levels because no significant alteration in these plasma lipid levels was found in our studies. Although one study suggests that D-4F administration increases HDL levels [33], others report no change in plasma cholesterol or HDL levels following D-4F administration [17, 18]. D-4F can increase the pre-beta fraction of HDL [22, 23], the fraction active in reverse cholesterol transport [34], but this fraction has not been reported to speed endothelial healing. HDL is postulated to stimulate EC migration through a receptor-mediated pathway [35], but 4F does not significantly increase migration under control conditions in vitro, arguing against a ligand-receptor mechanism. Instead, the beneficial effects of L-4F on EC migration in vitro are seen only in reducing the adverse effects of lysoPC. Thus, the most plausible explanation for the benefit in vivo is that D-4F ameliorates an inhibitory effect that accompanies hypercholesterolemia and arterial injury, both of which cause an oxidative stress.
Increased levels of lipid oxidation products accompany hypercholesterolemia. ApoA-I mimetics decrease the oxidation of LDL [12, 13], scavenge lipid hydroperoxides from LDL [36], and increase paraoxonase activity [22]. These properties can minimize the oxidative stress and the increased lysoPC levels seen with hypercholesterolemia. Furthermore, 4F has been shown to bind oxidized lipids with an affinity that is 4–6 orders of magnitude greater than that of apoA-I [28], and our in vitro studies suggest that 4F can bind lysoPC. Our study shows that D-4F decreases plasma lysoPC levels, either through decreased production or binding of lysoPC, reducing a potent anti-migratory agent. In fact, the plasma lysoPC level inversely correlates with endothelial healing. In addition, D-4F reduces measures of oxidative stress, including plasma TBARS and urinary 8-isoprostane levels, which inversely correlate with endothelial healing. Our results parallel those of previous work demonstrating that antioxidant administration decreases measures of oxidative stress and improves endothelial healing of arterial injuries in hypercholesterolemic mice [6].
ApoA-I mimetics have anti-inflammatory properties [37]. In our study, D-4F administration does not change the cellular infiltration or the percentage of macrophages in the injured artery as assessed by immunohistochemistry in our studies (data not shown). Similarly, D-4F administration does not produce a significant difference in tissue staining for oxLDL in the injured carotid arteries. This could be due to subtle differences between groups that are not detected by the relatively insensitive assessments used, or the magnitude of the injury may overwhelm the anti-inflammatory action of D-4F.
CONCLUSION
Hypercholesterolemia is accompanied by increased oxidative stress and lipid oxidation products that contribute to delayed reendothelialization of arterial injuries [6]. The current studies show that D-4F, an apoA-I mimetic, improves endothelial healing of arterial injuries, and suggest that it works through minimizing the oxidative stress and rise in lipid oxidation products induced by hypercholesterolemia. Understanding the mechanisms of delayed healing of arterial injuries allows the development of mechanism-based therapies, such as apoA-I mimetics, to promote endothelial healing. Administration of apoA-I mimetics has the potential to speed the healing of arterial injuries at angioplasty sites to minimize thrombogenicity and restenosis at these sites. Identifying treatments that can improve endothelial cell migration is one step toward improving the long-term patency of vascular interventions. Improved patency is clinically relevant, especially in patients with multiple comorbidities. Prolonged patency will decrease the need for re-intervention, as well as the potential morbidity and mortality associated with re-intervention. Improved long-term outcomes following intervention will improve patients’ quality of life.
Supplementary Material
(A) L-4F, but not scrambled peptide, decreases lysoPC-induced ROS production in EC. EC were incubated with DCF-DA for 20 min, washed twice, and incubated with L-4F (0.43 μmol/L) or scrambled peptide (0.43 μmol/L) for 3 hours. LysoPC (12.5 μmol/L) was added and after 15 min, fluorescence was measured. (B) L-4F does not inhibit lysoPC induced NAD(P)H oxidase activation. Confluent EC were incubated with medium or L-4F for 3 hours, then washed and incubated with lysoPC (12.5 μmol/L) for 15 min. Immunoblot analysis for phosphorylated p47phox was performed (top panel). Blots were reprobed for actin to verify equal loading of the lanes (bottom panel). Shown is a representative blot of three separate experiments. (C) L-4F does not inhibit lysoPC induced NAD(P)H oxidase subunit externalization. EC were incubated with L-4F for 3 hours and washed before addition of lysoPC (12.5 μmol/L) for 15 minutes. The cells were lysed and the cytosolic fraction was separated from the membrane fraction. Western blots were performed for p67phox (top panel) and p47phox (bottom panel).
HIGHLIGHTS.
Hypercholesterolemia leads to increased oxidative stress and lipid oxidation
Increased oxidative stress impairs arterial healing after injury
L-4F improves endothelial migration in vitro in presence of oxidized lipids
D4-F improves endothelial healing after carotid injury in hypercholesterolemic mice
ApoA-I mimetics may reduce thrombogenicity and restenosis of angioplasty sites
Acknowledgments
This project was supported by Grant Numbers RO1 HL064357, and F32 HL090205 from the National Heart, Lung, and Blood Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health. The National Heart, Lung, and Blood institute had no role in the study design, data collection, analysis and interpretation of the data, writing of the manuscript, or in the decision to submit this article for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stanley JC, Barnes RW, Ernst CB, Hertzer NR, Mannick JA, Moore WS. Vascular surgery in the United States: Workforce issues - Report of the Society for Vascular Surgery and the International Society for Cardiovascular Surgery, North American Chapter, Committee on Workforce Issues. J Vasc Surg. 1996;23:172–181. doi: 10.1016/s0741-5214(05)80050-3. [DOI] [PubMed] [Google Scholar]
- 2.Haudenschild CC, Schwartz SM. Endothelial regeneration. II. Restitution of endothelial continuity. Lab Invest. 1979;41:407–418. [PubMed] [Google Scholar]
- 3.Losordo DW, Isner JM, Diaz-Sandoval LJ. Endothelial recovery: the next target in restenosis prevention. Circulation. 2003;107:2635–2637. doi: 10.1161/01.CIR.0000071083.31270.C3. [DOI] [PubMed] [Google Scholar]
- 4.Murugesan G, Chisolm GM, Fox PL. Oxidized low density lipoprotein inhibits the migration of aortic endothelial cells in vitro. J Cell Biol. 1993;120:1011–1019. doi: 10.1083/jcb.120.4.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murugesan G, Fox PL. Role of lysophosphatidylcholine in the inhibition of endothelial cell motility by oxidized low density lipoprotein. J Clin Invest. 1996;97:2736–2744. doi: 10.1172/JCI118728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenbaum MA, Miyazaki K, Graham LM. Hypercholesterolemia and oxidative stress inhibit endothelial cell healing after arterial injury. J Vasc Surg. 2012;55:489–496. doi: 10.1016/j.jvs.2011.07.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seetharam D, Mineo C, Gormley AK, Gibson LL, Vongpatanasin W, Chambliss KL, Hahner LD, Cummings ML, Kitchens RL, Marcel YL, Rader DJ, Shaul PW. High-density lipoprotein promotes endothelial cell migration and reendothelialization via scavenger receptor-B type I. Circ Res. 2006;98:63–72. doi: 10.1161/01.RES.0000199272.59432.5b. [DOI] [PubMed] [Google Scholar]
- 8.Garner B, Waldeck AR, Witting PK, Rye KA, Stocker R. Oxidation of high density lipoproteins. II. Evidence for direct reduction of lipid hydroperoxides by methionine residues of apolipoproteins AI and AII. J Biol Chem. 1998;273:6088–6095. doi: 10.1074/jbc.273.11.6088. [DOI] [PubMed] [Google Scholar]
- 9.Barter PJ, Nicholls S, Rye KA, Anantharamaiah GM, Navab M, Fogelman AM. Antiinflammatory properties of HDL. Circ Res. 2004;95:764–772. doi: 10.1161/01.RES.0000146094.59640.13. [DOI] [PubMed] [Google Scholar]
- 10.Rubin EM, Krauss RM, Spangler EA, Verstuyft JG, Clift SM. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein AI. Nature. 1991;353:265–267. doi: 10.1038/353265a0. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, Zanotti I, Reilly MP, Glick JM, Rothblat GH, Rader DJ. Overexpression of apolipoprotein A-I promotes reverse transport of cholesterol from macrophages to feces in vivo. Circulation. 2003;108:661–663. doi: 10.1161/01.CIR.0000086981.09834.E0. [DOI] [PubMed] [Google Scholar]
- 12.Navab M, Hama SY, Cooke CJ, Anantharamaiah GM, Chaddha M, Jin L, Subbanagounder G, Faull KF, Reddy ST, Miller NE, Fogelman AM. Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: step 1. J Lipid Res. 2000;41:1481–1494. [PubMed] [Google Scholar]
- 13.Navab M, Hama SY, Anantharamaiah GM, Hassan K, Hough GP, Watson AD, Reddy ST, Sevanian A, Fonarow GC, Fogelman AM. Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: steps 2 and 3. J Lipid Res. 2000;41:1495–1508. [PubMed] [Google Scholar]
- 14.de Souza JA, Vindis C, Negre-Salvayre A, Rye KA, Couturier M, Therond P, Chantepie S, Salvayre R, Chapman MJ, Kontush A. Small, dense HDL 3 particles attenuate apoptosis in endothelial cells: pivotal role of apolipoprotein A-I. J Cell Mol Med. 2010;14:608–620. doi: 10.1111/j.1582-4934.2009.00713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suc I, Escargueil-Blanc I, Troly M, Salvayre R, Nègre-Salvayre A. HDL and ApoA prevent cell death of endothelial cells induced by oxidized LDL. Arterioscler Thromb Vasc Biol. 1997;17:2158–2166. doi: 10.1161/01.atv.17.10.2158. [DOI] [PubMed] [Google Scholar]
- 16.Ameli S, Hultgardh-Nilsson A, Cercek B, Shah PK, Forrester JS, Ageland H, Nilsson J. Recombinant apolipoprotein A-I Milano reduces intimal thickening after balloon injury in hypercholesterolemic rabbits. Circulation. 1994;90:1935–1941. doi: 10.1161/01.cir.90.4.1935. [DOI] [PubMed] [Google Scholar]
- 17.Navab M, Anantharamaiah GM, Hama S, Garber DW, Chaddha M, Hough G, Lallone R, Fogelman AM. Oral administration of an apo A-I mimetic peptide synthesized from D-amino acids dramatically reduces atherosclerosis in mice independent of plasma cholesterol. Circulation. 2002;105:290–292. doi: 10.1161/hc0302.103711. [DOI] [PubMed] [Google Scholar]
- 18.Navab M, Anantharamaiah GM, Reddy ST, Hama S, Hough G, Grijalva VR, Yu N, Ansell BJ, Datta G, Garber DW, Fogelman AM. Apolipoprotein A-I mimetic peptides. Arterioscler Thromb Vasc Biol. 2005;25:1325–1331. doi: 10.1161/01.ATV.0000165694.39518.95. [DOI] [PubMed] [Google Scholar]
- 19.Datta G, Chaddha M, Hama S, Navab M, Fogelman AM, Garber DW, Mishra VK, Epand RM, Epand RF, Lund-Katz S, Phillips MC, Segrest JP, Anantharamaiah GM. Effects of increasing hydrophobicity on the physical-chemical and biological properties of a class A amphipathic helical peptide. J Lipid Res. 2001;42:1096–1104. [PubMed] [Google Scholar]
- 20.Van Lenten BJ, Wagner AC, Navab M, Anantharamaiah GM, Hama S, Reddy ST, Fogelman AM. Lipoprotein inflammatory properties and serum amyloid A levels but not cholesterol levels predict lesion area in cholesterol-fed rabbits. J Lipid Res. 2007;48:2344–2353. doi: 10.1194/jlr.M700138-JLR200. [DOI] [PubMed] [Google Scholar]
- 21.Navab M, Anantharamaiah GM, Fogelman AM. The role of high-density lipoprotein in inflammation. Trends Cardiovasc Med. 2005;15:158–161. doi: 10.1016/j.tcm.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 22.Navab M, Anantharamaiah GM, Reddy ST, Hama S, Hough G, Grijalva VR, Wagner AC, Frank JS, Datta G, Garber D, Fogelman AM. Oral D-4F causes formation of pre-β high-density lipoprotein and improves high-density lipoprotein-mediated cholesterol efflux and reverse cholesterol transport from mcacrophages in apolipoprotein E-null mice. Circulation. 2004;109:3215–3220. doi: 10.1161/01.CIR.0000134275.90823.87. [DOI] [PubMed] [Google Scholar]
- 23.Troutt JS, Alborn WE, Mosior MK, Dai J, Murphy AT, Beyer TP, Zhang Y, Cao G, Konrad RJ. An apolipoprotein A-I mimetic dose-dependently increases the formation of prebeta1 HDL in human plasma. J Lipid Res. 2008;49:581–587. doi: 10.1194/jlr.M700385-JLR200. [DOI] [PubMed] [Google Scholar]
- 24.Chaudhuri P, Colles SM, Damron DS, Graham LM. Lysophosphatidylcholine inhibits endothelial cell migration by increasing intracellular calcium and activating calpain. Arterioscler Thromb Vasc Biol. 2003;23:218–223. doi: 10.1161/01.atv.0000052673.77316.01. [DOI] [PubMed] [Google Scholar]
- 25.van Aalst JA, Zhang DM, Miyazaki K, Colles SM, Fox PL, Graham LM. Role of reactive oxygen species in inhibition of endothelial cell migration by oxidized low-density lipoprotein. J Vasc Surg. 2004;40:1208–1215. doi: 10.1016/j.jvs.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 26.Rosenbaum MA, Miyazaki K, Colles SM, Graham LM. Antioxidant therapy reverses impaired graft healing in hypercholesterolemic rabbits. J Vasc Surg. 2010;51:184–193. doi: 10.1016/j.jvs.2009.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenbaum MA, Chaudhuri P, Graham LM. Hypercholesterolemia inhibits reendothelialization of arterial injuries by TRPC channel activation. J Vasc Sur. 2014 doi: 10.1016/j.jvs.2014.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Lenten BJ, Wagner AC, Jung CL, Ruchala P, Waring AJ, Lehrer RI, Watson AD, Hama S, Navab M, Anantharamaiah GM, Fogelman AM. Anti-inflammatory apoA-I-mimetic peptides bind oxidized lipids with much higher affinity than human apoA-I. J Lipid Res. 2008;49:2302–2311. doi: 10.1194/jlr.M800075-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghosh PK, Vasanji A, Murugesan G, Eppell SJ, Graham LM, Fox PL. Membrane microviscosity regulates endothelial cell motility. Nature Cell Biology. 2002;4:894–900. doi: 10.1038/ncb873. [DOI] [PubMed] [Google Scholar]
- 30.van Aalst JA, Burmeister W, Fox PL, Graham LM. α-Tocopherol preserves endothelial cell migration in the presence of cell-oxidized LDL by inhibiting changes in cell membrane fluidity. J Vasc Surg. 2004;39:229–237. doi: 10.1016/s0741-5214(03)01038-3. [DOI] [PubMed] [Google Scholar]
- 31.Vaziri ND, Bai Y, Yuan J, Said HL, Sigala W, Ni Z. ApoA-1 mimetic peptide reverses uremia-induced upregulation of pro-atherogenic pathways in the aorta. Am J Nephrol. 2010;32:201–211. doi: 10.1159/000316479. [DOI] [PubMed] [Google Scholar]
- 32.Liu J, Yao S, Wang S, Jiao P, Song G, Yu Y, Zhu P, Qin S. D-4F, an apolipoprotein A-I mimetic peptide, protects human umbilical vein endothelial cells from oxidized low-density lipoprotein-induced injury by preventing the downregulation of pigment epithelium-derived factor expression. J Cardiovasc Pharmacol. 2014;63:553–561. doi: 10.1097/FJC.0000000000000080. [DOI] [PubMed] [Google Scholar]
- 33.Chen X, Burton C, Song X, McNamara L, Langella A, Cianetti S, Chang CH, Wang J. An apoA-I mimetic peptide increases LCAT activity in mice through increasing HDL concentration. Int J Biol Sci. 2009;5:489–499. doi: 10.7150/ijbs.5.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.von Eckardstein A, Huang Y, Assmann G. Physiological role and clinical relevance of high-density lipoprotein subclasses. Curr Opin Lipidol. 1994;5:404–416. doi: 10.1097/00041433-199412000-00003. [DOI] [PubMed] [Google Scholar]
- 35.Kimura T, Sato K, Malchinkhuu E, Tomura H, Tamama K, Kuwabara A, Murakami M, Okajima F. High-density lipoprotein stimulates endothelial cell migration and survival through sphingosine 1-phosphate and its receptors. Arterioscler Thromb Vasc Biol. 2003;23:1283–1288. doi: 10.1161/01.ATV.0000079011.67194.5A. [DOI] [PubMed] [Google Scholar]
- 36.Datta G, Epand RF, Epand RM, Chaddha M, Kirksey MA, Garber DW, Lund-Katz S, Phillips MC, Hama S, Navab M, Fogelman AM, Palgunachari MN, Segrest JP, Anantharamaiah GM. Aromatic residue position on the nonpolar face of class a amphipathic helical peptides determines biological activity. J Biol Chem. 2004;279:26509–26517. doi: 10.1074/jbc.M314276200. [DOI] [PubMed] [Google Scholar]
- 37.Anantharamaiah GM, Mishra VK, Garber DW, Datta G, Handattu SP, Palgunachari MN, Chaddha M, Navab M, Reddy ST, Segrest JP, Fogelman AM. Structural requirements for antioxidative and anti-inflammatory properties of apolipoprotein A-I mimetic peptides. J Lipid Res. 2007;48:1915–1923. doi: 10.1194/jlr.R700010-JLR200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) L-4F, but not scrambled peptide, decreases lysoPC-induced ROS production in EC. EC were incubated with DCF-DA for 20 min, washed twice, and incubated with L-4F (0.43 μmol/L) or scrambled peptide (0.43 μmol/L) for 3 hours. LysoPC (12.5 μmol/L) was added and after 15 min, fluorescence was measured. (B) L-4F does not inhibit lysoPC induced NAD(P)H oxidase activation. Confluent EC were incubated with medium or L-4F for 3 hours, then washed and incubated with lysoPC (12.5 μmol/L) for 15 min. Immunoblot analysis for phosphorylated p47phox was performed (top panel). Blots were reprobed for actin to verify equal loading of the lanes (bottom panel). Shown is a representative blot of three separate experiments. (C) L-4F does not inhibit lysoPC induced NAD(P)H oxidase subunit externalization. EC were incubated with L-4F for 3 hours and washed before addition of lysoPC (12.5 μmol/L) for 15 minutes. The cells were lysed and the cytosolic fraction was separated from the membrane fraction. Western blots were performed for p67phox (top panel) and p47phox (bottom panel).