

Abstract
The multivariate nature of cancer necessitates multi-targeted therapy, and kinase inhibitors account for a vast majority of approved cancer therapeutics. While acute promyelocytic leukemia (APL) patients are highly responsive to retinoic acid (RA) therapy, kinase inhibitors have been gaining momentum as co-treatments with RA for non-APL acute myeloid leukemia (AML) differentiation therapies, especially as a means to treat relapsed or refractory AML patients. In this study GW5074 (a c-Raf inhibitor) and PP2 (a Src-family kinase inhibitor) enhanced RA-induced maturation of t(15;17)-negative myeloblastic leukemia cells and rescued response in RA-resistant cells. PD98059 (a MEK inhibitor) and Akti-1/2 (an Akt inhibitor) were less effective, but did tend to promote maturation-uncoupled G1/G0 arrest, while wortmannin (a PI3K inhibitor) did not enhance differentiation surface marker expression or growth arrest. PD98059 and Akti-1/2 did not enhance differentiation markers and have potential, antagonistic off-targets effects on the aryl hydrocarbon receptor (AhR), but neither could the AhR agonist 6-formylindolo(3,2-b)carbazole (FICZ) rescue differentiation events in the RA-resistant cells. GW5074 rescued early CD38 expression in RA-resistant cells exhibiting an early block in differentiation before CD38 expression, while for RA-resistant cells with differentiation blocked later, PP2 rescued the later differentiation marker CD11b; but surprisingly, the combination of the two was not synergistic. Kinases c-Raf, Src-family kinases Lyn and Fgr, and PI3K display highly correlated signaling changes during RA treatment, while activation of traditional downstream targets (Akt, MEK/ERK), and even the surface marker CD38, were poorly correlated with c-Raf or Lyn during differentiation. This suggests that an interrelated kinase module involving c-Raf, PI3K, Lyn and perhaps Fgr functions in a nontraditional way during RA-induced maturation or during rescue of RA induction therapy using inhibitor co-treatment in RA-resistant leukemia cells.
Keywords: Retinoic acid, Leukemia, Resistance, Inhibitors, c-Raf, Lyn
1. Introduction
Two pathways that are extensively targeted via kinase inhibitors are the Raf/MEK/ERK (MAPK) and PI3K/Akt signaling cascades, which are known to be activated (often simultaneously) by growth factor, cytokine or hormone stimulation, and are frequently subject to deregulation in malignant cells [1]. The MAPK pathway, once thought to be a three-tiered chute for transducing membrane-initiated growth factor signaling, can relay complex signals that promote proliferation, mitosis, differentiation, apoptosis, motility or other cell-specific functions. Potent MAPK inhibitors cannot function as stand-alone treatments since too many processes depend on this pathway. Meanwhile PI3K/Akt activation is associated with pro-survival and anti-apoptotic signaling. There is significant interest in employing PI3K [2] and Akt [3] inhibitors to overcome resistance and re-sensitize cells to apoptosis-inducing agents.
Retinoic acid (RA) is a morphogenic compound and dietary factor that exerts pro-differentiative and anti-proliferative effects in normal and malignant contexts, including breast, lung and prostate cancers [4,5]. Yet as a cancer therapeutic, RA falls short of its initial success as a potent differentiation induction therapy for acute promyelocytic leukemia (APL, FAB M3), in which it induces remission in 80–90% of all cases [6]. There is great interest in combining RA with other chemical agents, such as other differentiation-inducing compounds, conventional chemotherapies, or kinase inhibitors, to both enhance efficacy and overcome emergent RA resistance in non-APL acute myeloid leukemia (AML). Current clinical trials assess the effect of RA combined with kinase inhibitors such as dasatinib, HDAC inhibitors, or inhibitors of lysine-specific demethylase 1 (for example: NCT00892190, NCT00867672, NCT00995332, NCT02261779). Here, we tested the effects of PD98059 (MEK inhibition), GW5074 (c-Raf inhibition), wortmannin (PI3K inhibition), Akti-1/2 (Akt inhibition) and PP2 (Lyn inhibition) during RA-induced maturation in the non-APL AML patient-derived myeloblastic leukemia (FAB M2) cell line HL-60 [7,8] and two RA-resistant sublines.
The patient-derived HL-60 cell line (FAB M2) lacks the PML-RARα fusion protein pathognomonic for APL, rendering HL-60 an attractive model for investigating RA-induced mechanisms in a t(15;17)-negative context [9]. RA-treated HL-60 cells undergoing differentiation display increased CD38 and CD11b expression and G1/G0 cell cycle arrest. With a doubling time of 20–24 h, RA-treated HL-60 cells complete two cell divisions and are committed to granulocytic differentiation by 48 h [10,11]. Sustained activation of the c-Raf/MEK/ERK cascade persists for 48 h and beyond after RA treatment [12]. However, inhibiting MEK after RA-induced HL-60 that has completed one division cycle does not inhibit RA-induced differentiation [13], indicating that sustained MEK or ERK activation may be necessary only during the lineage-uncommitted, priming phase and not for the second (lineage commitment) division. c-Raf phosphorylation concurrent with reduced MEK/ERK activation is known [14–18].
Despite the sustained ERK activation that occurs in RA-treated HL-60 cells, noncanonical c-Raf function has emerged as a hallmark of this system. c-Raf propels RA-induced differentiation [12,19] but induced phosphorylation at the c-Raf activating sites S338 and Y340/Y341 cannot be detected in RA-treated HL-60 [20,21]. Instead, phosphorylation occurs at the S259 putative inhibitory site [22], the S621 putative stability site [23] and the S289/296/301 c-Raf sites [24,25]. The S289/296/301c-Raf sites are targets of ERK, but whether these sites are inhibitory [26] or activating [27] remains unclear (see Discussion). pS621c-Raf undergoes nuclear translocation and interacts with transcription factors in RA-induced HL-60 cells [20,28]. Phosphorylated S259 has been shown specifically to prevent c-Raf membrane localization [29,30] and may promote Ras-independent and membrane-independent functions of c-Raf.
An RA-inducible interaction between the Src-family kinase (SFK) Lyn and pS259c-Raf was reported by Congleton et al. (2012) [24]. Lyn and Fgr are the predominant SFKs in myeloid cells [31,32] and both are upregulated by RA treatment in HL-60 cells [11,24]. Lyn displays RA-inducible phosphorylation at both its activating SFK Y416 site, which on Lyn is Y396 [24,33] and its inhibitory Y507 site [34]. Meanwhile Fgr is not phosphorylated at the SFK Y416 site (Y400 on Fgr) after RA treatment [24], implicating Lyn as the primary active SFK in RA-treated HL-60, as well as in NB4 cells [35]. We recently reported that Lyn and PI3K exhibit RA-inducible interaction and phosphorylation, and both bind to the RA-upregulated surface marker CD38, which is known to propel differentiation [36]. A Lyn/PI3K interaction in the context of differentiation has been shown [36,37]. Thus despite the attractiveness of PI3K inhibitors to diminish cell survival signaling, PI3K inhibition may have a negative effect on RA-induced maturation. Akt is the downstream effector of PI3K, and interestingly, Akt is able to phosphorylate c-Raf at S259 [38].
We previously developed two sequentially emergent RA-resistant HL-60 cells by chronic RA-exposure. Both fail to upregulate CD11b expression, G1/G0 arrest and signaling factor expression/activation after RA treatment [25]. However, one RA-resistant cell line retains RA-inducible CD38 expression (R38+ HL-60) while the other has lost this marker as well (R38− HL-60). The SFK inhibitor PP2, which enhances RA-induced differentiation in wild-type HL-60 and NB4 cells [24,39], rescues differentiation in both R38+ and R38− RA-resistant HL-60 cells [25]. Using the kinase inhibitors PD98059, GW5074, wortmannin, and Akti-1/2, we found that the c-Raf inhibitor GW5074 also emerges as an augmenter of RA-induced differentiation in wild-type and RA-resistant HL-60 cells, and had a similar effect to PP2 as reported previously. In RA-resistant cells, GW5074 was more capable in rescuing early events in induced differentiation, whereas PP2 was more capable for rescuing late events, however contrary to expectation, the two were not synergistic.
2. Materials and methods
2.1. Cell lines and treatments
HL-60 cells, derived from the original patient isolates, were a generous gift of Dr. Robert Gallagher and maintained in this laboratory. Two retinoic acid (RA)-resistant HL-60 sublines (R38+ and R38−) were isolated as described previously [25]. Cell cultures were maintained in 1640 RPMI medium (Invitrogen, Carlsbad, CA) supplemented with heat-inactivated 5% fetal bovine serum (FBS; Hyclone, Logan, UT) and 1% antibiotic/antimycotic (Invitrogen) and grown at 37 °C in a 5% CO2 humidified environment. Cells were seeded at 0.2 × 106 for 48 h experiments or at 0.1 × 106 for 72 h experiments. Cell viability was monitored via Trypan blue exclusion. All-trans-retinoic acid (RA; Sigma, St. Louis MO) was added from a stock solution of 5 mM in ethanol to cell cultures at a final concentration of 1 μM. PD98059 (Cell signaling, Danvers MA) was added from a 10 mM stock solution in DMSO to a final concentration of 2 μM in culture. GW5074 (Sigma) was added from a 10 mM stock solution in DMSO to a final concentration of 2 μM in culture. Wortmannin (Calbiochem/EMD Chemicals, San Diego CA) was added from a 5 mM stock solution in DMSO to a final concentration of 1 μM in culture. Akti-1/2 (Calbiochem) was added from a 10 mM stock solution in DMSO to a final concentration of 1 μM in culture. PP2 (Calbiochem), obtained as a 10 mM solution in DMSO, was added to cultures at a final concentration of 10 μM. The AhR agonist 6-Formylindolo(3,2-b)carbazole (FICZ; Enzo Life Sciences, Exeter, United Kingdom), was added from a 100 μM stock solution in DMSO to a final concentration of 100 nM in culture.
2.2. CD38 and CD11b quantification
0.5 × 106 cells were centrifuged at 700 rpm for 5 min, and cell pellets were resuspended in 200 μl PBS containing 2.5 μl of phycoerythrin (PE)-conjugated anti-CD38 antibody and allophycocyanin (APC)-conjugated anti-CD11b antibody (BD Pharmingen, San Jose CA). Samples were incubated at 37 °C for 1 h, then analyzed by flow cytometry on a BD LSRII flow cytometer (BD Biosciences, San Jose CA). APC fluorescence (excitation using 633 nm, red laser) was collected with a 735 nm dichroic longpass and 660/20 nm bandpass filter. PE fluorescence (excitation at 488 nm, blue laser) was collected with a 550 nm dichroic longpass and 576/26 nm bandpass filter. Gates for untreated controls were set to exclude 95% of the live cell population peak.
2.3. G1/G0 arrest quantification
0.5 × 106 cells were centrifuged at 700 rpm for 5 min, and cell pellets were resuspended in 200 μl of cold (4 °C) hypotonic propidium iodide (PI) staining solution containing 50 μg/ml propidium iodide, 1 μl/ml Triton X-100, and 1 mg/ml sodium citrate in PBS. Samples were incubated overnight at 4 °C. Nuclei fluorescence was then analyzed on a BD LSRII using 488 nm excitation and collected with a 550 nm dichroic longpass and 576/26 nm bandpass filter. Doublets were excluded from the analysis.
2.4. Western blotting
15 × 106 cells were washed twice with PBS via centrifugation at 700 rpm for 5 min. For total lysate collection, cell pellets were resuspended in 200–350 μl M-PER lysis buffer (Thermo Scientific, Rockford IL) supplemented with protease and phosphatase inhibitor cocktails (Sigma) at 1:100 volume ratio. After incubation on ice for 30 min, lysates were cleared via centrifugation for 30 min at 13,000 rpm and DNA pellets were discarded. For collection of cytoplasmic and nuclear fractions, cell pellets were resuspended in 200 μl CER I buffer from the NE-PER extraction kit (Thermo Scientific), followed by subsequent kit instructions. The BCA Protein Assay kit (Thermo Scientific) was used to assess protein yield of cell lysates. Equal amounts of protein lysates were resolved by SDS-PAGE using 12% polyacrylamide gels (Bio-Rad, Hercules CA). Resolved lysate was then transferred onto PVDF membrane (Millipore, Billerica MA), followed by blocking with 5% dry milk w/v in PBS-Tween. Antibody dilutions were prepared as a 1:1000 dilution in 5% BSA w/v PBS-Tween. Blots were incubated with primary antibody overnight at 4 °C, washed and incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h at room temperature. The primary antibodies were specific against ERK1/2, phospho(T202/Y204)-ERK1/2, MEK1/2, phospho(T217/221)-MEK1/2, Raf1, phospho(S259)-Raf1, phospho(S289/296/301)-Raf1, Lyn, Fgr, pan-phospho(Y416)-SFK, phospho(Y507)-Lyn, Slp76, Vav1, phospho(Y458)-p85 PI3K, Akt, phospho(S473)-Akt, phospho(T308)-Akt, GAPDH, TBP (all from Cell Signaling, Danvers, MA), c-Cbl, AhR (Santa Cruz Biotechnol-ogy, Santa Cruz CA) and pS621c-Raf (Thermo Scientific).
2.5. Statistical analysis
p-Values between treatment group means were calculated using ANOVA within GraphPad software. Repeat Western blot data were quantified using ImageJ. The Pearson correlation coefficient between average quantified blot data is calculated following the formula
| (1) |
which determines the covariance cov(x, y) of two monotonically increasing/decreasing variables divided by the product of their standard deviations σxσy. ρ(x, y) > 0 indicates a positive correlation, ρ (x, y) = 0 indicates no correlation, and ρ (x, y) < 0 indicates a negative correlation, with ρ (x, y) = 1 or ρ (x, y) = −1 indicating an exact positive or negative correlation, respectively. For clustering analysis, the distances between variables within a dataset are ranked using an average linkage method, which follows the formula
| (2) |
where D(x, y) is the distance metric, in this case 1 − (x, y). Pearson correlation coefficients and hierarchical clustering analysis using the average linkage method were performed in MATLAB.
3. Results
3.1. Phenotypic maturation in response to retinoic acid and four kinase inhibitors
We first examined the effects of combined retinoic acid (RA) and kinase inhibitor treatment on phenotypic differentiation markers in wild-type HL-60 and two RA-resistant HL-60 cell lines, R38+ and R38−. Phenotypic changes were assessed at 48 h, which for this system is after onset of terminal differentiation [10,11]. Wild-type HL-60 exhibits increased CD38 and CD11b expressions and G1/G0 cell cycle arrest 48 h after RA treatment (Fig. 1A–C). R38+ RA-resistant cells display RA-inducible CD38 expression, an early differentiation marker, but do not have increased CD11b, a later differentiation marker, or G1/G0 arrest. R38− RA-resistant HL-60 cells fail to upregulate all three of these markers (CD38, CD11b, G1/G0 arrest) after RA treatment. The inhibitors PD98059 (MEK inhibition), GW5074 (c-Raf inhibition), wortmannin (PI3K inhibition) or Akti-1/2 (Akt inhibition) were then added with RA in co-treatments.
Fig. 1.

Phenotypic markers during RA and kinase inhibitor co-treatment in HL-60. Wild-type (WT), R38+ and R38−HL-60 cells were treated for 48 h with 1 μM RA, or RA combined with 2 μM PD98059 (PD), 2 μM GW5074 (GW), 1 μM wortmannin (Wo), or 1 μM Akti-1/2 (Akti) and analyzed by flow cytometry for (A) CD38 expression, (B) CD11b expression, or (C) G1/G0 cell cycle arrest. Error bars represent standard error. Asterisks for p-values of treatment group means compared to control indicate whether p < 0.0001 (****), p < 0.001 (***), p < 0.01 (**) or p < 0.05 (*). (A) CD38 is maximally expressed in WT and R38+ HL-60 and not inhibited by any kinase inhibitor treatment. GW5074 significantly increase CD38 expression in R38−cells. (B) CD11b is increased by RA and enhanced by GW5074 in WT HL-60 cells, while all other co-treatments reduced the RA-induced CD11b expression. GW5074 can significantly increase CD11b expression in R38+ and R38−. (C) GW5074 can enhance RA-induced G1/G0 arrest in WT HL-60 and rescue G1/G0 arrest in R38+ and R38−. Akti-1/2 and PD98059 also tended to increase G1/G0 arrest.
Cultures were treated with RA and PD98059 at a non-toxic concentration determined previously [12]. CD38, an early marker, is maximally expressed after 48 h of RA treatment in both wild-type and R38+ HL-60, but adding PD98059 failed to upregulate CD38 in the R38− cells (Fig. 1A). As previously reported [28], PD98059 reduced the RA-induced CD11b expression in wild-type HL-60, an indication of diminished maturation (Fig. 1B). Concordantly, CD11b expression (which is not induced by RA in the two RA-resistant HL-60 cell lines) remained unchanged by PD98059 + RA treatment. However, PD98059 + RA did result in slightly enhanced maturation-uncorrelated G1/G0 arrest (see Section 4) in wild-type and R38+ HL-60 cell lines (Fig. 1C).
The c-Raf inhibitor GW5074 had unexpected results. Co-treatment with this inhibitor strongly induced CD38 expression in the R38− cells. This is the greatest rescue of CD38 expression that we have seen in R38− cells compared to other treatments that have also increased CD38, such as 1,25-dihydroxyvitamin D3 [11] and PP2 [25]. GW5074 co-treatment seemed less capable at increasing CD11b expression in R38−, suggesting that the drug could target early but not late events of the defective differentiation program in R38− (Fig. 1A–B). In contrast, GW5074 greatly enhanced RA-induced CD11b expression in wild-type HL-60 cells (Fig. 1B). Combined RA and GW5074 treatment also lead to a significant increase in CD11b expression in R38+ cells. GW5074 + RA enhanced cell cycle arrest in wild-type HL-60, and to a lesser extent in the RA-resistant HL-60 cell lines (Fig. 1C). GW5074 was used at a concentration previously determined to not induce growth arrest when treated alone in wild-type HL-60 [21].
Dose response experiments with the PI3K inhibitor wortmannin in these cells indicated that concentrations as high as 2 μM do not enhance CD38 expression, CD11b expression or G1/G0 arrest (Supplemental fig. S1 A–B). Thus PI3K inhibition at 1 μM, which could reduce cell culture density (data not shown), could not promote RA-induced differentiation events in wild-type or RA-resistant HL-60 (Fig. 1A–C). Wortmannin + RA co-treatment had no effect on surface marker expression in RA-resistant HL-60 cells and reduced CD11b expression in RA-treated wild-type HL-60. Wortmannin + RA did not enhance G1/G0 arrest; in R38− wortmannin co-treatment actually decreased growth arrest.
Co-treatment with Akti-1/2 also did not promote surface marker expression, and reduced CD11b expression in RA-treated wild-type HL-60 (Fig. 1A–B). However, unlike wortmannin, Akti-1/2 + RA did cause a slight increase in G1/G0 cell cycle arrest in all three HL-60 cell lines (Fig. 1C). Dose response studies to Akti-1/2 indicated that doses as high as 10 μM induced G1/G0 growth arrest by 48 h in wild-type HL-60 cells, but CD38 and CD11b levels are not significantly affected with 5 μM Akti-1/2 alone (Supplemental fig. S1 C–E).
Overall wortmannin treatment emerged as the worst strategy for improving RA-induced maturation. PD98059 and Akti-1/2 could increase RA-induced G1/G0 arrest but could not improve surface marker expression and instead reduced the CD11b differentiation marker of RA-treated wild-type HL-60 cells. Combined GW5074 + RA slightly increased G1/G0 arrest, but to our surprise greatly enhanced CD38 expression in R38− HL-60 cells and enhanced CD11b expression in all three cell lines, the parental HL-60 and its progressively more resistant derivatives, R38+ and R38−. Thus in addition to the Src-family kinase inhibitor PP2 reported previously [25], GW5074 emerges as a kinase inhibitor effective at upregulating RA-inducible maturation-specific events in wild-type and RA-resistant HL-60 cells.
3.2. Correlation between signaling factors during induction with retinoic acid and five kinase inhibitors
We next investigated the trends in expression and activation of several kinases implicated in regulating RA-induced maturation. A diagram of a potential interrelated kinase module involving these factors, curated from literature and our own reports, is depicted in Fig. 2A. We assessed the cytoplasmic and nuclear expressions and activation of these kinases, which include c-Raf, MEK, ERK, p85 PI3K, Akt, Lyn, Fgr, c-Cbl and the transcription factor AhR. Wild-type, R38+ and R38− cells were treated with RA alone or with RA combined with one of the five inhibitors: PD98059, GW5074, wortmannin, Akti-1/2 or PP2. We assessed nuclear and cytoplasmic expressions and phosphorylation events at 48 h after RA addition, the time at which onset of terminal differentiation occurs in the wild-type cells.
Fig. 2.
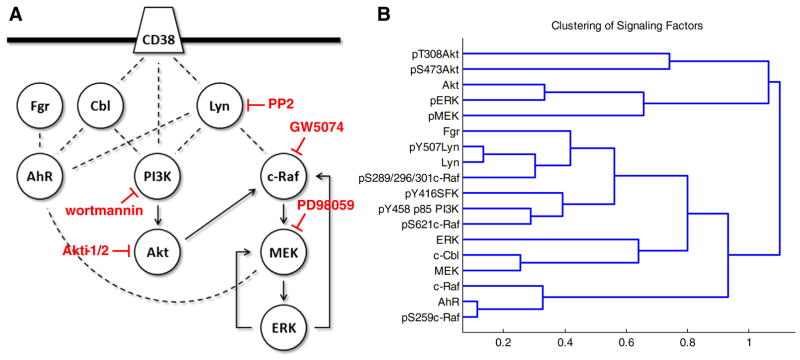
Signaling network of factors investigated in this study and hierarchical clustering. (A) Simplified diagram of the interactions and functional relationships between the proteins investigated in this study, curated from literature: Bunaciu and Yen 2013 Mol Can; Bunaciu et al. submitted; Yim et al. 2004 Biochem Biophys Res Commun; Congleton et al. 2012 Leukemia; Congleton et al. 2014 Cell Signal; Bunda et al. 2014 Oncogene; Dhillon et al. 2007 Oncogene; Steelman et al. 2011 Aging; Zimmermann and Moelling 1999. The targets affected by the inhibitors used in this study are indicated. (B) Repeat Western blot data was quantified using ImageJ and subject to hierarchical clustering analysis using the Pearson correlation coefficient as a distance metric and an average linkage method (see Section 2). Data includes both cytoplasmic and nuclear signaling factor expression and phosphorylation for wild-type (Supplemental figs. S2–7), R38+ (Supplemental figs. S2–7) and R38− (Supplemental figs. S2–7) treated with 1 μM RA or RA combined with 2 μM PD98059, 2 μM GW5074, 1 μM wortmannin, 1 μM Akti-1/2 or 10 μM PP2. Distances between clusters (1 — Pearson correlation coefficient) are indicated on the x-axis.
Repeat Western blot data for wild-type, R38+ and R38− HL-60 cells were quantified using ImageJ (Supplementary figs. S2–7). Repeat averages across all cell lines and treatments were subject to hierarchical clustering using the Pearson correlation coefficient as a distance metric and an average linkage method (Fig. 2B). Interestingly, total Akt expression, Akt phosphorylation at S473 and T308, and activated MEK (pMEK) and ERK (pERK) were all grouped into a cluster that was far removed from the rest of the proteins, which formed a large separate cluster (Fig. 2B). While total Akt expression levels were detectable, expression remained unchanged across combination treatments and Akt phosphorylation was also unchanged, and sometimes undetectable (Supplemental figs. S2–7). One exception to this was S473 Akt phosphorylation which was strongly induced by PP2 + RA in wild-type HL-60 (Supplemental fig. S2–7). Although cytoplasmic pERK decreased with PP2 + RA or GW5074 + RA treatments in RA-resistant HL-60 (Supplemental figs. S2–7), GW5074 sometimes induced ERK activation in wild-type HL-60 (Supplemental fig. S2–7).
In the large cluster containing the remainder of the signaling factors, the least correlated proteins from the rest of the group were AhR, c-Raf and pS259c-Raf (Fig. 2B), although these were highly correlated to each other. The next cluster down was separated into two branches, one of which contained proteins that were strongly cytoplasmic: c-Cbl, total MEK and total ERK. The next cluster embodies the Src-family kinases, c-Raf phosphorylated at either S621 or the S289/296/301 sites, and the active form of the regulatory subunit (pY458 p85) of PI3K. The Src-family kinase Lyn is highly correlated with phosphorylation at its Y507 inhibitory site, followed by pS289/296/301c-Raf and then Fgr. On a separate branch, pS621c-Raf, pY458 p85 PI3K and pY416SFK are grouped (Fig. 2B).
Overall the clustering reveals that certain c-Raf phosphorylation sites, Src-family kinases and phosphorylated PI3K are highly correlated signaling events, while active MEK/ERK and Akt are far removed from these events. This is somewhat surprising as both Akt and MEK/ERK supposedly lie downstream of the highly correlated factors (i.e. c-Raf and PI3K). This suggests that an interrelated kinase module involving c-Raf, PI3K, Lyn and Fgr functions in a nontraditional way during RA-induced maturation or during rescue of RA induction using inhibitor co-treatment.
3.3. Combined PP2 and GW5074 in RA-treated RA-resistant HL-60 cells
Among the agents tested, PP2 and GW5074 had the most prominent effects enhancing RA-induced differentiation, exhibiting different strengths for potentially different parts of the differentiation program, hence suggesting the possibility of synergistic effects. We next assessed how effectively combination GW5074 and PP2 treatment could rescue RA-induced maturation in the RA-resistant HL-60 cell lines R38+ and R38−. Wild-type HL-60 was not assessed since combined PP2 + RA treatment greatly maximizes all three phenotypic markers [24]. For clarity and comparison, the effects of PP2 and PP2 + RA treatment previously reported [25] are recapitulated here alongside GW5074, GW5074 + RA, PP2 + GW5074, and PP2 + GW5074 + RA for both R38+ and R38− RA-resistant HL-60 cells.
Surprisingly the combined use of PP2 and GW5074 with RA did not greatly enhance RA-induced differentiation compared to using a single agent in combination with RA. A striking result, however, was that GW5074 can greatly induce CD38 expression in R38− cells, even without RA (Fig. 3A) and even though CD38 is strongly regulated by a first intron RARE [40,41]. PP2 also upregulates CD38 to a lesser extent, while GW5074 or PP2 combined with RA further enhances CD38 expression compared to without RA. The GW5074-elicited response for CD38, an early differentiation marker, did not extend to the later differentiation marker CD11b. CD11b was weakly induced by GW5074 + RA in R38+ but not at all in R38− cells, consistent with progressive resistance. Also consistent with the progressive resistance, R38+ and R38− responded differently to GW5074 versus PP2. In R38+ HL-60 cells, comparing PP2 + RA versus GW5074 + RA, both drugs induced similar CD38 expression, but GW5074 + RA-induced CD11b expression is lower than for PP2 + RA treatment (Fig. 3A). Furthermore, comparing PP2 + RA versus GW5074 + RA for the more profoundly resistant R38− HL-60 cells, GW5074 + RA results in significantly higher CD38 expression, but CD11b expression fails to be induced while PP2 + RA induces only low CD11b expression, consistent with its ability to drive later differentiation events.
Fig. 3.

Phenotypic markers during combined GW5074, PP2 and RA treatment. R38+ and R38− HL-60 cells were treated for 48 h with RA, or RA combined with GW5074 (GW) and/or PP2 and analyzed by flow cytometry for (A) CD38 and CD11b expressions and (B) G1/G0 cell cycle arrest. PP2-treated R38+ and R38− HL-60 cells have been reported previously [25] but are recapitulated here for clear comparison. Error bars represent standard error. Asterisks for p-values of treatment group means compared to control indicate whether p < 0.0001 (****), p < 0.001 (***), p < 0.01 (**) or p < 0.05 (*). (A) GW5074-induced CD38 expression in higher than PP2 but GW5074-induced CD11b is reduced in R38+ and R38− cells. Combined PP2 + GW5074 with or without RA enhances markers even more in R38+ but actually decreases marker expression compared to GW5074 alone in R38−. (B) GW5074 does not appear to greatly increase the PP2-induced G1/G0 arrest in either RA-resistant HL-60 cell line.
Treatment with all three chemicals (PP2 + GW5074 + RA) in R38+ cells results in similar or slightly enhanced CD38 and CD11b expression compared to either PP2 + RA or GW5074 + RA, but there was no prominent enhancement conferred by using both GW5074 + PP2. In R38− HL-60 cells, PP2 + GW5074 + RA treatment actually resulted in decreased CD38 expression compared to GW5074 + RA, but still higher than PP2 + RA. However, CD11b levels were unchanged between PP2 + RA and PP2 + GW5074 + RA treatments. The apparent PP2 inhibition of GW5074-promoted CD38 expression was not dependent on RA since in R38− HL-60, GW5074 + PP2 treatment resulted in lower CD38 expression than GW5074 alone. This indicates that, although PP2 can improve RA response, PP2 treatment actually blunts the pro-differentiation effects of GW5074 in R38− cells, but not in R38+ cells. PP2 may ergo conflict with the early pro-differentiation effects of GW5074. And potentially consistent with this notion, in R38+ cells, where the differentiation defect is thought to be late, PP2 has no inhibitory effect on GW5074-enhanced CD38 or CD11b expression.
Overall PP2 was more strongly correlated with CD11b expression than GW5074, and PP2 actually diminished GW5074 effects in R38− cells but not R38+ cells. GW5074 did not enhance the PP2 or PP2 + RA-induced G1/G0 cell cycle arrest in R38+ HL-60 or R38− HL-60 cells (Fig. 3B). We also checked combined PP2 + RA treatment with wortmannin or Akti-1/2, but these had very little effect on CD38 expression, CD11b expression or G1/G0 cell cycle arrest (Supplemental fig. S1 H–K).
3.4. Signaling factor expression during combined PP2 and GW5074 treatment in RA-treated RA-resistant HL-60 cells
The ability of GW5074 to modulate PP2 effects on phenotypic conversion of cells motivated the identification of the signaling correlates. We next explored the effect of GW5074 on the total expression and activation of signaling factors rescued in PP2− or PP2 + RA-treated RA-resistant HL-60 cells. We focused on c-Raf and Src-family kinase events. GW5074 tended to diminish PP2 or PP2 + RA-induced c-Raf expression and Lyn expression at 48 h, but had little to no effect on Fgr expression (Fig. 4A). GW5074 did tend to decrease PP2 or PP2 + RA-induced c-Raf phosphorylation at S621, but did not diminish PP2 or PP2 + RA-induced S259 or S289/296/301 c-Raf phosphorylation. Nor did GW5074 affect Y416 Src-family kinase (Lyn Y397) phosphorylation in either RA-resistant HL-60 cell line. GW5074 may have in fact alleviated PP2-reduced ERK activation. Looking at CD38-associated factors c-Cbl, Vav1, and Slp76, GW5074 had little to no effect on these proteins (Fig. 4B). In R38− HL-60 cells, GW5074 may have increased the PP2− and PP2 + RA-induced expression of p47phox, a member of the ROS production pathway [42].
Fig. 4.

Quantified signaling factor expression during combined GW5074, PP2 and RA treatment and correlation analysis. 48 h Western blot data (at least three repeats) of total lysates were quantified using ImageJ and fold change to respective R38+ or R38− control calculated. Error bars represent standard error. p-value analysis cannot be performed on quantified blot data, which is nonlinear. Instead a representative blot is displayed beneath each graph. GAPDH loading controls were performed to ensure even loading (not shown). (A) Signaling factors for c-Raf and Src-family kinase events and activated ERK were assessed for R38+ or R38− cell treated with PP2, PP2 + RA (reported previously in [25]), GW5074 (GW), GW + RA, PP2 + GW and PP2 + GW + RA. Since quantified data is estimated from immunoblot images in which signal detection may or may not have been in the linear range, p-value analysis is not applicable to quantified blot data. (B) Signaling factors for CD38-associated factors c-Cbl, Vav1 and Slp76, the p47phox protein. (C) Pearson correlation coefficient matrix between MAPK and Src-family kinase signaling factor events from Fig. 4A and phenotypic results from Fig. 3A, i.e. for R38+ and R38− across treatments with PP2, PP2 + RA, GW5074, GW5074 + RA, PP2 + GW5074 and PP2 + GW5074 + RA. Colorbar: white (1) indicates positive correlation, mid-gray (0) indicates no correlation, black (−1) indicates negative correlation. (D) Hierarchical clustering analysis of data presented in Fig. 4A using the Pearson correlation coefficient as a distance metric and an average linkage method (see Section 2). Distances between clusters (1 — Pearson correlation coefficient) are indicated on the x-axis.
We calculated the Pearson correlation coefficient between the quantified repeat Western blot data and phenotypic markers across both RA-resistant cell lines and all combinations of GW5074 and/or PP2 with or without RA (Fig. 4C). The most striking (strongest) correlation observed is that between Fgr and CD11b expression, followed by the correlation between pS289/296/301c-Raf and G1/G0 arrest. However, the c-Raf and other Src-family kinase events all appear to exhibit a similar degree of correlation to differentiation markers CD38, CD11b, and G1/G0 arrest. Once again the notable standout is pERK, which tended to be negatively correlated with the RA-induced maturation markers.
Clustering of the signaling factors presented in Fig. 4A using the Pearson correlation coefficient and an average linkage method reveals that Y416 Src-family kinase (Lyn Y397) phosphorylation and activated ERK are correlated (Fig. 4D), but separated from the remaining factors. PP2-induced but GW5074-reduced events are linked more closely (Lyn expression, c-Raf expression and S621 phosphorylation) while those least affected by GW5074 treatment (Fgr expression, and S259 and S289/296/301 c-Raf phosphorylation) are successively more distant.
3.5. An AhR agonist fails to rescue phenotypic maturation in RA-resistant HL-60
Since the aryl hydrocarbon receptor (AhR) i) functionally interacts with molecular constituents assayed here, ii) regulates these signaling molecules to drive differentiation, and iii) is linked to phosphorylated c-Raf as described here (Fig. 2B), we sought to determine if AhR activation could rescue RA-induced differentiation in the resistant cells. If we assume that PD98059 and Akti-1/2 can exert their effects through a mechanism that antagonizes AhR (see Section 4), we queried whether an AhR agonist like 6-Formylindolo(3,2-b)carbazole (FICZ) could rescue differentiation in RA-treated R38+ and R38−. FICZ enhances RA-induced maturation in wild-type HL-60 cells [34]. We found that FICZ does not significantly augment CD38 or CD11b expression in either R38+ or R38− RA-resistant HL-60 cells (Fig. 5A–B). FICZ also failed to rescue RA-inducible G1/G0 cell cycle arrest (two repeats, Fig. 5C).
Fig. 5.

Treatment of wild-type, R38+ and R38− HL-60 with the AhR agonist FICZ. Wild-type (WT), R38+ and R38− were treated for 48 h with RA or RA and the AhR agonist FICZ and analyzed by flow cytometry for (A) CD38 expression, (B) CD11b expression, or (C) G1/G0 cell cycle arrest. Asterisks for p-values of treatment group means compared to control indicate whether p < 0.0001 (****), p < 0.001 (***), p < 0.01 (**) or p < 0.05 (*). FICZ treatment does not significantly rescues (A) CD38 expression, (B) CD11b expression or (C) G1/G0 cell cycle arrest in either RA-resistant cell line.
4. Discussion
4.1. Summary of results
Retinoic acid-induced leukemic cell differentiation is regulated by an ensemble of signaling molecules, and we previously found that the Src-family kinase (SFK) inhibitor PP2 enhances RA-induced differentiation in wild-type HL-60 cells and rescues differentiation in RA-resistant HL-60 cells [25]. Here we probed for mechanistic insight and tested four additional kinase inhibitors in the context of RA-treated wild-type or RA-resistant HL-60 cells: PD98059 (MEK inhibition), GW5074 (c-Raf inhibition) wortmannin (PI3K inhibition), and Akti-1/2 (Akt inhibition). We showed that GW5074 was effective at upregulating CD38 expression in RA-treated RA-resistant HL-60 cells. PD98059, wortmannin and Akti-1/2, however, all blunted RA-induced CD11b expression in wild-type HL-60. PD98059 and Akti-1/2 were only effective insofar as they could increase growth arrest in all three HL-60 cell lines, while the PI3K inhibitor wortmannin could not increase G1/G0 arrest. Considering the ensemble of wild-type parental and emergent resistant cell responses to RA with five different inhibitors, hierarchical clustering reveals a tightly coupled signaling module that incorporates Lyn and phosphorylated c-Raf, and also phosphorylated PI3K (Fig. 2B) which may be downstream of Lyn [36]. Close coupling of Lyn and c-Raf has been shown previously [11,24]. Activation of ERK and Akt were much less correlated to this network (Fig. 2B), and this is corroborated in the analysis restricted to the resistant cells treated with PP2 and/or GW5074 (with or without RA) where pERK was negatively correlated to CD11b expression and growth arrest (Fig. 4C).
4.2. Kinase inhibitors for combination therapy
Inhibitor treatments can be complicated by off-target effects, combinatorial effects, and unintended activating effects (either direct or indirect). PD98059, for example, is a flavonoid that can act as an antagonist of aryl hydrocarbon receptor (AhR) at the same concentrations used to inhibit MEK [43]. AhR has been implicated as a positive regulator of RA-induced HL-60 differentiation [14,34]. Akti-1/2 is a highly selective, noncompetitive Akt inhibitor, but can inhibit the AhR pathway through its off-target inhibition of CAMKIα activity [44,45]. Interestingly both PD98059 and Akti-1/2 have similar effects on RA-induced maturation: namely growth arrest without differentiation. PD98059 can trigger growth arrest in many cell types [46–48].
Not only may PD98059 and Akti-1/2 exert antagonistic effects on AhR function, but the SFK inhibitor PP2 can function instead as a strong AhR agonist [49]. We tested the AhR agonist FICZ, which potentiates differentiation in wild-type HL-60, but FICZ could not induce G1/G0 arrest nor propel CD38 and CD11b expression in the RA-resistant cells. This suggests that an AhR-related defect may exist in these cells, and may shed light on the nature of the SFK/AhR functional relationship. While i) the AhR-specific agonist FICZ failed to augment differentiation in RA-resistant HL-60, and while ii) the highly specific SFK inhibitor dasatinib has a weaker effect on differentiation in wild-type HL-60 [24], the potential off-target effect of PP2 may implicate that simultaneous SFK inhibition and AhR activation is necessary to promote RA-induced differentiation in cells exhibiting a potential AhR (or SFK) defect.
Raf inhibitor effects can be complicated since Raf proteins often suppress their own activation, and the classical Raf/MEK/ERK cascade is subject to complex feedback control. GW5074 has a high specificity for Raf kinases [45], but in general Raf inhibitors may actually promote c-Raf/c-Raf or c-Raf/B-Raf dimerization, leading to transactivation [50–52]. In one context GW5074 treatment activated c-Raf without downstream activation of ERK, and this mechanism was also Akt-independent [53]. Here we found that GW5074 impairs PP2-induced c-Raf expression and S621 phosphorylation (as well as Lyn expression) in R38+ and R38− RA-resistant cells, but PP2-induced pS259 and pS289/296/301 c-Raf phosphorylation (as well as Fgr expression) were unaffected.
4.3. Nontraditional c-Raf function and c-Raf inhibition compared to Lyn inhibition
ERK activation occurs as early as 4 h after RA treatment in HL-60, while c-Raf phosphorylation does not occur until after 12 to 24 h [13, 54]. c-Raf phosphorylation is MEK-dependent, suggesting a positive feedback loop [12,54,55]. Additionally, c-Raf exhibits RA-induced phosphorylation at its S259, S621 and S289/296/301 sites [20,24,25], none of which associate c-Raf with traditional Ras-initiated or membrane-initiated MAPK signaling. The S289/296/301 c-Raf sites are phosphorylated by ERK, and although their function remains unclear [26,27,56], these phosphorylation sites may sustain the activity of pS259c-Raf [27]. pS621c-Raf is localized in the nucleus of RA-induced HL-60 cells where it associates with the transcription factor NFATc3 at the promoter regions of the BLR1 (CXCR5) gene, which encodes a transmembrane protein that propels MAPK signaling [28]. Also, c-Raf phosphorylation without ERK activation can occur during RA-induced differentiation [25]. It is clear that in RA-treated HL-60 cells, c-Raf deviates from canonical MAPK signaling and appears to be downstream of MEK/ERK.
Whether GW5074 activates or inhibits c-Raf in the RA-induced maturation system is unknown. GW5074 does not affect PP2-induced S259 or S289/296/301 phosphorylation (putative c-Raf inhibitory sites), nor affect SFK Y416 phosphorylation, in either RA-resistant cell line. Despite a strong coupling between Lyn and c-Raf function, there are discrepancies between their respective inhibitors. PP2 could promote certain events like c-Raf and Lyn expression while GW5074 suppresses this expression. GW5074 also suppressed PP2-indcued c-Raf phosphorylation at S621, which may be an autophosphorylation and/or stability site [23,57] that correlates directly with c-Raf expression. Also, GW5074 seemed capable of enhancing PP2-induced CD38 expression in RA-resistant cells, yet the opposite was not necessarily true. PP2 actually blunted the GW5074-induced CD38 expression in R38− HL-60 cells. What can be distilled from these effects is that GW5074 may propel early differentiation events while PP2 can drive late events. Also, notable variable effects of PP2, GW5074 and their combination on RA-induced CD38 expression suggests that Lyn and c-Raf are relatively uncoupled from CD38 expression, which can be seen in Fig. 4C. CD38 itself can be uncoupled from late differentiation as evidenced by the R38+ HL-60 cell line. GW5074 did not augment PP2-induced cell cycle arrest (also a later maturation event), and could very minimally increase the PP2 + RA-induced CD11b expression in R38+ HL-60 cells. Although GW5074 can propel CD38 expression, GW5074 had little effect on the PP2-upregualted CD38-associated proteins c-Cbl, Vav1, Slp76 or the Src-family kinase Fgr.
4.4. Lyn Activity and its effects on PI3K and Akt
Lyn exerts conflicting roles in various contexts. In B cell receptor signaling Lyn plays both a positive and inhibitory role [58]. Lyn can either promote Akt phosphorylation [59] or inhibit Akt activation [60–64], and thus Lyn has variable anti- or pro-apoptotic effects. Lyn can associate with PI3K during differentiation [36,37] and can activate p85 PI3K [65]. Activated Lyn-PI3K-Akt signaling promotes myeloma proliferation [66] or drives ROS production [67]. Although Lyn and PI3K display enhanced interaction and phosphorylation during RA-induced maturation, one group found that enhanced Akt phosphorylation is not detectable until 72–96 h after RA induction in HL-60 cells [68]. Consistent with this, we could not detect RA-inducible changes in pT308 or pS473 Akt in cytoplasmic or nuclear fractions at the 48 h timepoint. However, PP2 did enhance S473 Akt phosphorylation in the wild-type cells, suggesting that in the HL-60 system, Lyn activity inhibits Akt activity. Akt activity is likely to be important for the function of mature granulocytes, but is not required for the onset of differentiation.
Although PP2 is a pan-SFK inhibitor, the suspected target in this system is Lyn. Lyn and Fgr are the predominant SFKs in myeloid cells [31,32] and although both are upregulated by RA, Lyn displays RA-inducible Y416 phosphorylation while Fgr does not [24,33]. However, the function of Lyn itself remains unclear. Congleton et al. (2014) [36] showed that Lyn kinase activity may be required for phosphorylation of two targets downstream of CD38: c-Cbl and PI3K. At the same time, PP2 enhances differentiation and blocks phosphorylation at the activating Y416 site on Lyn (Lyn Y397) in rescued RA-resistant HL-60 cells [24,25]. But in the wild-type case, combined RA + PP2 can “protect” Y416 phosphorylation, as well as protect ERK phosphorylation. Thus Y416 phosphorylation on Lyn seems correlated to differentiation in the wild-type cells but uncorrelated in the RA-resistant cells. In either case, Y416 phosphorylation is highly coupled to ERK activation as shown here (Fig. 4D) and by Congleton et al. (2012) [24]. Therefore like phosphorylated ERK, Lyn phosphorylation at Y416 may be dispensable for RA-induced maturation. Y416 phosphorylation can be present even when c-Src is folded into its repressive conformation [69]. Meanwhile inhibitory Y507 Lyn phosphorylation also seems to increase with RA-treatment and even further with RA + FICZ treatment in wild-type HL-60 [34], and in this study correlated with Lyn expression. These paradoxes suggest complicated Lyn regulation via multiple phosphorylation events, or possibly a structural role as a scaffold apart from activity. Note that Fgr tends to strongly parallel induced differentiation ([11] and this study) despite undetectable phosphorylation. It appears that SFKs have a significant regulatory role in governing response or resistance, but their functions may be multifaceted and context-dependent.
Overall both Lyn and c-Raf thus exhibit nontraditional (and poorly understood) phosphorylation during RA-induced or PP2-rescued differentiation in wild-type and RA-resistant HL-60 cells. Comparing the potential effects of GW5074 on early versus late RA-induced differentiation to the c-Raf phosphorylation sites either downregulated or not downregulated, S259 and S289/296/301 phosphorylation may be needed for early RA-induced differentiation events while S621 (inhibited by GW5074) is needed for late differentiation. We previously speculated that S259 might be an early differentiation event [11]. GW5074 can promote Y507 Lyn phosphorylation in wild-type HL-60 (Supplemental Fig. S2–7–7) but whether Y416 phosphorylation (which remains coupled to ERK activation) in this system correlates to an active form of Lyn is unclear.
5. Conclusions
Historically RA differentiation induction therapy has been successful for t(15;17)-positive acute promyelocytic leukemia (APL) patients, and emergent RA resistance is associated with mutation(s) in the PML-RARα fusion product [9]. However mutations can arise months after relapse, and in myeloid leukemia cell lines like HL-60 and NB4, RA resistance is not always correlated with mutation in PML-RARα or RARα. One recent study verified that leukemias, both with defining chromosomal translocations and those that display a normal karyotype, are driven by global random genetic events [70]. This stresses that multi-targeted therapy is a necessity for leukemia and other cancers, and specifically kinase inhibitors are attractive agents for enhancing the pro-differentiative and anti-proliferative effects of RA. We have screened five kinase inhibitors, and PP2 and GW5074 emerged as potent enhancers of RA differentiation induction therapy, capable of rescuing two progressively RA-resistant cell lines. These kinase inhibitors also served to probe the functionality of a kinase network involved in RA-induced maturation in t(15;17)-negative cells. We found that within an interrelated signaling module, Lyn and c-Raf function emerged as crucial to RA-induced maturation. The results have ramifications toward understanding nontraditional c-Raf and Lyn function, which were not strongly correlated with downstream events like ERK and Akt activation.
Supplementary Material
Acknowledgments
This work was supported by grants R01 CA033505, R01 CA152870 from NIH (A.Y.) and NIH/PS-OC (Shuler/A.Y./R.P.B.), NYSTEM NY Dept. Health (A.Y.), Cornell Vertebrate Genomics (VERGE) (R.P.B.), and CBET-0846876 from NSF (J.D.V./H.A.J.).
Footnotes
Financial support: this work was supported by grants R01 CA033505, R01 CA152870 from NIH (A.Y.) and NIH/PS-OC (Shuler/A.Y./R.P.B.), NYSTEM NY Dept. Health (A.Y.), Cornell Vertebrate Genomics (VERGE) (R.P.B.), and CBET-0846876 from NSF (J.D.V./H.A.J.).
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.cellsig.2015.03.014.
References
- 1.Steelman LS, Chappell WH, Abrams SL, Kempf RC, Long J, Laidler P, Mijatovic S, Maksimovic-Ivanic D, Stivala F, Mazzarino MC, Donia M, Fagone P, Malaponte G, Nicoletti F, Libra M, Milella M, Tafuri A, Bonati A, Bäsecke J, Cocco L, Evangelisti C, Martelli AM, Montalto G, Cervello M, McCubrey JA. Aging. 2011;3(3):192–222. doi: 10.18632/aging.100296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akinleye A, Avvaru P, Furqan M, Song Y, Liu D. J Hematol Oncol. 2013;6(1):88. doi: 10.1186/1756-8722-6-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martelli AM, Tazzari PL, Tabellini G, Bortul R, Billi AM, Manzoli L, Ruggeri A, Conte R, Cocco L. Leukemia. 2003;17(9):1794–1805. doi: 10.1038/sj.leu.2403044. [DOI] [PubMed] [Google Scholar]
- 4.Bushue N, Wan YY. Adv Drug Deliv Rev. 2010;62:1285–1296. doi: 10.1016/j.addr.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang X, Gudas LJ. Annu Rev Pathol: Mech. 2011;6:345–364. doi: 10.1146/annurev-pathol-011110-130303. [DOI] [PubMed] [Google Scholar]
- 6.Tallman MS, Altman JK. Blood. 2009;114(25):5126–5135. doi: 10.1182/blood-2009-07-216457. [DOI] [PubMed] [Google Scholar]
- 7.Fontana JA, Colbert DA, Deisseroth AB. Proc Natl Acad Sci U S A. 1981;78(6):3863–3866. doi: 10.1073/pnas.78.6.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dalton WT, Ahearn MJ, McCredie KB, Freireich EJ, Stass SA, Trujillo JM. Blood. 1988;71:242–247. [PubMed] [Google Scholar]
- 9.Gallagher RE. Leukemia. 2002;16:1940–1958. doi: 10.1038/sj.leu.2402719. [DOI] [PubMed] [Google Scholar]
- 10.Yen A, Forbes M, DeGala G, Fishbaugh J. Cancer Res. 1987;47:129–134. [PubMed] [Google Scholar]
- 11.Jensen HA, Bunaciu RP, Ibabao CN, Myers R, Varner JD, Yen A. PLoS One. 2014;9(6):e98929. doi: 10.1371/journal.pone.0098929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang J, Yen A. J Biol Chem. 2008;283(7):4375–4386. doi: 10.1074/jbc.M708471200. [DOI] [PubMed] [Google Scholar]
- 13.Yen A, Roberson MS, Varvayanis S, Lee AT. Cancer Res. 1998;58:3163–3172. [PubMed] [Google Scholar]
- 14.Bunaciu RP, Yen A. Cancer Res. 2011;71:2371–2380. doi: 10.1158/0008-5472.CAN-10-2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee M. Biochem Biophys Res Commun. 2006;350(2):450–456. doi: 10.1016/j.bbrc.2006.09.067. [DOI] [PubMed] [Google Scholar]
- 16.Li Z, Hosoi Y, Cai K, Tanno Y, Matsumoto Y, Enomoto A, Morita A, Nakagawa K, Miyagawa K. Biochem Biophys Res Commun. 2006;341(2):363–368. doi: 10.1016/j.bbrc.2005.12.193. [DOI] [PubMed] [Google Scholar]
- 17.Yamaguchi K, Richardson MD, Bigner DD, Kwatra MM. Cancer Chemother Pharmacol. 2005;56(6):585–593. doi: 10.1007/s00280-005-1030-3. [DOI] [PubMed] [Google Scholar]
- 18.Opavsky MA, Martino T, Rabinovitch M, Penninger J, Richardson C, Petric M, Trinidad C, Butcher L, Chan J, Liu PP. J Clin Invest. 2002;109(12):1561–1569. doi: 10.1172/JCI13971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yen A, Williams M, Platko JD, Der C, Hisaka M. Eur J Cell Biol. 1994;65(1):103–113. [PubMed] [Google Scholar]
- 20.Smith J, Bunaciu RP, Reiterer G, Coder D, George T, Asaly M. Exp Cell Res. 2009;315:2241–2248. doi: 10.1016/j.yexcr.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tasseff R, Jensen HA, Congleton J, Yen A, Varner JD. Investigation of the cRaf Interactome and Steady-state Multiplicity in Retinoic Acid-induced Differentiation of HL-60 Cells. Department of Chemical and Biomolecular Engineering, Cornell University; Ithaca NY: 2014. (Unsubmitted manuscript) [Google Scholar]
- 22.Morrison DK, Heidecker G, Rapp UR, Copeland TD. J Biol Chem. 1993;266(23):17309–17316. [PubMed] [Google Scholar]
- 23.Noble C, Mercer K, Hussain J, Carragher L, Giblett S, Hayward R, Patterson C, Marais R, Pritchard CA. Mol Cell. 2008;31(6):862–872. doi: 10.1016/j.molcel.2008.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Congleton J, MacDonald R, Yen A. Leukemia. 2012;26(6):1180–1188. doi: 10.1038/leu.2011.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jensen HA, Styskal LE, Tasseff R, Bunaciu RP, Varner JD, Yen A. PLoS One. 2013;8(3):e58621. doi: 10.1371/journal.pone.0058621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dougherty MK, Muller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, Conrads TP, Veenstra TD, Lu KP, Morrison DK. Mol Cell. 2005;17:215–224. doi: 10.1016/j.molcel.2004.11.055. [DOI] [PubMed] [Google Scholar]
- 27.Balan V, Leicht DT, Zhu J, Balan K, Kaplun A, Singh-Gupta V, Qin J, Ruan H, Comb MJ, Tzivion G. Mol Biol Cell. 2006;17:1141–1153. doi: 10.1091/mbc.E04-12-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Geil WM, Yen A. FEBS J. 2013 doi: 10.1111/febs.12693. http://dx.doi.org/10.1111/febs.12693. [DOI] [PMC free article] [PubMed]
- 29.Kubicek M, Pacher M, Abraham D, Podar K, Eulitz M, Baccarini M. J Biol Chem. 2002;277(10):7913–7919. doi: 10.1074/jbc.M108733200. [DOI] [PubMed] [Google Scholar]
- 30.Dhillon AS, Meikle S, Yazici Z, Eulitz M, Kolch W. EMBO J. 2002;21(1–2):64–71. doi: 10.1093/emboj/21.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Katagiri K, Katagiri T, Koyama Y, Morikawa M, Yamamoto T, Yoshida T. J Immunol. 1991;146(2):701–707. [PubMed] [Google Scholar]
- 32.Dos Santos C, Demur C, Bardet V, Prade-Houdellier N, Payrastre B, Récher C. Blood. 2008;111(4):2269–2279. doi: 10.1182/blood-2007-04-082099. [DOI] [PubMed] [Google Scholar]
- 33.Kropf PL, Wang L, Zang Y, Redner RL, Johnson DE. Leukemia. 2010;24(3):663–665. doi: 10.1038/leu.2009.267. [DOI] [PubMed] [Google Scholar]
- 34.Bunaciu RP, Yen A. Mol Cancer. 2013;12:39. doi: 10.1186/1476-4598-12-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Welch H, Maridonneau-Parini I. Oncogene. 1997;15:2021–2029. doi: 10.1038/sj.onc.1201356. [DOI] [PubMed] [Google Scholar]
- 36.Congleton J, Shen M, Macdonald R, Malavasi F, Yen A. Cell Signal. 2014;26(7):1589–1597. doi: 10.1016/j.cellsig.2014.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kamei T, Jones SR, Chapman BM, MCGonigle KL, Dai G, Soares MJ. Mol Endocrinol. 2002;16(7):1469–1481. doi: 10.1210/mend.16.7.0878. [DOI] [PubMed] [Google Scholar]
- 38.Zimmermann S, Moelling K. Science. 1999;286(5445):1741–1744. doi: 10.1126/science.286.5445.1741. [DOI] [PubMed] [Google Scholar]
- 39.Miranda MB, Redner RL, Johnson DE. Mol Cancer Ther. 2007;6:3081–3090. doi: 10.1158/1535-7163.MCT-07-0514. [DOI] [PubMed] [Google Scholar]
- 40.Kishimoto H, Hoshino S, Ohori M, Kontani K, Nishina H, Suzawa M, Kato S, Katada T. J Biol Chem. 1998;273:15429–15434. doi: 10.1074/jbc.273.25.15429. [DOI] [PubMed] [Google Scholar]
- 41.Drach J, McQueen T, Engel H, Andreeff M, Robertson KA, Collins SJ, Malavasi F, Mehta K. Cancer Res. 1994;54(7):1746–1752. [PubMed] [Google Scholar]
- 42.Kobayashi T, Tsunawaki S, Seguchi H. Redox Rep. 2001;6(1):27–36. doi: 10.1179/135100001101536003. [DOI] [PubMed] [Google Scholar]
- 43.Reiners JJ, Jr, Lee JY, Clift RE, Dudley DT, Myrand SP. Mol Pharmacol. 1998;53(3):438–445. doi: 10.1124/mol.53.3.438. [DOI] [PubMed] [Google Scholar]
- 44.Gilot D, Giudicelli F, Lagadic-Gossmann D, Fardel O. Chem Biol Interact. 2010;188:546–552. doi: 10.1016/j.cbi.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 45.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. Biochem J. 2007;408(Pt 3):297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moon DO, Park C, Heo MS, Park YM, Choi YH, Kim GY. Int Immunopharmacol. 2007;7(1):36–45. doi: 10.1016/j.intimp.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 47.Hoshino R, Tanimura S, Watanabe K, Kataoka T, Kohno M. J Biol Chem. 2001;276(4):2686–2692. doi: 10.1074/jbc.M006132200. [DOI] [PubMed] [Google Scholar]
- 48.Yamaguchi K, Tomita H, Sugano E, Nakazawa T, Tamai M. Jpn J Ophthalmol. 2002;46(6):634–639. doi: 10.1016/s0021-5155(02)00618-4. [DOI] [PubMed] [Google Scholar]
- 49.Frauenstein K, Tigges J, Soshilov AA, Kado S, Raab N, Fritsche E, Haendeler J, Denison MS, Vogel CF, Haarmann-Stemmann T. Arch Toxicol. 2014 doi: 10.1007/s00204-014-1321-8. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. Nature. 2010;464(7287):427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJ, Stokoe D, Gloor SL, Vigers G, Morales T, Aliagas I, Liu B, Sideris S, Hoeflich KP, Jaiswal BS, Seshagiri S, Koeppen H, Belvin M, Friedman LS, Malek S. Nature. 2010;464(7287):431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 52.Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C, Marais R. Cell. 2010;140(2):209–2. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chin PC, Liu L, Morrison BE, Siddiq A, Ratan RR, Bottiglieri T, D’Mello SR. J Neurochem. 2004;90(3):595–608. doi: 10.1111/j.1471-4159.2004.02530.x. [DOI] [PubMed] [Google Scholar]
- 54.Hong HY, Varvayanis S, Yen A. Differentiation. 2001;68(1):55–66. doi: 10.1046/j.1432-0436.2001.068001055.x. [DOI] [PubMed] [Google Scholar]
- 55.Yen A, Varvayanis S. Vitro Cell Dev Biol Anim. 2000;36(4):249–255. doi: 10.1290/1071-2690(2000)036<0249:raiaop>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 56.Hekman M, Fischer A, Wennogle LP, Wang YK, Campbell SL, Rapp UR. FEBS Lett. 2005;579(2):464–468. doi: 10.1016/j.febslet.2004.11.105. [DOI] [PubMed] [Google Scholar]
- 57.Mischak H, Seitz T, Janosch P, Eulitz M, Steen H, Schellerer M, Philipp A, Kolch W. Mol Cell Biol. 1996;16(10):5409–5418. doi: 10.1128/mcb.16.10.5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu Y, Harder KW, Huntington ND, Hibbs ML, Tarlinton DM. Immunity. 2005;22(1):9–18. doi: 10.1016/j.immuni.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 59.Bates RC, Edwards NS, Burns GF, Fisher DE. Cancer Res. 2001;61(13):5275–5283. [PubMed] [Google Scholar]
- 60.Zagozdzon R, Bougeret C, Fu Y, Avraham HK. Int J Oncol. 2002;21:1 347–1 352. [PubMed] [Google Scholar]
- 61.Ma P, Vemula S, Munugalavadla V, Chen J, Sims E, Borneo J, Kondo T, Ramdas B, Mali RS, Li S, Hashino E, Takemoto C, Kapur R. Mol Cell Biol. 2011;31(19):4052–4062. doi: 10.1128/MCB.05750-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pogue SL, Kurosaki T, Bolen J, Herbsy R. J Immunol. 2002;165(3):1300–1306. doi: 10.4049/jimmunol.165.3.1300. [DOI] [PubMed] [Google Scholar]
- 63.Li HL, Davis WW, Whiteman EL, Birnbaum MJ, Puré E. Proc Natl Acad Sci U S A. 1999;96(12):6890–6895. doi: 10.1073/pnas.96.12.6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Negro R, Gobessi S, Longo PG, He Y, Zhang ZY, Laurenti L, Efremov DG. Blood. 2012;119:6278–6287. doi: 10.1182/blood-2012-01-403162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pleiman CM, Hertz WM, Cambier JC. Science. 1994;263(5153):1609–1612. doi: 10.1126/science.8128248. [DOI] [PubMed] [Google Scholar]
- 66.Iqbal MS, Tsuyama N, Obata M, Ishikawa H. Biochem Biophys Res Commun. 2010;392(3):415–420. doi: 10.1016/j.bbrc.2010.01.038. [DOI] [PubMed] [Google Scholar]
- 67.Zhu QS, Xia L, Mills GB, Lowell CA, Touw IP, Corey SJ. Blood. 2006;107(5):1847–1856. doi: 10.1182/blood-2005-04-1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Matkovic K, Brugnoli F, Bertagnolo V, Banfic H, Visnjic D. Leukemia. 2006;20(6):941–951. doi: 10.1038/sj.leu.2404204. [DOI] [PubMed] [Google Scholar]
- 69.Irtegun S, Wood RJ, Ormsby AR, Mulhern TD, Hatters DM. PLoS One. 2013;8(7):e71035. doi: 10.1371/journal.pone.0071035. (3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, Kandoth C, Fulton RS, McLellan MD, Dooling DJ, Wallis JW, Chen K, Harris CC, Schmidt HK, Kalicki-Veizer JM, Lu C, Zhang Q, Lin L, O’Laughlin MD, McMichael JF, Delehaunty KD, Fulton LA, Magrini VJ, McGrath SD, Demeter RT, Vickery TL, Hundal J, Cook LL, Swift GW, Reed JP, Alldredge PA, Wylie TN, Walker JR, Watson MA, Heath SE, Shannon WD, Varghese N, Nagarajan R, Payton JE, Baty JD, Kulkarni S, Klco JM, Tomasson MH, Westervelt P, Walter MJ, Graubert TA, DiPersio JF, Ding L, Mardis ER, Wilson RK. Cell. 2012;150(2):264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.