

Abstract
The measurement of not only the location but also the organization of molecules in live cells is crucial to understanding diverse biological processes. Polarized light microscopy provides a nondestructive means to evaluate order within subcellular domains. When combined with fluorescence microscopy and GFP-tagged proteins, the approach can reveal organization within specific populations of molecules. This unit describes a protocol for measuring the architectural dynamics of cytoskeletal components using polarized fluorescence microscopy and OpenPolScope open-access software (www.openpolscope.org). The protocol describes installation of linear polarizers or a liquid crystal (LC) universal compensator, calibration of the system, polarized fluorescence imaging, and analysis. The use of OpenPolScope software and hardware allows for reliable, user-friendly image acquisition to measure and analyze polarized fluorescence.
Keywords: polarized fluorescence, cytoskeleton, image analysis, OpenPolScope
Introduction
Traditionally, the molecular organization of cellular structures was determined by measuring the differential phase shift, or retardance, of intrinsically birefringent structures without dyes or labels (Inoué and Bajer, 1961). Polarized light microscopy revealed some of the first glimpses of spindle dynamics and movement of large organelles in cytosol. A limitation in the initial applications of the method however is that the arrangement and identity of specific molecules could not be determined. Polarized fluorescence microscopy was then developed which exploits the dipole moment of fluorescent molecules, including fluorescent proteins (FP) fused to proteins of interest. With this approach, both the position and orientation of specific molecules can be measured. With appropriate care given to the design of the fusion protein so that the FP is rotationally constrained, both the anisotropy (amount of order) and the azimuth (average orientation of ensemble of molecules) of proteins of interest can be monitored in live cells and through time-lapse imaging.
The protocol in this unit describes how to acquire and analyze images in which linear polarized light either excites or is detected from the fluorophore in GFP to determine the orientation of GFP dipoles that report order and alignment of specific molecules in cells. As an example, the use of the system is illustrated using the septin cytoskeleton, which forms a higher order structure at the site of cell division in Saccharomyces cerevisiae. Septin filaments that constitute the assembly have previously been shown to be highly organized throughout the cell cycle, with a 90° transition in organization just prior to cytokinesis. The method of detecting and analyzing fluorescence anisotropy is applicable in theory to any collection of molecules that can be constrained either at the level of the fluorophore association with the protein of interest or due to the configurational constraints of the assembled structure (i.e., a lattice). The septin assembly in yeast can serve as an excellent positive control for a new polarized fluorescence system or even as a calibration of imaging systems. Strains for use as controls are available from the authors.
Basic Protocol 1: Polarized Fluorescence Microscopy Analysis using OpenPolScope Software
Overview: This protocol is used to determine the prevailing orientations of fluorophore dipoles associated with molecules in living cells. It is described using GFP as a fluorescent tag, however any FP or rigidly associated dye can be used to assess anisotropy in live cells. After presenting the acquisition protocol, we discuss considerations and approaches for analyzing the acquired images. While other image acquisition and analysis platforms can be used, our protocol assumes images are acquired and analyzed using the OpenPolScope plugins to Micro-Manager and ImageJ, which are open access microscopy software freely available on the internet: www.micro-manager.org; rsb.info.nih.gov/ij/; openpolscope.org. The measured intensity varies as a cosine square of the angle between the prevailing orientation of the dipoles and orientation of the excitation polarization. To calculate the anisotropy and orientation of polarized fluorescence in every resolved image point (pixel), a stack of 5 images is acquired. Each image represents all the fluorescence emitted by fluorophores that are excited by light that is linearly polarized at orientations 0, 45, 90 and 135 degrees. The fifth image resamples 0 degree, to allow for the correction of fluorescence bleaching that occurs during the acquisition of the image stack. After correcting for fluorescence bleaching, camera pixels for each orientation are fit to a cosine square function to calculate the resulting anisotropy and orientation for the ensemble of molecules contributing to that pixel. Spatial statistics can be employed to determine both the variability of orientations within a structure and between structures in a population of cells. Once calculated, anisotropy and orientation values can be used along with knowledge of how the fluorophore was constrained to model cytoskeletal organization (as in for example, (DeMay et al., 2011b).
In the protocol described here, we recommend to use polarized excitation light and no polarizer in the emission/imaging path of the microscope. Hence, all emitted fluorescence contributes to each of the acquired images, while their anisotropy is measured by the variation of total fluorescence with excitation polarization. This approach enables a higher efficiency of fluorescence excitation and detection, compared to a setup that uses unpolarized excitation and polarization analysis of the emitted fluorescence. We recommend the detection of all emitted fluorescence to achieve high efficiency, low bleach rate, and low photo-damage in the sample. Further discussions on the relative merits of configurations that use variable polarizers in the excitation path only, in the emission/imaging path only, and parallel polarizers in both excitation and emission paths are discussed in DeMay et al., 2011b.
Materials
Slide with sheet polarizer with known transmission orientation, preferably parallel to an edge.
Overnight culture of Saccharomyces cerevisiae
Media with low fluorescence for imaging
Agarose
Depression slides
VALAP
Coverslips for mounting S. cerevisiae sample
Wide-field microscope. For best results, use strain-free oil immersion objective lenses and condenser optics (any brand)
Liquid crystal (LC) based universal polarizer with mounting adapter and electronic controller with operating wavelength of 450–700nm hardware can be purchased from OpenPolScope Resource.
--Alternative to LC: five linear polarized filters (Chroma 21033a) in filter wheel (Ludl Electronic Products Ltd 99A075) and UV blocker 420nm LP emission filter 32nm (Chroma E420LPv2-32)
Digital CCD camera supported by Micro-Manager software
ImageJ (http://imagej.nih.gov/)
Micro-Manager (www.micro-manager.org) (Edelstein et al., 2010)
OpenPolScope software (full installation, www.openpolscope.org)
The polarization of the excitation light can be controlled either by a liquid crystal based universal polarizer or a filter wheel equipped with appropriately rotated linear polarizers. OpenPolScope provides support for acquiring polarized fluorescence images using both of these modes. We first describe the use of the LC universal polarizer, which has certain advantages, as it can minimize polarization aberrations introduced by optical components in the light path. The filter wheel with polarizers, on the other hand, permits the use of broadband illumination light, even white light, while the LC universal polarizer requires narrowband monochromatic light of typically not more than 30 nm spectral bandwidth. The center wavelength, however, can vary between 450 and 700 nm for the LC universal polarizer.
LC Compensator Installment
The liquid crystal universal polarizer, available from the OpenPolScope Resource at MBL, is placed in the illumination light path of the microscope. For illumination, one can either use the epi- or transmitted light path. The transmitted light path has two advantages: (1) it avoids the use of a dichroic mirror in the imaging path, and (2) the LC polarizer can also be used for birefringence or diattenuation measurements of unlabeled material (Mehta et al., 2013). The detrimental effect of the dichroic mirror in the imaging path arises from its property as a partial polarizer and contributor to spurious instrument bias. However, the bias can be minimized by the application of an instrument correction procedure available in the OpenPolScope software. Therefore, if the setup is only used for polarized fluorescence imaging, the use of the epi-illumination path can improve the overall efficiency of the setup and artifacts can be corrected.
List of components and their specifications:
Bright light source for excitation light (typically arc lamp or high luminance LEDs)
-
Band-pass interference filter for illumination light path. Required center wavelength, spectral bandwidth, and discrimination efficiency depend on the fluorophores and microscope setup used:
Center wavelength is determined by excitation spectrum of fluorophores.
Spectral bandwidth must not overlap with band-pass of emission filter and less than 30 nm when using LC universal polarizer.
Discrimination efficiency (suppression of light outside spectral bandwidth) must be better than 10−6 for light that is in the band-pass of the emission filter.
LC universal polarizer placed in location that has minimal temperature fluctuations (away from arc lamp) and no UV (λ < 400 nm) exposure. After passing through polarizer, light should not bounce off mirrors and should pass through a minimum of optical elements to avoid polarization aberrations.
If epi-illumination path is chosen, the specifications of the dichroic mirror in the fluorescence cube affect the sensitivity and efficiency of polarized fluorescence measurements. Dichroic mirrors with similar spectral characteristics can have quite different polarization performance specifications. Typically, manufacturers do not design dichroic mirrors with polarization specifications in mind. However, modern design software for specifying the dielectric layers for dichroic mirrors can include polarization performance and it is worthwhile to ask the manufacturer about suitability for polarization applications.
Alternative implementation with filter wheel in excitation light path
Depending on the shape and placement of available openings in the light path, it may be optimal to place linear polarizers in a filter wheel. In this scenario, the filter wheel is rotated between images at each orientation. If a filter wheel is to be used, five linear polarizers must be fitted into a filter wheel at four distinct angles. Using the stage as a reference, polarizers should be fitted at the following angles: 0°, 135°, 90°, 45°, and 0° again for correction of fluorescence bleaching. To properly orient the linear polarizers relative to the stage, the slide with sheet polarizers is placed on stage and used as a polarization analyzer. In turn, each polarizer in the filter wheel is carefully rotated until extinction is achieved with the sheet polarizer on a slide that is appropriately oriented for each filter wheel position. It might be necessary to remove the filter wheel from the light path to rotate one of its polarizers, resulting in a lengthy process of step-by-step rotation and testing the amount of light passing through the slide analyzer. After the orientation of one polarizer is calibrated, the slide is rotated 45° and the orientation of the next polarizer in the filter wheel is adjusted. If the slide cannot be rotated on the stage, it can be exchanged for another slide which has the polarization analyzer rotated by 45° with respect to the previous slide. Once the polarizers are properly fitted, and the filter wheel is installed in the filter wheel controller, a UV blocker must be added between the light source and filter wheel to prevent bleaching the polarizers.
Preparing yeast sample with septin-GFP construct
Septin-GFP expressing yeast can serve as a control for instrument set-up as the signal is robust and reproducible(DeMay et al., 2011a; DeMay et al., 2011b).
Create a 1.4% agarose gel pad with appropriate low fluorescent media for growing S. cerevisiae cells.
-
Melt agarose in microwave or in an eppendorf tube on a heat block and aliquot 200 ul onto a clean depression slide before immediately putting a second slide orthogonal to the first on top to form the pad.
For strains expressing Cdc12conGFP, low fluorescent SC media supplemented with 50 μg/ml G418 added to media is appropriate to use. Add ~ 5 ul of log-phase S. cerevisiae cells to center of gel pad for imaging and apply coverslip. A small weight or pressure can be applied after coverslip is added to limit the movement of cells. The coverslip should be sealed with VALAP.
Polarized Fluorescence Imaging
-
1
In Micro-Manager, open PolAcquisition plugin and set file path to appropriate user and session folder. Set imaging to fluorescence, and wavelength to excitation center wavelength used. Under the fluorescence tab, select the anisotropy process.
The anisotropy formula calculates (Imax−Imin)/(Imax+Imin) in each pixel, based on the bleach corrected fluorescence intensity values measured for excitation polarization 0°, 135°, 90°, and 45°(DeMay et al., 2011b). Instead of the anisotropy, one can also calculate the ratio Imax/Imin for each pixel. While the anisotropy values vary between 0 (isotropic) and 1(fully aligned), the ratio varies between 1 (isotropic) to infinity (fully aligned). -
2
In addition to bleach correction, intensity values are also corrected for an elevated black level. To measure the black level, block the light path to the camera (e.g. move light path to the eye-piece), and acquire a (black) image, using the same exposure time and gain value that are used for a regular fluorescence acquisition. Determine the mean intensity and enter its value as the black level for the microscope system used. The black level field is under the Parameters tab.
-
3
It is important to calibrate the liquid crystal settings for the microscope system used. This can be done using the slide with sheet polarizer as a specimen having known orientation. The slide acts as a polarization analyzer for the polarization state produced by the LC. We first assume that the LC polarizer is used in a trans-illumination system.
After focusing on the specimen, orient the transmission axis of the sheet of polarizer orthogonal to the polarization state that you are calibrating. For example, to calibrate 0° (horizontal) polarization, orient the sheet polarizer so that is transmits vertically (90°) polarized light.
Select a region of interest (ROI) with in the sheet polarizer.
In PolAcquisition, under the LC tab select the Calibration tab select the Setting dropdown, which is orthogonal to the transmission axis of the selected ROI. In the example above, the dropdown setting is 0°, while the transmission axis of the sheet polarizer is 90°. Use Single ROI Calibration option and click the Calibrate button. The calibration routine seeks to minimize the transmitted intensity, automatically optimizing the LC settings for horizontal excitation polarization in the specimen plane.
Repeat the above procedure for each of the four orientations of the excitation light.
-
4
For calibrating a LC universal polarizer that is located in the epi-illumination path, a separate light detector or camera behind the slide with sheet polarizer on the sample stage is needed. This extra camera records the intensity passing through the sheet polarizer. If the readout of the extra detector or camera is not available to Micro-Manager, the readout can still be used manually to calibrate the LCs by observing the intensity minimum by eye.
-
5
After calibration of a trans-illumination system, with the polarizer sheet still on the sample stage, click Check under the Controls tab. Verify that the orientation box at the bottom shows the orientation of the transmission axis of the polarizer sheet. The anisotropy must be near 1.0.
-
6
By convention, we choose zero degree azimuth to correspond to horizontal orientation in an image. To make the calculated azimuth of 0° correspond to horizontal orientation, the reference angle in the Pol-plugins needs to be set correctly (Figure 2). The reference angle can have any value between 0° and 180° and depends on the mutual orientations between the camera and the polarizers. In a trans-illumination system, the correct value is best determined by imaging the edge of a sheet polarizer or other material, whose morphological feature has a known orientation with respect to the polarization axis. In an epi-illumination system, a sample with known polarized fluorescence needs to be used. After setting the correct reference angle, rotating the polarizer or fluorescent sample counter-clockwise should change the azimuth from 0° (polarization axis horizontal), to 45° (diagonal), 90° (vertical), 135° (other diagonal), and 180° (horizontal again). As polarization has no direction, 0° and 180° are equivalent and both correspond to horizontal orientation. To estimate the angle of morphological features, you can use the angle tool in ImageJ.
-
7
In addition to the reference angle, it is also important to determine if the check box for “mirror in light path” needs to be set. This can be determined by rotating the polarizer or fluorescent sample on the stage and taking measurements every 45°. Like described under point 5, the sequence of calculated azimuth values needs nominally to be 0°, 45°, 90°, 135°, and 180°. Small deviations of a few degrees are possible because of aberrations in the light path or the linear polarizer alignments in the filter wheel. However, if the sequence is 0°, 135°, 90°, 45°, 180°, checking “mirror in light path” will flip the 2 diagonal values.
-
8
Take images of cells within the field using the “SM” option in the PolAcquisition. To create multi-acquisition images use the “Series” option. This will automatically trigger the acquisition of 5 fluorescence images for the 4 different orientations and a bleach correction image. All images will automatically save in the defined session folder.
Multi-acquisition images are ideal for observing cytoskeletal assembly in live cells. In the Micro-Manager/OpenPolScope system, there are options for: z-stacks, time series, and multiple xy positions. Depending on the microscope set-up, the user can take advantage of several or all options.
Figure 2.
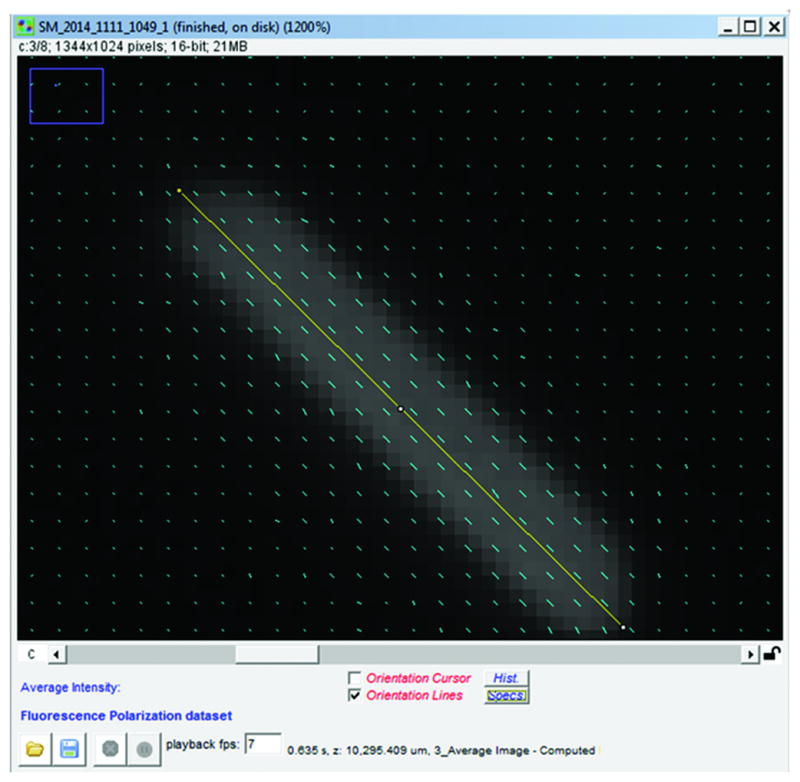
GFP crystal image acquired using PolAcquisition with adjusted orientation reference and black level. Orientation lines have been projected over average fluorescence image of crystal. Yellow line was drawn along long axis of crystal to gauge how well orientation lines lie parallel to crystal long axis.
Image processing
-
9
Open the PolAnalyzer Micro-manager plugin. Again, set the path to the appropriate user and session folder and open the acquired “SM” dataset. All the parameters (black level, wavelength, etc.) related to the dataset will be automatically loaded into the plugin.
-
10
To background correct the image, process the image using an “isotropic” region and a “specimen” region under the fluorescence tab (Figure 3). The use of two regions is to ensure accurate correction in case of differential photobleaching in the soluble pool of tagged proteins and the assembled structure. This is a formal possibility if the structure is a tightly-packed, ordered assembly such as the septin cortex.
When performing this step be sure to select an “isotropic” region of background. Generally, with fluorescently labelled samples, a region within the cytoplasm, but outside of the structure of interest is used. The “specimen” region should be an ROI within the structure, or one of the structures, of interest. Be sure to select all correction options under “isotropic” and “specimen” corrections. -
11
Run the process using the Process button, which will overwrite the previous computed images with the new background corrected images. The raw images are not altered during processing.
Figure 3.

Average fluorescence image with ROIs used for background correction using PolAnalyzer. A. Average fluorescence image with ROI used for isotropic correction. The ROI was drawn in a cytoplasmic area known to have isotropic (background) signal. B. PolAnalyzer settings used for isotropic correction. C. Average fluorescence image with ROI used for specimen correction. The ROI was drawn within the structure of interest, which will be observed for anisotropy. D. PolAnalyzer settings used for specimen correction.
Visualization
-
12
From the image data window the OpenPolScope Orientation Lines plugin can be used to visualize organization, and export orientation data (Figure 4) by using the “Specs” option.
-
13
Select a ROI (if required of the sample) and use the “Specs” option on the image data window to select options in the Orientation Lines plugin for visualization (Figure 4).
Figure 4.
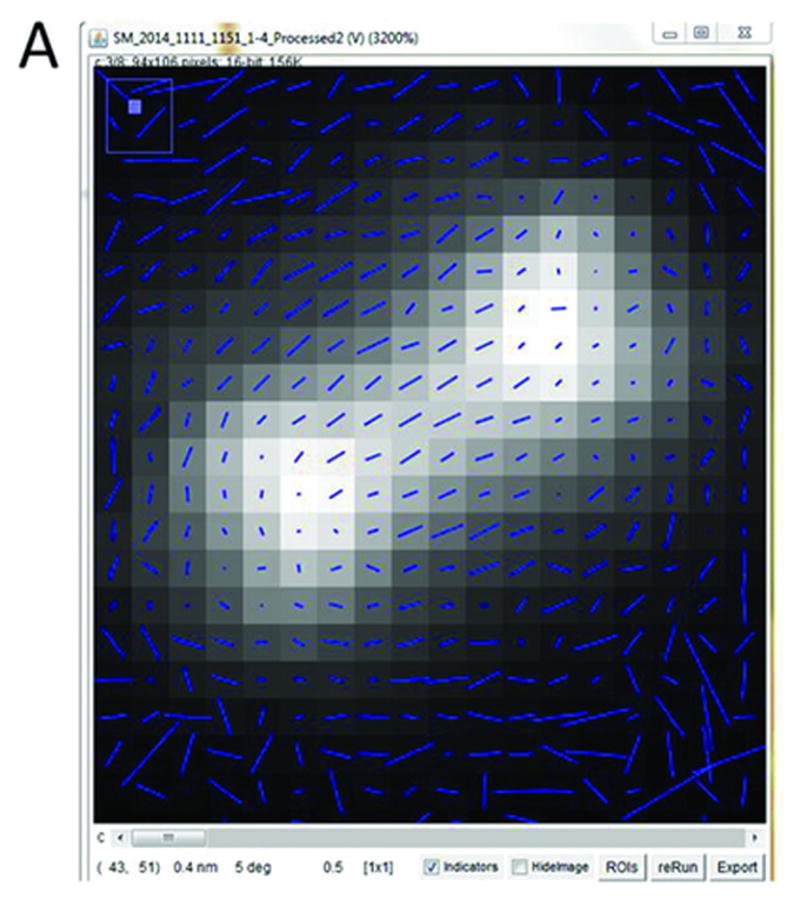
Pixel-by-pixel map of septin structure at yeast bud neck. Average fluorescence image with orientation lines overlaid. Length of lines is proportional to magnitude of anisotropy, and lines indicate prevailing orientations of GFP dipoles within individual pixels. Orientation indicators window with settings used to create pixel-by-pixel map of septin hourglass.
Computing statistics
It is often important to look at statistical properties of the anisotropy and orientation over multiple pixels or grid that defines a specific structure. Two distinct approaches to computing the statistics can be taken, depending on the question being asked.
Histograms of intensity, orientation, or anisotropy can be computed with PolAnalyzer to examine the dominant intensity, orientation, or anisotropy of the structure. However, the mean and variance of these histograms, or ‘linear statistics’ do not convey the mean orientation or mean anisotropy of the structure. To illustrate the pitfalls of interpreting anisotropy data (which is circular in nature) with linear statistics, consider a structure with equal number of pixels that have 0°, 45°, 90°, and 135° degree orientation, as well as equal intensity and equal anisotropy. It is clear that overall the structure is isotropic, but taking a linear average will assign the structure an orientation of 90° and finite anisotropy.
We have chosen to implement the circular statistics such that the computed mean anisotropy and orientation are the same as if the camera pixel was the shape and size of the structure being analyzed. However, the implemented circular statistics is applied to the calculated anisotropy and orientation data only, which are the result of previous calculations that might have included instrument, bleach and other corrections. The variance is computed as 1- (mean anisotropy/peak anisotropy). The variance is defined in this fashion because a high variance in angular distribution of dipoles reduces the mean anisotropy, and vice versa. More detailed discussing of the calculation of circular statistics are in DeMay et al., 2011a.
When using the data for statistical calculations using either of the approaches described above, be mindful of the orientation of the structure within the field of view relative to the axis of excitation. Generally, you will need to refer the measured orientations relative to cell landmarks or geometry before making biological interpretations.
Following steps describe how to export the anisotropy and orientation information to an excel sheet.
When exporting orientation data using a 1×1pixel grid, the values for all the pixels within the selected ROI will be exported. The values in this case represent all the pixels; however, based on options, pixel values can be weighed against anisotropy and average fluorescence values. Using a 1×1 grid does not compute circular statistics, which requires a minimum of 2×2.
After selecting the grid size, select the option “show values in Results table” to export the data whenever the Orientation Lines are computed.
To export data from a multi-dimensional dataset, under PolAnalyzer’s option tab select “Don’t process – Only walk through Dataset” and use the Play/Walk option. The process will walk the dataset through its dimensions and in doing so compute the Orientation Lines for each dimension and also export the data into a Results table. Once this “Walk” is completed, the data can be saved into an excel file from the results table. To obtain physically interpretable mean and variance of the orientation over the structure, circular statistics need to be used (Fisher, 1993).
REAGENTS AND SOLUTIONS
VALAP (Vaseline, Lanolin, Paraffin mixture)
Equal parts by weight:
Vaseline
Lanolin
Paraffin
Slowly melt to mix
Paraffin can be replaced by bees wax, to achieve a softer consistency at room temperature and a slightly lower melting temperature, which is about 50 – 60°C.
COMMENTARY
Background Information
First observing the natural birefringence of the mitotic spindle and other biological structures, Runnström and Schmidt paved the way for the field of polarized light microscopy in biology (Runnström, 1928; Schmidt, 1924; Schmidt, 1937). With this new field came new biological questions, and the microscopes improvement to better decipher variations in biological organization (Inoué and Dan, 1951; Inoué and Hyde, 1957; Swann and Mitchison, 1950). This led to an extensive study of the dynamic architecture of the mitotic spindle, and thus, it was the first time a biological structure was characterized on a submicroscopic level (Inoué, 1952; Inoué, 1953; Inoué, 1964; Salmon, 1975; Sato et al., 1975).
A major improvement to the polarized light microscope was the addition of a computer controlled liquid crystal compensator in what became known as the “new pol-scope” (Oldenbourg and Mei, 1995). This allowed for high spatial and temporal resolution in imaging specimen birefringence within an entire field of view. The ability to measure the dynamic molecular organization of biological structures greatly expanded the imaging capabilities in the field. This allowed the Pol-Scope to be used for imaging structures previously not visible above noise, such as single microtubules (Oldenbourg et al., 1998; Tran et al., 1995).
Although extensively studied with polarized light microscopy, mitotic spindles are not the only biological structure, whose architectural dynamics was revealed by the technique. Insight into the organization of septin higher order structures has resulted from polarized fluorescence imaging. Unlike microtubules, which contribute the lion share to the birefringence of the mitotic spindle, higher order septin structures are one of many components that contribute to the birefringence measured near the cell surface. Therefore, yeast and other cells were genetically manipulated to co-express septin with green fluorescent protein (GFP) whose dipole orientation was then used to indicate the ensemble molecular organization of septin structures (DeMay et al., 2011a; DeMay et al., 2011b; Vrabioiu and Mitchison, 2007). With the need to label septin structures to measure molecular organization, the dipole orientation of GFP was exploited to create a “read-out” for septin organization in vivo. Using the dipole orientation of GFP as a marker for the orientation of target molecules is a technique that can be applied to various cellular structures and model organisms.
The OpenPolScope software (www.openpolscope.org) was developed to meet the need to better understand submicroscopic molecular organization of biological and chemical samples across various fields.. The goal of the open access software is to provide modern techniques for polarized light microscopy throughout the scientific fields and communities. With easily accessible software, and user-friendly interface, OpenPolScope promotes interdisciplinary collaboration and further development of acquisition and analysis capabilities.
Critical Parameters and Troubleshooting
Sensitivity
The optimized system will have the ability to detect small differences in signal intensity between the different polarization angles of light. The highest sensitivity will be achieved by carefully considering all hardware components, including the magnification and NA of the objective lens, sensitivity of the CCD or other detector, efficiency of the fluorescence filter cube, and performance of the light source. As for most applications in fluorescence microscopy, using a system that optimally excites and harvests photons from the signal of interest with minimal background will yield the most robust analysis of anisotropy of cellular structures.
Speed and dynamic structures
The rotating filter wheel of linear polarizers will be slower than the LC in switching polarization orientations, therefore it is critical to consider any movement in the sample being analyzed that could occur between frames during acquisition. If the object moves, this can lead to spurious anisotropy that is a motion artifact. This will result from changes in fluorescence intensity that are not due to prevailing orientations of GFP molecules that are optimally exciting/emitting at some polarization angle. Instead, the changes are due to the lateral motion of a fluorescent object that was present and absent in images associated with different polarization settings and therefore appears to have a polarization dependent fluorescence.
Resolution
While implementing polarized fluorescence on a widefield microscope, it is critical to keep in mind that the anisotropy and orientation calculated for a given pixel represents an ensemble of molecules and thus is the average orientation of all the molecules. For example, in the analysis of septins, the measured orientation was almost always orthogonal to the cell axis regardless of using different septin-conGFP constructs that should place the GFP at a variety of different positions relative to the septin filament axis. Through known structures of septins and modeling, we have inferred that symmetry in the septin structure leads to two supplementary angles being formed in each subassembly (DeMay et al., 2011b) that can be any number of unique angles but they always average to 90 degrees or zero degrees. Using multiple different constrained constructs (discussed below) can help clarify if there is underlying symmetry in the organized assembly that is being averaged or if the molecules are in fact all oriented the same way in, for example, a polarized structure. Alternatively, imagine two orthogonal arrays of cytoskeletal filaments that are equal in proportions. These populations, while ordered, would appear isotropic because at the resolution of the microscope, the two populations of orientations will “cancel” each other out and mask both of the actual underlying orientations.
Engineering rigid probes
An absolutely critical component to successfully implementing polarized fluorescence microscopy is to design fluorescent proteins or dyes that are sufficiently constrained so as to report a fixed orientation. In some cases, the structures of interest are sufficiently ordered that engineering a constrained tag is not necessary because the FP is sufficiently confined simply by the architecture of the assembly. In many cases, probes will need to be engineered that are sufficiently rigid that the orientation of the dipole can actually be captured and measured on a reasonable timescale for conventional cameras and microscopes. A few tricks for designing constrained tags can be gleaned from septins (DeMay et al., 2011a; DeMay et al., 2011b; Vrabioiu and Mitchison, 2007), but for any new protein of interest, sufficient constraining will need to be empirically determined and pathway specific controls will need to be performed for functionality. For septins, trimming 3–4 amino acids from the N-terminus of GFP and fusing this to the C-terminal predicted coiled-coil (CC) makes a sufficiently constrained construct. A series of variants that sequentially trim off individual residues of the CC, which would be predicted to move the GFP by ~100 degrees assuming an alpha helical secondary structure, were made in hopes of determining the precise position of the GFP in space. This analysis was confounded by the symmetry issues of septin filaments as mentioned above, however were still informative when combined with modeling.
If there is structure information known, the internal insertion of GFP, sandwiched within the peptide, in a region that is constrained by other aspects of the secondary and tertiary structure of the protein is likely to be sufficiently constrained. Placing tags in different positions in the protein combined with structural information and modeling can bring powerful insight into structural rearrangements that may happen in vivo and not be detectable using conventional microscopy approaches.
Anticipated Results
When imaging biological structures with unknown organizations, be mindful that each pixel represents an ensemble of molecules (see Resolution section under Critical Parameters and Troubleshooting). The anisotropy and orientation of the measured values can be affected by several factors depending on the underlying organization of the biological structure. For example, if multiple, but distinctly, ordered populations of molecules exist, but are disproportionate in size, the measured anisotropy and orientation values will reflect the dominant population, and not capture the secondary, minority population even if it is highly ordered. Alternatively, the presence of an ordered and disordered population within the same pixel will result in decreased anisotropy and lead to an underestimation of the degree of order in the oriented population. Finally, as noted above, two highly ordered assemblies that are orthogonal to one another may appear isotropic despite being highly ordered. It can be useful in such a situation to examine the structure from a different viewpoint to potentially capture the molecules in an arrangement where the signals from the two populations do not interfere with one another. For the yeast septin ring, this view was a cross section of the ring compared to an en face view of the hourglass structure. Alternative views can also be important for accurate modeling.
Time Considerations
The time necessary to carry out polarized fluorescence imaging is highly dependent on the model organism being used and the biological process being imaged. In the case of imaging the reorientation process of septins in S. cerevisae no more than one and a half hours is needed for a single cell (the doubling time of S. cerevisae). Depending on the model system being used and the biological process of interest imaging may require more or less time.
Figure1.
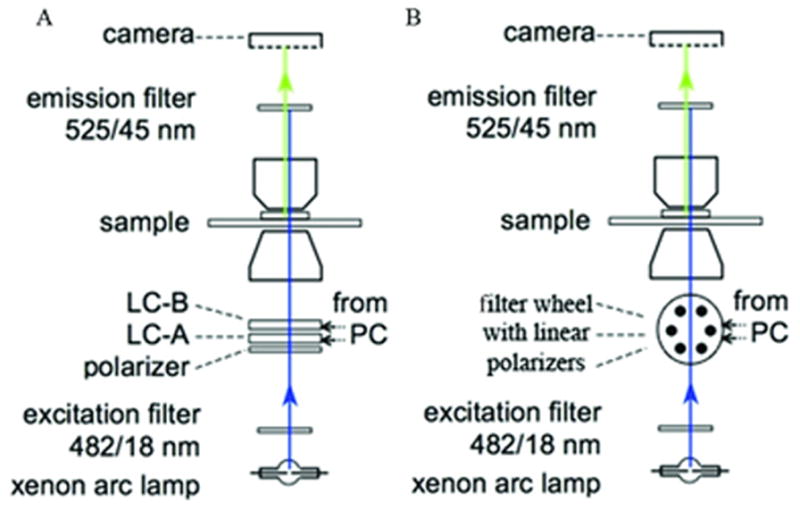
Schematic of polarized fluorescence microscope light path. Light path set up for polarized fluorescence imaging using an LC (A) or linear polarizers (B). Adapted from DeMay et al., 2011b.
Acknowledgments
The authors acknowledge support by the Marine Biological Laboratory through fellowships to the Human Frontier Science Program fellowship LT000096/2011-C to SBM, NSF grant MCB 1212400 awarded to ASG, Lemann, Spiegel, and Colwin MBL fellowships to ASG, and Nikon Research Award to ASG, NIH grant EB002583 awarded to RO.
The development of the OpenPolScope software was started in 2010 during a sabbatical of RO in the laboratory of Ron Vale at UCSF. The OpenPolScope team at MBL (AV, SBM, RO) thanks Arthur Edelstein and Nico Stuurman at UCSF for their input throughout the project.
Footnotes
INTERNET RESOURCES
www.openpolscope.org: Open-access software for polarized light microscopy imaging and analysis.
www.micro-manager.org: Open-access imaging software for microscope automation (Edelstein et al., 2010).
rsb.info.nih.gov/ij/: Public domain image processing software.
http://openpolscope.org/pages/Downloads.htm: OpenPolScope protocol and downloads.
Literature Cited
- DeMay BS, Bai X, Howard L, Occhipinti P, Meseroll RA, Spiliotis ET, Oldenbourg R, Gladfelter AS. Septin filaments exhibit a dynamic, paired organization that is conserved from yeast to mammals. The Journal of cell biology. 2011a;193:1065–1081. doi: 10.1083/jcb.201012143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMay BS, Noda N, Gladfelter AS, Oldenbourg R. Rapid and quantitative imaging of excitation polarized fluorescence reveals ordered septin dynamics in live yeast. Biophysical journal. 2011b;101:985–994. doi: 10.1016/j.bpj.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein A, Amodaj N, Hoover K, Vale R, Stuurman N. Computer control of microscopes using μManager. Current protocols in molecular biology. 2010:14.20. 11–14.20. 17. doi: 10.1002/0471142727.mb1420s92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher N. Statistical analysis of circular data. Cambridge University Press; Cambridge: 1993. [Google Scholar]
- Inoué S. The effect of colchicine on the microscopic and submicroscopic structure of the mitotic spindle. Exp Cell Res Suppl. 1952;2:305–318. [Google Scholar]
- Inoué S. Polarization optical studies of the mitotic spindle. Chromosoma. 1953;5:487–500. doi: 10.1007/BF01271498. [DOI] [PubMed] [Google Scholar]
- Inoué S. Organization and function of the mitotic spindle. In: Allen RD, Kamiya N, editors. Primitive Motile Systems in Cell Biology. Vol. 549. Academic Press, Inc; New York: 1964. p. 598. [Google Scholar]
- Inoué S, Bajer A. Birefringence in endosperm mitosis. Chromosoma. 1961;12:48–63. doi: 10.1007/BF00328913. [DOI] [PubMed] [Google Scholar]
- Inoué S, Dan K. Birefringenoe of the dividing cell. Journal of Morphology. 1951;89:423–455. [Google Scholar]
- Inoué S, Hyde WL. Studies on depolarization of light at microscope lens surfaces II. The simultaneous realization of high resolution and high sensitivity with the polarizing microscope. The Journal of biophysical and biochemical cytology. 1957;3:831–838. doi: 10.1083/jcb.3.6.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta SB, Shribak M, Oldenbourg R. Polarized light imaging of birefringence and diattenuation at high resolution and high sensitivity. Journal of Optics. 2013;15:094007. doi: 10.1088/2040-8978/15/9/094007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenbourg R, Mei G. New polarized light microscope with precision universal compensator. Journal of microscopy. 1995;180:140–147. doi: 10.1111/j.1365-2818.1995.tb03669.x. [DOI] [PubMed] [Google Scholar]
- Oldenbourg R, Salmon E, Tran P. Birefringence of single and bundled microtubules. Biophysical journal. 1998;74:645–654. doi: 10.1016/S0006-3495(98)77824-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runnström J. Die Veränderungen der Plasmakolloide bei der Entwicklungserregung des Seeigeleies. Protoplasma. 1928;4:388–514. [Google Scholar]
- Salmon E. Spindle Microtubules: Thermodynamics of in vivo assembly and role in chromosome movement*. Annals of the New York Academy of Sciences. 1975;253:383–406. doi: 10.1111/j.1749-6632.1975.tb19216.x. [DOI] [PubMed] [Google Scholar]
- Sato H, Ellis GW, Inoué S. Microtubular origin of mitotic spindle form birefringence. Demonstration of the applicability of Wiener’s equation. The Journal of cell biology. 1975;67:501–517. doi: 10.1083/jcb.67.3.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt W. Die Bausteine des Tierkörpers in polarisiertem Licht. Bonn 1924 [Google Scholar]
- Schmidt WJ. Die Doppelbrechung von karyoplasma, zytoplasma und metaplasma. Vol. 11. Gebrüder Borntraeger; 1937. [Google Scholar]
- Swann M, Mitchison J. Refinements in polarized light microscopy. Journal of Experimental Biology. 1950;27:226–237. doi: 10.1242/jeb.27.2.226. [DOI] [PubMed] [Google Scholar]
- Tran P, Salmon E, Oldenbourg R. Quantifying single and bundled microtubules with the polarized light microscope. The Biological Bulletin. 1995;189:206–206. doi: 10.1086/BBLv189n2p206. [DOI] [PubMed] [Google Scholar]
- Vrabioiu AM, Mitchison TJ. Symmetry of septin hourglass and ring structures. Journal of molecular biology. 2007;372:37–49. doi: 10.1016/j.jmb.2007.05.100. [DOI] [PubMed] [Google Scholar]