

Abstract
We have previously found that adhesion fibroblasts exhibit lower apoptosis and higher protein nitration as compared with normal peritoneal fibroblasts. In this study, we sought to determine whether the decreased apoptosis observed in adhesion fibroblasts is caused by lower caspase-3 activity due to an increase in caspase-3 S-nitrosylation. For this study, we have utilized primary cultures of fibroblasts obtained from normal peritoneum and adhesion tissues of the same patient(s). Cells were treated with increasing concentrations of peroxynitrite and cell lysates were immunoprecipitated with anti-caspase-3 polyclonal antibody. The biotinylated proteins were detected using a nitrosylation detection kit. Caspase-3 activity and apoptosis were measured by colorimetric and TUNEL assays, respectively. Our results showed that caspase-3 S-nitrosylation is significantly higher in adhesion fibroblasts as compared with normal peritoneal fibroblasts. This increase in S-nitrosylation resulted in a 30% decrease in caspase-3 activity in adhesion fibroblasts. Peroxynitrite treatment resulted in a dose response increase in caspase-3 S-nitrosylation, leading to a decrease in caspase-3 activity and apoptosis in normal peritoneal fibroblasts. We conclude that S-nitrosylation of caspase-3 is the reason for its decreased activity and subsequent decrease in apoptosis of adhesion fibroblasts. The mechanism by which caspase-3 S-nitrosylation occurs is not fully understood. However, the role of hypoxia in the formation of peroxynitrite via superoxide production may suggest a possible mechanism.
Postoperative adhesions result from the normal peritoneal wound healing response and develop in the first 3–5 days after injury.1 Adhesions are a major cause of infertility, abdominal pelvic pain, and small bowel obstruction. Subsequent surgeries are also likely to be prolonged and potentially incur greater risks of intraoperative/postoperative complications.2 Despite their overbearing influence on surgical care, the mechanisms by which injury to the peritoneum triggers the inflammatory response leading to postoperative adhesion development remain poorly understood.
Many studies have shown that inflammatory lesions are characterized by hypoxia.3,4 Previous experimental studies further indicated that hypoxia can modulate apoptosis in different types of cell lines including fibroblasts.5,6 Fibroblasts established from adhesion tissues are more resistant to hypoxia-induced apoptosis than fibroblasts established from normal peritoneum. In fact, the relative resistance to apoptosis by activated adhesion fibroblasts under hypoxic conditions has been proposed as an important cause of development of adhesions following abdominal surgery.6,7 Apoptosis is a tightly regulated molecular process that removes excess or unwanted cells from organisms. There are at least two signaling pathways known to trigger apoptosis. The first is mediated by interactions of membrane receptors and ligands, such as Fas ligand or tumor necrosis factor-α (TNF-α). The second pathway is triggered by exogenous stimuli such as inflammation, hypoxia, radiation, and chemotherapeutic drugs for which the death signal is transmitted through the mitochondria. Dysregulated apoptosis may be involved in the pathogenesis of diseases such as cancer, neurodegeneration, autoimmunity, and postoperative adhesions.7,8 The apoptotic program is executed by the caspase family of cysteine proteases that are expressed as inactive zymogens in cells and cleaved to form active tetrameric enzymes. Initiator caspases (e.g., caspases-8, -9, and -10) activate downstream effector caspases (e.g., caspases-3, -6, and -7), which in turn cleave specific cellular targets, resulting in cell death.9 In cells, caspase-3 has been shown to play an important role as a downstream member of the protease cascade, where various cell death pathways converge into the same effector pathway.10 Upon activation of the protease cascade, the caspase-3 proenzyme is proteolytically cleaved into the p17 and p12 subunits, which then heterodimerize to form the active enzyme.11 Considering that caspases are key mediators of apoptosis, identifying any regulatory modifications of these proteases is necessary to elucidate mechanisms of cellular balancing between survival and death. Reactive radicals, produced after oxygen (O2) supply is restored, have been implicated in a number of signal transduction pathways. Among the important radicals produced during hypoxia is the bioregulatory molecule nitric oxide (NO).12 NO plays an important role in cellular regulatory mechanisms, such as vasodilation, inhibition of platelet aggregation, and modification of proteins.13–15 NO exists in different redox states: NO − , NO+, and NO−.16 Furthermore, superoxide (O2• −) reacts with NO to produce peroxynitrite (ONOO−).17 It is well known that high concentrations of NO are proapoptotic and cytotoxic for different cells.18,19 In contrast, low concentrations of NO have been shown to be protective against apoptosis.20–22 In addition to the known cGMP-dependent effects, NO modifies proteins containing a cysteine residue via S-nitrosylation.15 S-nitrosylation is a posttranslational modification involving the attachment of NO to cysteine residues or transition metals.23 Important S-nitrosylation targets are the caspase proteins. Some investigators have suggested that S-nitrosylation of the catalytic site cysteine in caspases serves as an on/off switch regulating caspase activity during apoptosis in endothelial cells, lymphocytes, and trophoblasts.24,25
We have previously identified phenotypic differences between fibroblasts isolated from normal peritoneum and adhesion tissues from the same patient(s). Specifically, adhesion fibroblasts have lower apoptosis and higher protein nitration as compared with normal peritoneal fibroblasts.6,26 The hypothesis to be tested is that increased caspase-3 S-nitrosylation leads to the loss of its activity which subsequently decreases apoptosis in adhesion fibroblasts.
MATERIALS AND METHODS
Source and culture of human fibroblasts
Normal parietal peritoneal tissue from the anterior abdominal wall lateral to midline incision and adhesion tissue were excised from patients undergoing laparotomy for pelvic pain, at the initiation of the surgery following entry into the abdominal cavity as previously described.7,27 Normal peritoneum was at minimum 3 in. from any adhesions. Subjects did not have an active pelvic or abdominal infection and were not pregnant. All patients gave informed written consent to tissue collection, which was conducted under a protocol approved by the Wayne State University Institutional Review Board. Harvested tissue samples from five women were immediately placed in standard media (Dulbecco’s modified Eagle’s medium [DMEM] medium containing 10% fetal bovine serum [FBS], 2% penicillin and streptomycin). Tissues were cut into small pieces in a sterile culture dish and transferred into another fresh T-25 flask with 3 mL of dispase solution (2.4 U/mL; GIBCO BRL Invitrogen, Carlsbad, CA). The flasks were incubated overnight at 37 °C in an environ-shaker (LAB-LINE Instruments Inc., Dubuque, IA). The samples were then centrifuged for 5 minutes at 1,400μg, transferred into a fresh T-25 flask with prewarmed DMEM medium, and placed in 37 °C incubator (95% air and 5% CO2); out-growth of fibroblasts generally took 2 weeks.
Once confluence was reached, the cells were transferred to 90 mm2 tissue culture dishes and cultured in standard media with 10% FBS. Thereafter, the confluent dishes were subcultured by trypsinization (1 : 3 split ratios). Studies were conducted using passage three to five cells to maintain comparability.
Treatment of human normal peritoneal and adhesion fibroblasts with peroxynitrite
Cells (2×106) were treated with increasing concentrations of ONOO− (0, 0.5, 1.0, and 1.5 mM) for 24 hours. All experiments were performed in triplicate.
Treatment of human normal peritoneal and adhesion fibroblasts with anti-Fas immunoglobulin M (IgM)
Cells (2×106) were treated with 50 ng/mL of anti-Fas IgM or isotype-matched IgM as a control for 24 hours. All experiments were performed in triplicate.
Measurement of apoptosis (TUNEL assay) by flow cytometry
Apoptosis of treated and control normal peritoneal and adhesion fibroblasts was assessed using the TUNEL technique as described by the Promega Apoptosis Detection System and as we have described previously.6,7 Positive controls were performed by treating cells with DNase I (1 mg/mL) in TdT buffer for 10 minutes at room temperature before incubation with a biotinylated nucleotide. Flow cytometry data are presented as percent of apoptotic cells.6,7
Caspase-3 activity
Chemicon’s Caspase-3 Colorimetric Activity Assay Kit (Chemicon International, Temecula, CA) was used and provides a simple and convenient means for assaying the activity of caspases that recognize the sequence DEVD. The assay is based on spectophotometric detection of the chromophore p-nitroaniline (pNA) after cleavage from the labeled substrate DEVD-pNA. The free pNA can be quantified using a spectrophotometer or a microtiter plate reader at 405 nm. Comparison of the absorbance of pNA from an apoptotic sample with an uninduced control allows determination of the fold increase in caspase-3 activity. Cells (2×106) were harvested and lysed in 300 μL of cell lysis buffer included with the kit, and concentrations were equalized for each sample set. Subsequently, 150 μg of cell lysate was combined with substrate reaction buffer containing 30 μg of caspase-3 substrate, acetyl-DEVD-p-nitroaniline (Ac-DEVD-pNA). This mixture was incubated for 1 hour at 37 °C, and then absorbance was measured with a plate reader (Ultramark, Bio-Rad, Hercules, CA) by detection of the chromophore pNA after cleavage from the labeled substrate DEVD-pNA. Background reading from cell buffers and substrate should be subtracted from the readings of samples before calculating the increase in caspase-3 activity.
Detection of S-nitrosylation of caspase-3
Peroxynitrite (0.5 μM) treated normal and adhesion fibro-blast cell lysates were immunoprecipitated with anti-caspase-3 polyclonal antibody conjugated with protein A/G plus agarose beads. Immunoprecipitated caspase-3 zymogen was released by boiling the beads at 95 °C for 5 minutes. Biotinylated proteins were separated by SDS-PAGE and detected using nitrosylation detection reagent I (HRP) according to the manufacturer’s protocol (S-Nitrosylated Protein Detection Kit, Cayman Chemical, Ann Arbor, MI).
RESULTS
Peroxynitrite treatment inhibited caspase-3 activity in normal peritoneal and adhesion fibroblasts
To determine whether the effect of ONOO− treatment resulted from blockade of caspase-3 activity, we measured caspase-3 catalytic activity in normal peritoneal and adhesion fibroblasts before and after ONOO− treatment under normal and hypoxic conditions. Treatment of fibroblasts with OONO− caused a dose-dependent caspase-3 activity decrease (Figure 1). There was a significant difference in caspase-3 activity between normal peritoneal and adhesion fibroblasts (p < 0.0001). Adhesion fibroblasts exhibited significantly lower caspase-3 activity as compared with normal peritoneal fibroblasts (Figure 1, p < 0.0001). This finding was consistent with our previously reported study that characterizes the adhesion phenotype with significantly lower apoptosis.6,7
Figure 1.
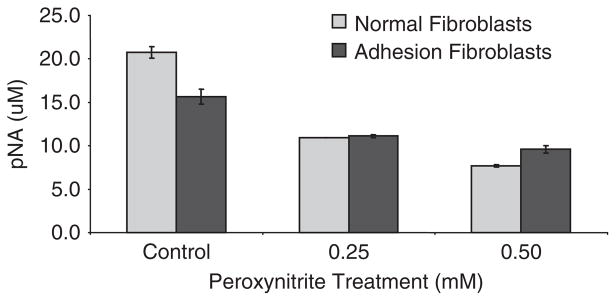
Caspase-3 activity was measured in human normal peritoneal and adhesion fibroblasts isolated from the same patient treated with increasing concentrations of peroxynitrite.
Additionally, treatment with 0.5 mM ONOO−, as evaluated by TUNEL assay, resulted in a decrease in apoptosis in both normal peritoneal (20.5%) and adhesion fibroblasts (4.12%), but to a larger extent in normal fibroblasts (Figure 2). These results suggest that ONOO− reduced apoptosis and inhibited the activation of caspase-3 in both types of fibroblasts. We next examined whether S-nitrosylation was involved in the activation of caspase-3.
Figure 2.

Fluorescent activated cell sorting analysis of TUNEL assay showing that apoptosis is higher in fibroblasts of normal peritoneum (41.8% apoptotic cells) than adhesions (6.00% apoptotic cells) of the same patient. Peroxynitrite treatment markedly decreased number of apoptotic cells in normal peritoneal (21.3%) and caused a small decrease in adhesion fibroblasts (1.88%).
Inhibition of caspase-3 activity by S-nitrosylation in normal peritoneal and adhesion fibroblasts
The decrease in caspase-3 activity in response to OONO− treatment may be due to S-nitrosylation of caspase-3. To further clarify whether caspase-3 is S-nitrosylated by OONO−, we measured S-nitrosylation of caspase-3 using a S-nitrosylation detection kit. Treatment with 0.5 mM ONOO− resulted in a significant increase in S-nitrosyla-tion of procaspase-3 (p=0.0052 for normal peritoneal fibroblasts, p=0.0011 for adhesion fibroblasts) and caspase-3 (p=0.0035 for normal peritoneal fibroblasts, p=0.0025 for adhesion fibroblasts), confirming that the caspase-3 in fibroblasts is susceptible to S-nitrosylation (Figure 3A–C). There was no significant change in the level of procaspase-3 and active caspase protein in response to ONOO− treatment (Figure 3A). These results further support our hypothesis that the inhibition of caspase-3 activity by S-nitrosylation is one of the major mechanisms by which NO can inhibit apoptosis in both normal and adhesion fibroblasts under hypoxic condition.
Figure 3.

Total and S-nitrosylated levels of procaspase-3 and caspase-3 in normal peritoneal and adhesion fibroblasts in response to 0.25 and 0.5 μM peroxynitrite treatment (A–C).
Denitrosylation of caspase-3 in normal peritoneal and adhesion fibroblasts
Denitrosylation of procaspase-3 and activated caspase-3 in normal peritoneal and adhesion fibroblasts was significantly increased in response to 50 ng/mL of anti-Fas IgM or isotype-matched IgM as control (Figure 4). In order to determine whether denitrosylation of caspase-3 also affected caspase-3 activity in normal and adhesion fibroblasts, cells were treated with 50 ng/mL of anti-Fas IgM or isotype-matched IgM as a control. The level of caspase-3 activity was significantly higher in the treated cells (Figure 5).
Figure 4.

Denitrosylation of procaspase-3 and activated caspase-3 in normal peritoneal and adhesion fibroblasts in response to 50 ng/mL of anti-Fas immunoglobulin M (IgM) (CLONE ch-11) or isotype-matched IgM control treatment. S-nitrosylated caspase-3 was measured with the S-nitrosylation protein detection assay kit.
Figure 5.

Caspase-3 activity in normal peritoneal and adhesion fibroblasts treated with 50 ng/mL of anti-Fas immunoglobulin M (IgM) or isotype-matched IgM as a control.
DISCUSSION
Hypoxia is an important trigger for the function of fibro-blast cells during peritoneal healing and development of postoperative adhesions. As we have previously shown, hypoxia is the main trigger for the development of the adhesion phenotype in which adhesion fibroblasts manifested a marked decrease in apoptosis.6 We have evidence to believe that hypoxia exerts its effect through the production of O2• −, which binds to NO and forms OONO−.28 Indeed, we have shown that protein nitration, the fingerprint of ONOO− formation, was higher in adhesion as compared with normal peritoneal fibroblasts.26 In addition to the potential role of OONO− in protein nitration, there is increasing evidence implicating OONO− in inhibiting caspase-3–mediated apoptosis.29 Indeed, caspases are known to contain sensitive thiol moieties, and ONOO− has been shown to induce S-nitrosylate and inactivate isolated recombinant caspase-3 enzyme.30
The role of NO and OONO− in the S-nitrosylation reaction is still not clear, and little is known about the oxidative actions of OONO− which seems to be more important than the nitrosative effect of OONO−.31,32 Few S-nitrosylated proteins have been identified, and it is unknown if protein S-nitrosylation/denitrosylation is a component of signal transduction cascades. Caspase-3 zymogens were found to be S-nitrosylated on their catalytic-site cysteine in unstimulated human cell lines and denitrosylated upon activation of the Fas apoptotic pathway.33 S-nitrosylation is reversible and a seemingly specific posttranslational modification that regulates the activity of several signaling proteins.23 Some investigators have suggested that S-nitrosylation of the catalytic site cysteine in caspases serves as an on/off switch regulating caspase activity during apoptosis in endothelial cells, lymphocytes, and trophoblasts.24,34 The overall content of S-nitrosylated molecules was increased after exposure of human cells to ONOO−, indicating a possible role in the long term signal transduction of hypoxia. These data showed an increase in overall S-nitrosylation by acetylcholine-induced endothelial NO synthase (eNOS) activation.35 Additionally others have demonstrated an increase in S-nitrosylation of specific targets, such as the catalytic subunit p17 of caspase-3, p21ras, and thioredoxin, by application of shear stress, which was accompanied by an augmented activity of p21ras and thioredoxin.36
In the present study we have demonstrated that exposure of normal peritoneal fibroblasts to OONO− induced the S-nitrosylation of caspase-3, which led to the inactivation of caspase-3 and hence, lower apoptosis. In contrast, our results also indicated that denitrosylation, induced by treatment with anti-Fas IgM, significantly increased caspase-3 activity. These findings are consistent with the fact that altering the balance of nitrosylation/denitrosylation affects caspase-3 activity and subsequent programmed cell death. It is uncertain whether the effects observed were due entirely to ONOO− or from intermediates derived from the decomposition of ONOO− at physiological pH such as the hydroxyl radical (OH • ), nitrogen dioxide (NO2•), or the nitryl cation ( ).37 One possible mechanism of caspase-3 S-nitrosylation is through ONOO− breakage into nitrite ( ) and nitrate ( ) by which reduces to NO occurs via the nitrite-reductase reaction: . The reduction of ions to NO is realized by electron-donor systems with the participation of NADH, NADPH, flavoproteins, and cytochrome oxidase in mitochondria and by NADH, NADPH, flavoproteins, and cytochrome P-450 in the endoplasmic reticulum. In erythrocytes, the reduction of to NO is catalyzed by electron-donor systems with participation of NADH, NADPH, flavoproteins, and deoxy-hemoglobin.
Proapoptotic caspase-3 consists of two subunits, p12 and p17, that form a heterodimer to build the active protease. Subunit p17 contains a reactive cysteine at position 163 in the catalytic center that is essential for enzyme activity. NO and ONOO− inhibit the p17 subunit via S-nitrosylation of this critical cysteine.22 Thus, the increased S-nitrosylation of the caspase-3 zymogen and p17 subunit after exposure to ONOO− contributes to the anti-apoptotic effect of NO in adhesion fibroblasts. The anti-apoptotic effects of NO, apart from cGMP signaling pathway, can also be mediated through a number of mechanisms independent of cGMP signaling including S-nitrosylation.38 By detection of nitrosylated caspase-3 directly, we confirmed that NO and ONOO− inhibit apoptosis by preventing the activation of caspase-3 via S-nitrosylation. We suggested that S-nitrosylation of caspase-3 by NO and ONOO− is a novel cGMP-independent anti-apoptotic mechanism in adhesion fibroblasts.
Our results show that S-nitrosylation levels of both pro- and cleaved caspase-3 after ONOO− treatment of normal peritoneal fibroblasts are higher than untreated adhesion fibroblasts; however, the apoptosis level of ONOO−-treated normal peritoneal fibroblasts is higher than untreated adhesion fibroblasts. These findings may implicate the involvement of other signaling pathways or mechanisms besides the S-nitrosylation of caspase-3.
It has been known that the effects of NO on apoptosis are not only stimulatory but also inhibitory as shown in this study.39 It has been the subject of discussion as to why the same molecule should perform both these opposite tasks, and it is thought that biological conditions such as the redox state, concentration, exposure time and the combination with O2, O2• −, and other molecules determine the net effects of NO on apoptosis.40 In our experimental condition, these factors applied to the cells may affect S-nitrosylation of caspase-3. Also, NO is implicated in both apoptotic and necrotic cell death depending on the NO chemistry and the cellular biological redox state.40 Targeting free radical production, specifically O2• − formation, may be a potential therapeutic intervention for the elimination of the adhesion fibroblasts during peritoneal healing.
Acknowledgments
This study was supported in part by NIH grant number NIH 1RO1 GM069941-01A3 to G.M. Saed.
References
- 1.Diamond MP, Hershlag A. Adhesion formation/reformation. Prog Clin Biol Res. 1990;358:23–33. [PubMed] [Google Scholar]
- 2.Diamond MP, Freeman ML. Clinical implications of post-surgical adhesions. Human Reprod Update. 2001;7:567–76. doi: 10.1093/humupd/7.6.567. [DOI] [PubMed] [Google Scholar]
- 3.Kokura S, Yoshida N, Yoshikawa T. Anoxia/reoxygenation-induced leukocyte–endothelial cell interactions. Free Radic Biol Med. 2002;33:427–32. doi: 10.1016/s0891-5849(02)00852-3. [DOI] [PubMed] [Google Scholar]
- 4.Lewis JS, Lee JA, Underwood JC, Harris AL, Lewis CE. Macrophage responses to hypoxia: relevance to disease mechanisms. J Leukoc Biol. 1999;66:889–900. doi: 10.1002/jlb.66.6.889. [DOI] [PubMed] [Google Scholar]
- 5.Smith SC, Baker PN, Symonds EM. Increased placental apoptosis in intrauterine growth restriction. Am J Obstet Gynecol. 1997;177:1395–401. doi: 10.1016/s0002-9378(97)70081-4. [DOI] [PubMed] [Google Scholar]
- 6.Saed GM, Diamond MP. Apoptosis and proliferation of human peritoneal fibroblasts in response to hypoxia. Fertil Steril. 2002;78:137–43. doi: 10.1016/s0015-0282(02)03145-x. [DOI] [PubMed] [Google Scholar]
- 7.Saed GM, Abu-Soud HM, Diamond MP. Role of nitric oxide in apoptosis of human peritoneal and adhesion fibroblasts after hypoxia. Fertil Steril. 2004;82 (Suppl 3):1198–205. doi: 10.1016/j.fertnstert.2004.04.034. [DOI] [PubMed] [Google Scholar]
- 8.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science (New York, NY) 1995;267:1456–62. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 9.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science (New York, NY) 1998;281:1312–6. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 10.Kumar S. ICE-like proteases in apoptosis. Trends Biochem Sci. 1995;20:198–202. doi: 10.1016/s0968-0004(00)89007-6. [DOI] [PubMed] [Google Scholar]
- 11.Nicholson DW, Ali A, Thornberry NA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 12.Stuehr DJ. Mammalian nitric oxide synthases. Biochimica Biophysica Acta. 1999;1411:217–30. doi: 10.1016/s0005-2728(99)00016-x. [DOI] [PubMed] [Google Scholar]
- 13.Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med. 1993;329:2002–12. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 14.Nathan C. Nitric oxide as a secretory product of mammalian cells. FASEB J. 1992;6:3051–64. [PubMed] [Google Scholar]
- 15.Stamler JS, Simon DI, Osborne JA, Mullins ME, Jaraki O, Michel T, Singel DJ, Loscalzo J. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci USA. 1992;89:444–8. doi: 10.1073/pnas.89.1.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stamler JS, Singel DJ, Loscalzo J. Biochemistry of nitric oxide and its redox-activated forms. Science. 1992;258:1898–902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- 17.Pryor WA, Squadrito GL. The chemistry of peroxynitrite: a product from the reaction of nitric oxide with superoxide. Am J Physiol. 1995;268 (Part 1):L699–722. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- 18.Brune B, Mohr S, Messmer UK. Protein thiol modification and apoptotic cell death as cGMP-independent nitric oxide (NO) signaling pathways. Rev Physiol Biochem Pharmacol. 1996;127:1–30. doi: 10.1007/BFb0048263. [DOI] [PubMed] [Google Scholar]
- 19.Sarih M, Souvannavong V, Adam A. Nitric oxide synthase induces macrophage death by apoptosis. Biochem Biophys Res Commun. 1993;191:503–8. doi: 10.1006/bbrc.1993.1246. [DOI] [PubMed] [Google Scholar]
- 20.Mannick JB, Asano K, Izumi K, Kieff E, Stamler JS. Nitric oxide produced by human B lymphocytes inhibits apoptosis and Epstein–Barr virus reactivation. Cell. 1994;79:1137–46. doi: 10.1016/0092-8674(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 21.Genaro AM, Hortelano S, Alvarez A, Martinez C, Bosca L. Splenic B lymphocyte programmed cell death is prevented by nitric oxide release through mechanisms involving sustained Bcl-2 levels. J Clin Invest. 1995;95:1884–90. doi: 10.1172/JCI117869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dimmeler S, Haendeler J, Nehls M, Zeiher AM. Suppression of apoptosis by nitric oxide via inhibition of interleukin-1beta-converting enzyme (ICE)-like and cysteine protease protein (CPP)-32-like proteases. J Exp Med. 1997;185:601–7. doi: 10.1084/jem.185.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stamler JS, Lamas S, Fang FC. Nitrosylation. The prototypic redox-based signaling mechanism. Cell. 2001;106:675–83. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 24.Mannick JB, Schonhoff C, Papeta N, Ghafourifar P, Szibor M, Fang K, Gaston B. S-Nitrosylation of mitochondrial caspases. J Cell Biol. 2001;154:1111–6. doi: 10.1083/jcb.200104008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dash PR, Cartwright JE, Baker PN, Johnstone AP, Whitley GS. Nitric oxide protects human extravillous trophoblast cells from apoptosis by a cyclic GMP-dependent mechanism and independently of caspase 3 nitrosylation. Exp Cell Res. 2003;287:314–24. doi: 10.1016/s0014-4827(03)00156-3. [DOI] [PubMed] [Google Scholar]
- 26.Saed GM, Zhao M, Diamond MP, Abu-Soud HM. Regulation of inducible nitric oxide synthase in post-operative adhesions. Hum Reprod. 2006;21:1605–11. doi: 10.1093/humrep/dei500. [DOI] [PubMed] [Google Scholar]
- 27.Muschel RJ, Bernhard EJ, Garza L, McKenna WG, Koch CJ. Induction of apoptosis at different oxygen tensions: evidence that oxygen radicals do not mediate apoptotic signaling. Cancer Res. 1995;55:995–8. [PubMed] [Google Scholar]
- 28.Cudd A, Fridovich I. Electrostatic interactions in the reaction mechanism of bovine erythrocyte superoxide dismutase. J Biol Chem. 1982;257:11443–7. [PubMed] [Google Scholar]
- 29.Lau A, Arundine M, Sun HS, Jones M, Tymianski M. Inhibition of caspase-mediated apoptosis by peroxynitrite in traumatic brain injury. J Neurosci. 2006;26:11540–53. doi: 10.1523/JNEUROSCI.3507-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He L, Lemasters JJ. Heat shock suppresses the permeability transition in rat liver mitochondria. J Biol Chem. 2003;278:16755–60. doi: 10.1074/jbc.M300153200. [DOI] [PubMed] [Google Scholar]
- 31.Ji Y, Neverova I, Van Eyk JE, Bennett BM. Nitration of ty-rosine 92 mediates the activation of rat microsomal glutathi-one S-transferase by peroxynitrite. J Biol Chem. 2006;281:1986–91. doi: 10.1074/jbc.M509480200. [DOI] [PubMed] [Google Scholar]
- 32.Steffen M, Sarkela TM, Gybina AA, Steele TW, Trasseth NJ, Kuehl D, Giulivi C. Metabolism of S-nitrosoglutathione in intact mitochondria. Biochem J. 2001;356 (Part 2):395–402. doi: 10.1042/0264-6021:3560395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mannick JB, Hausladen A, Liu L, Hess DT, Zeng M, Miao QX, Kane LS, Gow AJ, Stamler JS. Fas-induced caspase denitrosylation. Science. 1999;284:651–4. doi: 10.1126/science.284.5414.651. [DOI] [PubMed] [Google Scholar]
- 34.Rössig L, Fichtlscherer B, Breitschopf K, Haendeler J, Zeiher AM, Mülsch A, Dimmeler S. Nitric oxide inhibits caspase-3 by S-nitrosylation in vivo. J Biol Chem. 1999;274:6823–6. doi: 10.1074/jbc.274.11.6823. [DOI] [PubMed] [Google Scholar]
- 35.Gow AJ, Chen Q, Hess DT, Day BJ, Ischiropoulos H, Stamler JS. Basal and stimulated protein S-nitrosylation in multiple cell types and tissues. J Biol Chem. 2002;277:9637–40. doi: 10.1074/jbc.C100746200. [DOI] [PubMed] [Google Scholar]
- 36.Hoffmann J, Dimmeler S, Haendeler J. Shear stress increases the amount of S-nitrosylated molecules in endothelial cells: important role for signal transduction. FEBS Lett. 2003;551:153–8. doi: 10.1016/s0014-5793(03)00917-7. [DOI] [PubMed] [Google Scholar]
- 37.Whiteman M, Armstrong JS, Cheung NS, Siau JL, Rose P, Schantz JT, Jones DP, Halliwell B. Peroxynitrite mediates calcium-dependent mitochondrial dysfunction and cell death via activation of calpains. FASEB J. 2004;18:1395–7. doi: 10.1096/fj.03-1096fje. [DOI] [PubMed] [Google Scholar]
- 38.Maejima Y, Adachi S, Morikawa K, Ito H, Isobe M. Nitric oxide inhibits myocardial apoptosis by preventing caspase-3 activity via S-nitrosylation. J Mol Cell Cardiol. 2005;38:163–74. doi: 10.1016/j.yjmcc.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 39.Maejima Y, Adachi S, Ito H, Nobori K, Tamamori-Adachi M, Isobe M. Nitric oxide inhibits ischemia/reperfusion-induced myocardial apoptosis by modulating cyclin A-associated kinase activity. Cardiovasc Res. 2003;59:308–20. doi: 10.1016/s0008-6363(03)00425-5. [DOI] [PubMed] [Google Scholar]
- 40.Arstall MA, Sawyer DB, Fukazawa R, Kelly RA. Cytokine-mediated apoptosis in cardiac myocytes: the role of inducible nitric oxide synthase induction and peroxynitrite generation. Circulation Res. 1999;85:829–40. doi: 10.1161/01.res.85.9.829. [DOI] [PubMed] [Google Scholar]