

Abstract
Neutrophil extracellular traps (NETs) are released, as neutrophils die in vitro, in a process requiring hours, leaving a temporal gap for invasive microbes to exploit. Functional neutrophils undergoing NETosis have not been documented. During Gram-positive skin infections, we directly visualized live PMN in vivo rapidly releasing NETs, which prevented bacterial dissemination. NETosis occurred during crawling thereby casting large areas of NETs. NET-releasing PMN developed diffuse decondensed nuclei ultimately becoming devoid of DNA. Cells with abnormal nuclei displayed unusual crawling behavior highlighted by erratic pseudopods and hyperpolarization consistent with the nucleus being a fulcrum for crawling. A combined requirement of Tlr2 and complement mediated opsonization tightly regulated NET release. Additionally live human PMN developed decondensed nuclei and formed NETS in vivo and intact anuclear neutrophils were abundant in Gram-positive human abscesses. Therefore early in infection, non-cell death NETosis occurs in vivo during Gram-positive infection in mice and humans.
Gram-positive bacteria such as Staphylococcus aureus1–4 and Streptococcus pyogenes5 are highly invasive pathogens causing severe skin infections and sepsis in humans. Neutrophils are critical for host survival and NETs may represent a novel defense mechanism6–9. In vitro NETs are a cell-death event occurring hours after initial stimulation10,11; however questions remain concerning NETosis in vivo. Conventional PMN killing involves cell recruitment, emigration, chemotax and phagocytosis of microbes12,13. However chasing individual microbes is ineffective to deal with rapidly disseminating Gram-positive pathogens. Utilizing both phagocytosis and NETosis might be counter-productive if neutrophils engulf bacteria and then lyse releasing all their bacterial contents. Thus non-lytic NETosis may prevent bacteria from escaping following phagocytosis. To date NETosis has only been observed in immobile and incapacitated cells. In vitro, we described rapid NET release (< 10 min) without cell lysis, however the fate and function of these PMN could not be evaluated14,15. In this study we hypothesize that rapid NET release may be carried out in vivo by live viable PMN.
To understand the relationship between conventional PMN-mediated and NET-mediated host defense, we directly visualize PMN behavior during Gram-positive skin infections in mice and humans. Using spinning-disk confocal intravital microscopy (SDCIM) surprising insights, not in line with the current NETosis paradigm, were revealed. Cell death and lytic release of NETs were not observed in vivo, nor was nuclear condensation and budding off en bloc, observed in red blood cell enucleation16. Instead, live functional PMN crawled, phagocytosed and rapidly NETosed simultaneously, resulting in widespread tissue NETs that limited bacterial dissemination. NETosis occurred in emigrated PMN during localized infection and this was tightly regulated through Tlr2 and complement mediated opsonization. NETosing PMN crawled with erratic pseudopod formation and extreme cell morphologies due to the loss of the nuclear structure acting as a fulcrum for the cytoskeleton17,18. Importantly, human neutrophils released NETs in an in vivo model and patients with acute Gram-positive abscesses demonstrated intact NETosing PMN identical to the animal model. Here we describe an alternate fate of in vivo NETosing PMN that is distinct from cell-death NETosis.
Results
Gram-positive bacteria induced rapid NET formation in vivo
S. aureus infected mouse skin was imaged using SDCIM and the cell-impermeable DNA specific dye SYTOX Orange was used to visualize the NETs (Method 1) (Fig. 1a). We observed sheets of DNA, in contrast to the thin strands of in vitro NETs (15 nm to 100 nm)6,11,19 (Fig. 1a and Supplementary Video 1). Chemokine induced sterile inflammation recruited PMN but NETs were not released (Fig. 1a), despite undergoing the same surgical procedure. Emigrated PMN crawled through infected tissues thereby widening NET distribution (Supplementary Video 1). NETs were inducible between 1 × 106 to 1 × 108 colony-forming units (CFUs). Acute infection did not result in dead PMN as demonstrated by lack of SYTOX uptake.
Figure 1.

Rapid in vivo NETosis during acute Gram-positive bacterial infections is directly visualized in vivo. (a) Method 1: NET release was visualized as extracellular DNA (SYTOX Orange, white arrows) in vivo during infection (S. aureus, Xen29), but not during sterile inflammation (MIP-2 superfusion) (neutrophils are green, while NETs are red)(6 mice). (b) Method 2: NET quantification using fluorochrome conjugated histone or neutrophil elastase specific antibodies following live S. aureus, S. pyogenes or killed bacteria (S. aureus Xen8.1). Control animals received fluorochrome conjugated IgG isotype control (n = 4 for each group, n = 3 for dead bacteria). (c) Temporal NET tissue accumulation with PMN images removed for clarity of NETs. (d) Method 3: In vivo PMN nuclei pre-stained with the cell-permeable DNA dye (SYTO 60, blue) during sterile inflammation (left) or during infection with GFP-S. aureus (GFP-USA300)(right). A PMN with a normal nucleus is circled in blue, while a PMN with a diffuse nucleus is circled in green. NETs are demonstrated during infection with a white arrow. NET release is quantified by a ratio of extracellular DNA to intracellular DNA. (e) Impaired histone release in Tlr2−/− and C3−/− animals (n = 3 each group). (f) Impaired DNA release in wild type, Tlr2−/− and C3−/− mice (n = 3 for all groups). NET area was determined using Volocity imaging software (* = P < 0.05 for treatment versus control, or treatment versus sterile inflammation, or knockout versus wild type, # = P < 0.05 for treatment versus C3−/−).
NETs were not visible in blood vessels within the infected skin; therefore muscle was imaged for improved visualization of the intravascular compartment (Supplementary Fig. 1a) NETosis was not observed, consistent with the view that systemic infection is required for intravascular NETs. Emigrated PMN (peritoneal) had increased NET release, without cell lysis, compared to non-emigrated PMN (marrow) exposed to bacteria in an in vitro assay (Supplementary Fig. 1b). We hypothesize that emigration may enhance DNA release and prevent NET spread through the blood, thereby decreasing bystander damage.
NETs are rapidly released from the nucleus
To confirm authentic in vivo NETs (histones + granular proteins + DNA9,20), a second assay was developed using specific fluorescently conjugated antibodies to visualize NET constituents (Method 2). Significant release of histones and neutrophil elastase occurred within minutes in response to Gram-positive human pathogens (S. aureus and S. pyogenes) (Fig. 1b,c). Negligible NETs occurred during sterile inflammation. Histone stained areas expanded in large sheets (Fig. 1c, Supplementary Fig. 2a and Supplementary Video 2) identical to extracellular DNA. Injection of dead, washed bacteria resulted in NET release (Fig. 1b), demonstrating innate recognition is sufficient independent of active toxin release.
Mitochondrial DNA NETs exist21,22, however in vivo mitochondrial imaging showed unaltered intracellular mitochondria during infection (Supplementary Fig. 2a,b). This does not exclude mitochondrial DNA, however the nucleus seems to be the major source of NETs in our model of infection.
Using a third technique, in vivo PMN nuclei were pre-stained with a cell-permeable DNA specific dye (SYTO 60, Method 3) allowing nuclear imaging throughout NETosis. Without infection, SYTO 60 is restricted to normal multilobar distinct nuclei however, abnormal PMN with indistinct diffuse nuclear remnants were observed during infection (Fig. 1d). The ratio of extracellular to intracellular DNA was quantified to demonstrate the increase in NETs during infection (Fig. 1d).
NET formation is tightly regulated and requires both Tlr2 and C3
NETs may cause bystander injury, therefore we hypothesized that release is tightly regulated. Two critical systems involved in host defense against S. aureus are Tlr2 and complement23. NETs were absent in Myd88−/− animals demonstrating Toll dependence (Supplementary Fig. 3a). Furthermore, both Tlr2−/− and C3−/− animals were incapable of releasing histones (Fig. 1e) or nuclear DNA (Fig. 1f) despite recruiting normal numbers of PMN (Supplementary Fig. 4a). Normal mouse serum restored NETosis in C3−/− animals indicating the importance of opsonization (Fig. 1f). As Tlr2−/− mice have intact complement and the C3−/− mice have intact Tlr2, it is clear that neither of these molecules is sufficient to cause NETs in isolation. Collectively these data suggest a multi-tiered regulation for which Tlr2 and complement are essential but not individually sufficient.
Tlr2 stimulation did not induce NETs (Supplementary Fig. 3c). In preliminary experiments, the C3a receptor played a greater role in NET production compared to the C5a receptor (Supplementary Fig. 3b), however, recombinant C3a alone or in addition to PAM-3-CYS (Tlr2 stimulant) had no effect on NETosis (Supplementary Fig. 3c), revealing that purified molecules could not mimic a pro-bono Gram-positive infection.
Viable NET-forming PMN Remain Functional in vivo
PMN did not take-up SYTOX (indicating death) during NETosis. We pre-labeled the neutrophil nucleus in vivo (Method 3) to understand if NETosing neutrophils continue to function. We observed a stark contrast between normal and NETosing PMN using SDCIM and 3D reconstruction (Fig. 2a). To visualize the innermost structures, we have adjusted the levels of 3D transparency. Non-NETosing PMN have intact, fully intracellular nuclei (left panel). The NETosing PMN has released a NET that surrounds the entire outer membrane (middle panel)(Fig. 2a). Here the NET is made transparent in order to see the underlying PMN. Remarkably, the NETosing PMN remains viable and continues to chemotax towards a GFP-bacterium (Supplementary Video 3). Further 3D images of PMN releasing intracellular DNA are shown (Supplementary Fig. 5a).
Figure 2.

PMN are viable and functional during nuclear breakdown and chromatin decondensation. PMN and nuclei were evaluated in vivo following live S. aureus (GFP-USA300). (a) A normal PMN that has captured a live bacterium (green) is shown by 2D imaging (top left) and contrasted by a NET-forming PMN (top middle) that is chemotaxing towards a GFP-bacterium (green arrow) while releasing DNA. Beneath each 2D spinning disk image is a panel of confocal 3D reconstruction using various degrees of transparency to distinguish the DNA in relation to the PMN outer membrane. The NETosing PMN in this image (middle panels) can be observed crawling toward the live bacteria in Supplementary Video 3. Four-color spinning disk confocal and 3D reconstruction reveals a PMN (yellow, Alexa 750 conjugated GR-1 specific antibody) with a diffuse nucleus (cell permeable SYTO 60, blue) releasing an extracellular NET (cell impermeable SYTOX Orange, red) while retaining live S. aureus (GFP-bacteria, green). (b) Three nuclear phenotypes were visualized; normal, diffuse, or absent. Top panels show extracellular membrane and nuclear staining, while the lower panels show the nucleus alone. (c) 3D-reconstruction reveals the morphology of each nuclei group. The 3D image rendering results in an artificial white reflection on the cell surface that does not represent an authentic fluorescent stain or nuclei. (d) Phagocytosis of live GFP-staphylococcus by neutrophils was quantified in wildtype, Tlr2−/− or C3−/− mice. The ability for neutrophils with decondensed or absent nuclei to phagocytose was quantified in (e) wildtype, Tlr2−/− or C3−/− animals. (n = 3 experiments per group). (** = P < 0.01 and *** = P < 0.001 for phagocytosis versus no phagocytosis)
To directly visualize PMN releasing intracellular DNA that becomes a NET we developed a 4-color SDCIM strategy combining the cell permeable DNA dye SYTO 60 and the cell impermeable dye SYTOX Orange. Live PMN were visualized that contained diffuse nuclear staining with concurrent release of NETs. 3D imaging confirmed that the PMN with extracellular DNA (red) contained a diffuse abnormal nucleus (right panel)(Fig 2a). PMN that crawled while releasing NETs were quantified for velocity, meandering index and crawling track (Supplementary Fig. 5b–d and Video 4). Therefore NETosis is not limited to immobile and incapacitated cells.
Live NETosing PMN develop decondensed nuclei and become anuclear
Neutrophil nuclear swelling and changes in morphology predict chromatin decondensation and nuclear envelope breakdown6,14,19,24,25. We observed three different nuclear morphologies of emigrated PMN exposed to S. aureus. Normal nuclei were distinct demarcated multilobar rings with uniform DNA-staining (Fig. 2b, Supplementary Fig. 6a and Video 5). The normal nucleus was malleable, but retained its structure during crawling. Diffuse nuclei lacked structure and had a heterogeneous, lower intensity staining pattern consistent with breakdown of the nuclear envelope, chromatin decondensation and intracellular chromatin dispersion (Fig. 2b, Supplementary Fig. 6b and Supplementary Video 5). Anuclear PMN had absent nucleic acid staining (Fig. 2b, Supplementary Fig. 6b and Supplementary Video 5). These observations were not due to photobleaching as all three nuclear phenotypes were observed in the same fields of view, over the same duration and laser intensity. To confirm structural nuclear differences, we used confocal z-stacks and 3D reconstruction (Fig. 2c). Completely anuclear PMN by 2D imaging were completely devoid of a nucleus on 3D reconstruction and no longer contained any stainable nucleic acids suggesting these cells had completed the NETosing process (Fig. 2c).
GFP-histone transgenic mice (Tg(HIST1H2BB/EGFP)1Pa) confirmed in vivo anuclear PMN (Supplementary Fig. 7a). The PMN on the left has a normal nucleus, whereas the PMN on the right has no detectable nucleus. PMN with normal and diffuse nuclei continued to crawl within the tissues. Flow cytometry of untreated animals reveal all PMN are uniformly and brightly positive for a nucleus prior to infection (Supplementary Fig. 7b). Bone marrow chimera experiments (GFP-expression restricted to hematogenous cells) support a subpopulation of PMN within infected tissue capable of surviving and functioning without a nucleus (Supplementary Fig. 7c).
To understand if NETosis affects other host-defense functions we quantified phagocytosis of live bacteria. In wild type animals approximately 50% of PMN phagocytosed bacteria (Fig. 2d). Although NETosis and phagocytosis were closely linked, phagocytosis alone was not sufficient to induce NETosis as Tlr2−/− PMN took up bacteria without nuclear changes (Fig. 2e). C3−/− mice could not phagocytose and did not have altered nuclei, or make NETS, however this impairment was reversed with normal serum demonstrating the importance of opsonization (Fig. 2e).
Unusual cellular phenotypes of NETosing PMN
Unique patterns of crawling and cell morphologies were observed during NETosis. Normal PMN with multilobar nuclei crawl with a single lead pseudopod (Fig. 3a). In contrast PMN with diffuse nuclei demonstrate hyperpolarization and multiple pseudopods (Fig. 3a). To test if the loss of the nucleus explains the bizarre crawling phenotype, we studied a well-defined induced anuclear human PMN cytoplast26–29 (Fig. 3a). Indeed cytoplasts exhibit crawling behaviors similar to NETosing PMN suggesting that the intact nucleus provides the structural fulcrum for normal crawling similar to other mobile cells17,18. Crawling was further analyzed in Tlr2−/− and C3−/− PMN. As anticipated Tlr2−/− and C3−/− PMN crawled with a normal single lead pseudopod and maintained morphology similar to non-NETosing PMN with normal nuclei (Fig. 3a).
Figure 3.

NET-forming PMN display a novel crawling phenotype in vivo related to nuclear structure. (a) 2D images of in vivo PMN with a normal nuclei, NET-forming cells with diffuse nuclei and an in vitro generated anuclear human PMN (cytoplast). Contouring analysis of each cell is shown beneath the image. The PMN outer membrane is traced every 30 s (PMN in the middle image with white arrow is traced). Typical crawling phenotypes are shown for Tlr2−/− and C3−/− animals. (b) Cell polarities in relation to nuclear morphology were quantified. (c) The relationship of pseudopod formation of crawling PMN compared to their nuclear architecture or between wild type, Tlr2−/− and C3−/− mice. (d) PMN velocity and meandering index during live staphylococcus infection. (e) Cellular tracking during live staphylococcus infection (*** = P < 0.001, ** = P < 0.01, * = P < 0.05 compared to sterile inflammation and ### = P < 0.001, # = P < 0.05 compared to wild type).
Polarization is normal for crawling cells and in vitro these cells become 1.5 to 2× longer than their width, the limitation of this value being the width of the nucleus. However in infected tissue PMN that had diffuse nuclei became hyperpolarized (Fig. 3b and Supplementary Fig. 4b). Both the NETosing PMN and the cytoplasts generated multiple pseudopods simultaneously, often at obtuse angles (90–180 °), a phenotype never observed in sterile inflammation. During sterile inflammation, the majority of PMN crawled with one dominant pseudopod, which rarely split to yield 2 pseudopods at an acute angle (< 90 °). Infection increased pseudopod formation, which was more common in cells with altered nuclei (Fig. 3c). Tlr2−/− and C3−/− PMN did not form multiple pseudopods. Despite having abnormal nuclei, hyperpolarization and multiple pseudopods, wild type PMN crawled with normal velocity and moved with purpose (i.e. a high meandering index)(Fig. 3d,e). We tracked individual PMN and found that NETosing PMN with diffuse nuclei crawled with high velocity and meandering index, while fully anuclear PMN crawled slowly (Supplementary Fig. 8a–c). Together these data suggest that structural nuclear breakdown is related to aberrant, but functional chemotaxis, similar to anuclear human PMN cytoplasts. By contrast, despite normal nuclei, Tlr2−/− and C3−/− had reduced crawling velocities and moved without purpose in random directions (Fig. 3d,e).
NETs are critical to contain an acute invasive infection in vivo
We followed staphylococcus skin infections over time using minimally invasive non-surgical luminescent imaging. Currently no specific inhibitor of NETosis exists, so exogenous DNase treatment was used to breakdown NETs. Animals treated with or without DNase had comparable S. aureus levels at 1 h, but DNase treatment decreased luminescence within 4 h (Fig. 4a–b). DNase did not alter bacterial growth in culture (data not shown). Disruption of NETs resulted in significant increase in bacteremia at 4 h and a decrease of skin CFUs at 24 h, indicating that bacteria can rapidly escape the portal of entry in the absence of NET trapping (Fig. 4c,d).
Figure 4.

NETs are essential to limit acute S. aureus dissemination. (a) Photon imaging of live luminescent S. aureus (Xen8.1, 1 × 108 CFU per 100 μl saline injection) within the mouse skin at 1 h and 4 h. (b) Live luminescent S. aureus were quantified within the skin of mice pretreated with either DNase (1,000U i.p.) or saline (i.p.) at 1 h, 4 h and 8 h post-infection (6 mice). (c) Bacterial dissemination from the skin to the blood at 4 h post-infection (Xen8.1) (6 mice). (d) CFUs grown from a 3.5 mm skin biopsy of the injection site at 24 h (6 mice). (* = P < 0.05).
We examined dissemination in mutant mice that lacked NETosis. Wild type, Tlr2−/− and C3−/− animals received intraperitoneal injections of live S. aureus and blood CFUs were determined at 5 h. Compared to wild type mice, both Tlr2−/− and C3−/− mice had impaired ability to contain S. aureus dissemination (Supplementary Fig. 4c). Notably C3−/− which were deficient in both phagocytosis and NETosis had more severe bacteremia than did Tlr2−/− which could phagocytose but not NETose.
Human PMN release NETs in vivo and become anuclear
We investigated PMN from human subjects diagnosed with Gram-positive abscesses (Supplementary Table 1). Fresh abscess aspirates were imaged using transmission electron microscopy. In all samples, we visualized normal PMN, PMN with degraded nuclear envelopes, decondensed and dispersed chromatin (diffuse nuclei) and PMN without any intracellular DNA or chromatin (anuclear)(Fig. 5a–e). Normal PMN had abundant heterochromatin and multilobar nuclei, while NETosing cells were at various stages of nuclear envelope breakdown and NET release. Granulated cells with diffuse nuclear staining contained nuclear envelope remnants and diffuse chromatin throughout the cytoplasm however structurally intact nuclei were not present (Fig. 5b). PMN without a nucleus but still containing limited dispersed DNA and chromatin were a common finding on EM supporting our in vivo observation that PMN with diffuse nuclear staining have dissolved the nucleus and contain some free chromatin resulting in a diffuse DNA pattern of fluorescence (Fig. 5c). Of note, PMN with diffuse or completely absent nuclear material still contain granules, suggesting that they retain the necessary components to kill bacteria through conventional mechanisms.
Figure 5.
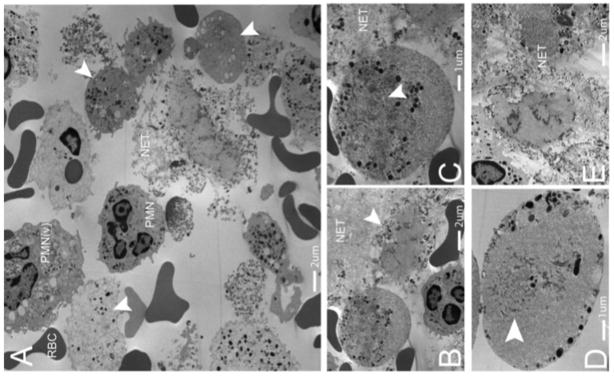
NETosis occurs in human abscesses due to Gram-positive bacterial infections. Five human patients presenting with Gram-positive abscesses were evaluated. Transmission electron microscopy was performed on freshly obtained clinical samples. (a) The abscesses contained intact neutrophils (PMN), red blood cells (RBCs), activated neutrophils with vesicles in the cytoplasm (PMNv), as well as numerous anuclear neutrophils with cytoplasmic granules and nuclear vesicles (arrowheads). (b) A typical anuclear neutrophil is demonstrated undergoing NET formation (arrowhead) and a second cell is shown, enlarged in (c), following nuclear envelope breakdown with dispersed chromatin and nuclear vesicles. The remnants of the nuclear envelope are highlighted (arrowhead). NETs are identified by the prototypical ‘beads on a string’ appearance on EM. (d) An anuclear neutrophil with nuclear envelope breakdown decondensed and dispersed chromatin (arrowhead), with vesicles and granules fusing with the outer plasma membrane (arrowhead). (e) Late stage neutrophils that have released chromatin and granules into the extracellular space.
NETs were identified by their typical beads-on-a-string EM appearance. To understand how NETs are released without cell lysis we imaged diffuse and anuclear PMN at higher magnification. We observed intracellular vesicles containing DNA fusing with the outer membrane resulting in delivery of NETs to the extracellular space (Fig. 5d). Electron microscopy of the abscesses also identified the late stages of NETosis (Fig. 5e).
The abscess aspirates were subjected to immunofluorescence microscopy, which demonstrated normal and anuclear neutrophils and NETs (Fig. 6a). Anuclear PMN stain positively for CD45 and CD66b but do not stain for intracellular DNA and NETs are visualized surrounding these cells (Fig. 6a). Live abscess PMN were injected directly into mouse skin to visualize NETosis in the identical manner used for mouse intravital microscopy. NETs were visualized before and after exogenous DNase treatment (Fig. 6b). A large percentage of human abscess neutrophils had decondensed nuclei with approximately 10% being anuclear (Fig. 6d), just like in mouse PMN.
Figure 6.

Immunofluorescence imaging of NETosis during human abscess formation. (a) Fresh abscess aspirates were stained with a PE-conjugated CD66b specific antibody, a PerCP-conjugated anti-CD45 specific antibody and SYTOX Green. The white arrow highlights an anuclear PMN adjacent to a NET that is outlined by a dotted line. The yellow arrow highlights a normal PMN with multilobar condensed nuclei. (b) Fresh live abscess aspirates stained with PE-conjugated CD16 specific antibody were directly injected into mouse skin to mimic our in vivo experiments and nuclei were pre-labeled with SYTO 60. NETs were visualized and quantified using the NET ratio. Exogenous DNase decreased visible NETs and decreased the NET ratio. (c) Normal PMN were stained with PE-conjugated CD16 specific antibody and SYTO 60. PMN were injected into mouse skin alone or with GFP-S. aureus. A NET is being released from the PMN stimulated with bacteria and the GFP-staphylococcus can be seen attached to the NET. NET release by in vivo human PMN is quantified as NET ratio (** P < 0.01 for untreated versus treated). (d) PMN nuclei were quantified in vivo from uninfected normal human PMN, human PMN infected with S. aureus and in human abscess PMN.
Normal neutrophils isolated from healthy volunteers were injected into the mouse skin with or without live GFP-staphylococcus. Normal multilobar nuclei were observed in greater than 90% of human PMN in the absence of infection and these cells did not release NETs (Fig. 6c,d). However in the presence of infection, a large proportion of human PMN developed decondensed nuclei, released NETs and became anuclear (Fig. 6c,d).
Discussion
Despite ample in vitro NET research, fundamental questions about in vivo NETosis have remained unanswered, resulting in skepticism about the physiological relevance. For example, it has been suggested that NETs may just be an explosive necrosis, apoptosis or a process of dying10,11,30,31. Moreover it has been demonstrated that NET-formation is slow, requiring hours and that NETosing PMN are non-functional thereby nullifying other critical roles that PMN play in host defense. As a result, highly invasive Gram-positive pathogens, like S. aureus and S. pyogenes, could disseminate prior to cell death NETosis, easily slipping by the incapacitated PMN. As multiple bacteria upregulate DNase and escape NETs32–35 it is critical that the enucleating PMN maintain conventional killing capacity. We found that viable PMN rapidly form NETs while crawling and that phagocytosis could occur at any point during NETosis. Anuclear PMN contained bacteria implying that phagolysosome maturation and NET release can be separately compartmentalized, such that bacteria cannot escape from inside the cell during NET release.
Another critique of NETs has been the inability to observe the remnants of NETosing PMN in vivo. Here we directly observed PMN releasing DNA and altering their nuclear structures. Interestingly only a subgroup of neutrophils became anuclear while a larger population of NETosing PMN degraded the nucleus but continued to have diffuse intracellular DNA. Our data demonstrates the existence of viable anuclear PMN consistent with the common histological finding of suppurative or necrotizing inflammation. Death of the NETosing PMN is inevitable and likely results in clearance by resident macrophages; therefore they may not be observed at later time points in tissue histology.
The nucleus is important for de novo protein synthesis and it would seem reasonable that loss of this organelle would lead to rapid cell death. However, anuclear platelets and red blood cells survive for long periods and red blood cells survive enucleation16,36. Clearly, a terminally differentiated cell with a short half-life (8 h) should be able to survive a brief period without a nucleus. Indeed, physical removal of the nucleus from human PMN does not affect their conventional killing ability as these cytokineplasts crawl, transmigrate, phagocytose staphylococci and kill them via NO27–29,37 thus providing an interesting analogy to NETosing PMN. Clearly, temporary anuclear survival may be essential in this innate immune cell.
NETosis may lead to bystander tissue damage. Certainly vascular NETs can cause small vessel vasculitis38, hepatitis15, thrombosis39, or lupus nephritis40 and pulmonary NETs contribute to the pathophysiology of cystic fibrosis41,42. We observed a tight regulation of NET production, potentially required to limit collateral damage.
Finally it has been questioned how NETs are catapulted22 or forcibly expulsed in solid tissues. Our imaging reveals subtle release of DNA, analogous to a novel method of nuclear envelope budding and vesicular release of NETs in vitro14. We found that vesicular release of NETs permits the PMN to maintain membrane integrity that allows for continued function.
Together our data reveal that the physiological fate of NETosing PMN is not immediate cell death and that the release of NETs covered in elastase and histones is an important immune response to infection in vivo.
Methods and Materials
SYTOX Orange, SYTOX Green, SYTO 60, MitoTracker Deep Red, and Xenon Alexa Fluor 594 Goat IgG Labeling kit were from Invitrogen. Other reagents used include DNase (Sigma), MIP-2 (R&D Systems) and DRAQ5 (Biostatus Ltd). Antibody to histone H2A.X (M-20), antibody to neutrophil elastase (M-18) and goat polyclonal IgG were from Santa Cruz Biotechnology. Antibody to GR-1 (clone RB6-8C5, Ablab) was conjugated to Alexa Fluor 488 (Molecular Probes, Invitrogen) or Alexa Fluor 750 (Ablab) and PE-conjugated antibody to GR-1 was obtained from BD Biosciences. PE and FITC-conjugated antibody to Ly6G (clone 1A8) was obtained from Biolegend. PE-conjugated antibody to human CD16 (clone 3G8), PE-conjugated antibody to human CD66b (clone G10F5) and PerCP-conjugated antibody to human CD45 (clone HI30) were purchased from Biolegend.
Green fluorescence protein (GFP) reporter vector construction and GFP labelling of S. aureus (MRSA, USA300)
The GFP reporter vector was constructed by gene fusion of the promoterless GFP+ with a truncated S. aureus protein. Briefly, the GFP+ (a GFP variant with increased fluorescence) coding sequence with an in-frame polylinker was cleaved with KpnI/SpeI from the plasmid pMUTIN-GFP+ 43 and subcloned into an E. coli-S. aureus shuttle vector pBT244 at the restriction site created by XbaI/KpnI. The recombinant plasmid contained the promoterless gfp+ reporter gene, designated pBT2-gfp+, was replicated in E. coli and verified with PCR, sequencing and restriction analysis. The genomic DNA of a CA-MRSA USA300 local clinical strain (USA300-2406) was digested with KpnI/EcoRI. The resultant KpnI-EcoRI fragments were inserted into pBT2-gfp+ plasmid at KpnI/EcoRI restriction site and transformed into E. coli DH5α. The clone with the brightest green fluorescence was selected and the plasmid was purified. The gene sequencing revealed that a 1.8kb KpnI-EcoRI fragment containing a truncated SAUSA300_1226 gene which encodes a homoserine dehydrogenase45 was fused with the promoterless GFP+ in the subsequent plasmid, designated pBT2-gfp+ fused. This plasmid was further transformed into the laboratory chemical mutant strain S. aureus RN4220 by electroporation using tryptic soy agar (TSA) plates containing chloramphenicol. The pBT2-gfp+fused plasmids purified from RN4220 were then used for labelling of the clinical CA-MRSA (USA300) S. aureus isolate by electroporation and under chloramphenicol selection46.
Animals
C57BL/6J, Complement-3 deficient (C3−/−) and transgenic GFP fusion to histone H2B (Histone-GFP) (B6.Cg-Tg (HIST1H2BB/eGFP) 1Pa/J) mice were purchased from The Jackson Laboratory. Myd88−/− and Tlr2−/− mice were provided by Prof. Shizuo Akira (Osaka University, Japan). Dr. T.Graf (Barcelona, Spain) provided the Tg(LysMeGFP). Mice (20–35 g, 6–10 weeks old) were maintained in a pathogen-free environment. The mice had access to food and water ad libitum. All procedures performed were approved by the University of Calgary Animal Care Committee and were in accordance with the Canadian Guidelines for Animal Research.
Bacteria Strains
Three S. aureus strains were used in this study. S. aureus (Xen8.1 and Xen29), known pathogenic strains that have been used in soft tissue, biofilm and sepsis models, were purchased from Caliper Life Sciences. The GFP-transgenic CA-MRSA USA300-2406 was developed as previously described above. Staphylococcus Xen29 and Xen8.1 were grown in LB broth (200 μg ml−1 kanamycin) and CA-MRSA was grown in brain heart infusion BHI (chloramphenicol 20 μg ml−1) at 37 °C overnight. Bacteria were subcultured in fresh LB or BHI and incubated for 2 h in a 37 °C shaker to obtain bacteria in mid-log phase growth prior to injection. Streptococcus pyogenes was obtained from ATCC (ATCC19615). S. pyogenes was grown on a sheep blood plate overnight at 37 °C, and then a single colony from the blood agar plate was inoculated into 25 ml fresh BHI broth for 18 h at 37 °C.
Surgical procedures
Mice were anaesthetized (10 mg kg−1 xylazine hydrochloride and 200 mg kg−1 ketamine hydrochloride) and body temperature was maintained using a rectal probe and heating pad. The mice were pretreated with intradermal MIP-2 (0.2 μg) diluted in sterile normal saline or MIP-2 tissue superfusion (5 nM). The right jugular vein was cannulated to administer additional anesthetic and fluorescent dyes. The microcirculation of the dorsal skin was prepared for microscopy as previously described47. Briefly, after shaving the mouse’s back, a midline dorsal incision was made extending from the tail region up to the level of the occiput. The skin was separated from the underlying tissue, remaining attached laterally to ensure the blood supply remained intact. The area of skin was then extended over a viewing pedestal and secured along the edges using 4.0 sutures. The loose connective tissue lying on top of the dermal microvasculature was carefully removed by dissection under an operating microscope. The exposed dermal microvasculature was immersed in isotonic saline and covered with a coverslip held in place with vacuum grease. Alexa Fluor 488, Alexa Fluor 649, PE or FITC conjugated GR-1 specific antibody (10 μl per mouse i.v.) or the neutrophil specific conjugated antibody to Ly6G (clone 1A8, Biolegend) was used to visualize neutrophils.
Three different methods were used to visualize NETs in vivo. Method 1 used the membrane impermeable SYTOX Orange dye (diluted 1:1,000 with sterile saline, 100 μl per mouse i.v.). MIP-2 superfusion (5 nM) was initiated 30 min prior to S. aureus administration (Xen29, DNase positive, 1 × 108 CFU in 100 μl sterile saline i.d.). Method 2 used Xenon labeled histone specific antibody (M-20) or neutrophil elastase specific antibody (M-18) (20 μg per mouse i.v.). The Xenon labeling kit was modified from manufacturer’s instructions for in vivo experiments. Preliminary experiments were performed using various concentrations of component A and component B of the Xenon kit. Optimal visualization was found with 7.5 μl component A and 10 μl of antibody incubated at room temperature for 5 min, without the use of component B. Control IgG (20 μg per mouse i.v.) was labeled using the same Xenon kit and modified procedure. MIP-2 (0.2 μg, i.d.) was administered 60 min prior to S. aureus (Xen8.1, DNase negative, 1 × 108 CFU in 100 μl of sterile saline, i.d.). For method 3 mice received an i.p injection of the cell-permeable nucleic acid dye SYTO 60 (5 μl of stock 5 mM diluted in 200 μl PBS, i.p.) 16 h prior to intravital microscopy or i.v. administration during surgery. MIP-2 (0.2 μg, i.d.) was administered 60 min prior to the clinical CA-MRSA transgenic-GFP bacteria (USA300-2406, 1 × 108 CFU in 100 μl of sterile saline, i.d.).
Following baseline visualization, all bacteria were directly administered into the field of view using a tuberculin needle (1 × 108 CFU in 100 μl of sterile saline, i.d.). Three to six separate areas were visualized in each experiment in order to minimize selection bias. For killed bacteria experiments (Xen8.1) bacteria was heated for 30 min at 65 °C and washed twice with sterile saline (n = 3).
Spinning-Disk Confocal Intravital Microscopy (SDCIM)
Spinning-disk confocal intravital microscopy was performed using an Olympus BX51 (Olympus) upright microscope equipped with a 20×/0.95 XLUM Plan Fl water immersion objective. The microscope was equipped with a confocal light path (WaveFx, Quorum) based on a modified Yokogawa CSU-10 head (Yokogawa Electric Corporation). Laser excitation at 488, 561, 649 and 730 nm (Cobalt), was used in rapid succession and fluorescence in green, red and blue channels was visualized with the appropriate long pass filters (Semrock). Exposure time for all wavelengths was constant at 1 s. Sensitivity settings were maintained at the same level for all experiments. A 512×512 pixels back-thinned EMCCD camera (C9100-13, Hamamatsu) was used for fluorescence detection. Volocity Acquisition software (Improvision Inc.) was used to drive the confocal microscope. Images captured using the spinning disk were processed and analyzed in Volocity 4.20. NET area was quantified using the Volocity software.
Bone Marrow Chimera Animals
Recipient C57BL/6 mice were irradiated with 2 doses of 5 Gy (Gammacell 40 137Cs γ-irradiation source; Nordion International). An interval of 3 h was allowed to pass between the first and second irradiations. Transgenic histone-GFP bone marrow was isolated from mice euthanized by spinal cord displacement and 8 × 106 donor bone marrow cells were injected into the tail vein of recipient irradiated mice. The mice were kept in microisolator cages for 8 weeks to allow full humoral reconstitution. This protocol previously confirmed that 99% of leukocytes in Thy1.1 and Thy1.2 congenic recipient mice were from donor bone marrow48.
Neutrophil Behavior
Individual neutrophils were manually tracked to determine velocity, distance, displacement, pseudopod formation and cell contouring. Cells that remained within the field of view for 5 min were examined. Volocity software was used to calculate velocity, distance and displacement.
Bacterial Dissemination
S. aureus (Xen8.1) were grown to mid-log phase, washed and suspended in saline. Mice were injected i.p. with 1 × 108 CFUs of the bacterial preparation in a volume of 200 uL. At 5–6 h after inoculation, mice were anaesthetized and whole blood was collected. Serial dilutions were cultured on LB agar plates overnight at 37 °C. To directly assess the effect of NETs on bacterial dissemination mice received a dorsal subcutaneous live mid-log phase inoculation of staphylococcus (Xen8.1, 1 × 108 CFU in 100 μl injection of saline). 1 h post bacterial administration mice received an injection of either DNase (1,000 U) or saline into the infected skin. Dissemination was quantified by obtaining blood via a cardiac puncture in anaesthetized animals at 4 h post-bacterial inoculation, diluting it 1:1 with PBS and plating it on LB kanamycin for 24 h.
In vivo Bacterial Monitoring
An IVIS Imaging System 200 (Caliper Life Sciences) was used to quantify luminescent live staphylococcus (Xen8.1) over time. Live mid-log phase Staphylococcus aureus (Xen8.1, 1 × 108 CFU in 100 μl injection of saline) was administered subcutaneously into shaved mice. Animals were anesthetized with 1.5% isoflurane while in the imaging chamber. Photons were measured during a 30 s exposure. Total photon emissions from uniform area of each mouse were quantified by using the Living Image software (Caliper Life Sciences). Mice were imaged at 1 h, 4 h and 8 h post-bacterial inoculation. At 24 h, the mice were euthanized and a skin biopsy (3.5 mm punch) was obtained from within the injection site. The tissue was homogenized and plated on LB agar plates with kanamycin and grown overnight to quantify CFU.
Human PMN Isolation
Whole blood was collected into acid citrate dextrose (1:5 blood, v/v). Erythrocytes were removed using dextran sedimentation followed by two rounds of hypotonic lysis. Neutrophils were isolated from the resulting cell suspension using Ficoll-Histopaque density centrifugation. PMN were stained ex vivo using PE-conjugated CD16 specific antibody and SYTO 60 (1:1,000 dilution) for 15 min prior to directed intradermal injection into the mouse skin prep. Ethical approval for obtaining healthy human volunteer blood was provided by The University of Calgary research ethics committee and each human subject provided informed consent.
Human Abscesses
The infectious disease specialty consultation service of the Division of Infectious Diseases in the Dept. of Medicine, Alberta Health Services – Calgary and Area, identified patients who were clinically diagnosed with cellulitis and secondary abscesses due to Gram-positive bacteria requiring wound aspiration as part of routine medical care. All patients were fully reviewed by a physician with specialty training in infectious diseases. Specimens were sent for routine Gram-stain and culture. Remaining samples were used for electron microscopy, immunofluorescence and in vivo spinning disk confocal experiments with consent of each patient. The University of Calgary research ethics committee approved the use of discarded human clinical samples.
Transmission Electron Microscopy
Fresh clinical aspirates were fixed with 2.5% glutaraldehyde. The cells were spun into a pellet using a microcentrifuge. After washing three times, the pellets were postfixed in 1% osmium tetroxide in cacodylate buffer for 1 h, dehydrated through a graded series of acetone, and embedded in epoxy-based resin. Ultrathin sections were cut in a Reichert-Jung Ultracut E microtome using a diamond knife and stained with 4% aqueous uranyl acetate and Reynolds’ lead citrate. The sections were observed in a Hitachi H7650 transmission electron microscope at 80 kV, and images were taken with an AMT 16000 digital camera mounted on the microscope microscope14.
Immunofluorescence Microscopy
Slides (smears) were prepared from clinical aspirates and allowed to air dry. Slides were fixed in ice-cold acetone for 10 min and dried. Slides were then blocked for 2 h at room temperature in PBS containing 2% BSA followed by staining for 1 h with a cocktail of fluorescently-labeled Ab together with SYTOX Green. Slides were washed and coverslips mounted with ProLong Gold antifade reagent (Invitrogen). Slides were imaged using a spinning-disk confocal microscope consisting of an Olympus IX81 inverted microscope (Olympus) coupled to a confocal light path (WaveFx; Quorum Technologies) based on a modified Yokogawa CSU-10 head (Yokogawa Electric Corporation). Excitation by each of 491-, 561-, and 642-nm laser wavelengths (Cobolt) were sequentially controlled and merged into a single optic cable using an LMM5 laser merge module (Spectral Applied Research) and fluorescence was visualized through band pass emission filters (Semrock) driven by a Modular Automation Controller (Ludi Electronic Products, Ltd.) and detected with a 512×512 pixels back-thinned EMCCD camera (C9100-13, Hamamatsu). Volocity Acquisition software (V5.2.1) (Improvision Inc.) was used to drive the confocal microscope
Statistics
Data were analyzed using GraphPad Prism (version 4, GraphPad Software Inc.). Reported values are expressed as mean ± s.e.m unless otherwise described. We assessed the statistical significance of the difference between 2 sets of data using an unpaired, 2-tailed t test. Where the difference between more than 2 sets of data was analyzed, we used a one-way analysis of variance, followed by Bonferroni multiple-comparisons test. All P values of less than 0.05 were considered statistically significant.
Supplementary Material
Live emigrated neutrophils rapidly form NETs in vivo within Gram-positive infected skin. Spinning disk confocal intravital microscopy was performed using SYTOX Orange to visualize extracellular DNA. Neutrophils (green) chemotax through tissue NETs (red) in response to S. aureus (Xen29). PMN do not take up the vital dye SYTOX. Neutrophils are hyperpolarized and form multiple pseudopods. White arrows demonstrate aberrant PMN crawling throughout NETs with hyperpolarization and multiple pseudopods.
Live emigrated neutrophils rapidly release extracellular histones as a component of NETs during a Gram-positive infection. PE-conjugated histone specific antibody demonstrates extracellular histone proteins (red) as a major component of NET structure. Live PMN chemotax throughout the tissue in response to S. aureus (Xen8.1).
Live emigrated PMN undergo NETosis while crawling during a Gram-positive infection. In vivo PMN nuclei were pre-labeled with the cell-permeable dye SYTO 60. The NETosing PMN continues to crawl toward the live GFP-staphylococcus. Released NETs surround the PMN. At the conclusion of the video we changed to z-stack imaging and this PMN is highlighted in Figure 2a. During the 3D imaging this PMN eventually engulfs the GFP-bacteria seen in the video.
Live PMN form NETs while crawling in vivo. Four-color spinning disk confocal intravital microscopy was used to visualize NET release from live PMN in response to Gram-positive skin infection. A PMN (yellow) that is crawling while releasing NETs is circled. Extracellular DNA NETs are visualized using the cell-impermeable DNA dye SYTOX Orange. GFP-staphylococcus can be seen within this crawling PMN. A PMN with retained intracellular nuclear dye (SYTO 60, blue) can be seen crawling to the right of the highlighted NETTing PMN. This cell is not releasing NETs.
In vivo intracellular imaging demonstrates PMN with normal, diffuse and absent nuclei during a Gram-positive skin infection. Normal nuclei are easily distinguished by their distinct multilobar morphology. Anuclear PMN are alive and continue to migrate. Examples are demonstrated of anuclear PMN engorged with GFP-staphylococcus as well as anuclear PMN devoid of bacteria. A PMN with an abnormal diffuse nucleus is highlighted for its multiple and erratic pseudopod formation and hyperpolarization. NETs are visualized within the area of anuclear PMN.
Acknowledgments
The pBT2 was a gift from R. Brückner (University of Kaiserslautern, Germany). We thank D. Knight and C. Baddick for technical assistance and ongoing support. We acknowledge P. Colarusso and the support staff of the Snyder Institute Live Cell Imaging Facility for assisting in the image capturing and analysis (Canada Foundation for Innovation funded). We also thank the University of Calgary Flow Cytometry Facility and L. Kennedy for their assistance. We are grateful to P. Forsyth for use of the IVIS 200. We thank the physicians, nurses and support staff in the Division of Infectious Diseases in the Dept. of Medicine, Alberta Health Services-Calgary and Area for assistance in obtaining clinical samples. We thank E. Yung, C. Horn, K. Nelson and K. Headley from Calgary Lab Services for assistance with the electron microscopy experiments.
The Canadian Institute of Health Research (CIHR) provided the operating grants to support this work. P. Kubes is an Alberta Innovates – Health Solutions (AIHS) Scientist, Canada Research Chair, and the Snyder Chair in Critical Care Medicine. B. Petri received an AIHS postdoctoral fellowship. B. Yipp is a Clinical Scholar (Department of Critical Care Medicine, Calgary), an AIHS clinical fellow and a CIHR fellow. During a portion of this study he received salary support from the Rockefeller University by Grant Award Number UL1RR024143 from the National Center for Research Resources (NCRR), a component of the US National Institutes of Health (NIH) and NIH Roadmap for Medical Research. The contents are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH.
Footnotes
The authors declare no conflict-of-interest or competing financial interests.
Author Contributions
B.G.Y. and B.P. designed the overall study, performed experiments, analyzed the data and wrote the manuscript, D.S. designed, performed and analyzed the TEM experiments, C.N.J. designed, performed and analyzed the immunofluorescence microscopy experiments, B.N.V.S performed intravital imaging, L.D.Z performed intravital imaging, Xenogen and dissemination experiments and analyzed the data, K.P. performed human neutrophil experiments, M.A. performed in vitro NETs assays, K.W. developed the transgenic bacteria, H.C.M helped develop the imaging analysis protocols, S.E.M and A.B.C designed and performed the human cytoplast experiments, K.Z. designed and supervised the development of the transgenic bacteria and provided expert advice on microbiology, J.C. designed the clinical experiments, supervised the acquisition of patient samples and clinical histories and provided expert advice on infectious diseases and microbiology, P.K. provided overall supervision, analyzed the data and wrote the manuscript.
References
- 1.Kollef MH, Micek ST. Methicillin-resistant Staphylococcus aureus: a new community-acquired pathogen? Curr Opin Infect Dis. 2006;19:161–168. doi: 10.1097/01.qco.0000216627.13445.e2. [DOI] [PubMed] [Google Scholar]
- 2.Skrupky LP, Micek ST, Kollef MH. Bench-to-bedside review: Understanding the impact of resistance and virulence factors on methicillin-resistant Staphylococcus aureus infections in the intensive care unit. Crit Care. 2009;13:222. doi: 10.1186/cc8028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Angus DC, et al. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 4.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 5.Cole JN, Barnett TC, Nizet V, Walker MJ. Molecular insight into invasive group A streptococcal disease. Nat Rev Microbiol. 2011;9:724–736. doi: 10.1038/nrmicro2648. [DOI] [PubMed] [Google Scholar]
- 6.Li P, et al. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. 2010;207:1853–1862. doi: 10.1084/jem.20100239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nauseef WM. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev. 2007;219:88–102. doi: 10.1111/j.1600-065X.2007.00550.x. [DOI] [PubMed] [Google Scholar]
- 8.Segal AW. How neutrophils kill microbes. Annu Rev Immunol. 2005;23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brinkmann V, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 10.Brinkmann V, Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol. 2007;5:577–582. doi: 10.1038/nrmicro1710. [DOI] [PubMed] [Google Scholar]
- 11.Fuchs TA, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176:231–241. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee WL, Harrison RE, Grinstein S. Phagocytosis by neutrophils. Microbes Infect. 2003;5:1299–1306. doi: 10.1016/j.micinf.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 13.Flannagan RS, Cosio G, Grinstein S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol. 2009;7:355–366. doi: 10.1038/nrmicro2128. [DOI] [PubMed] [Google Scholar]
- 14.Pilsczek FH, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol. 2010;185:7413–7425. doi: 10.4049/jimmunol.1000675. [DOI] [PubMed] [Google Scholar]
- 15.Clark SR, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nature medicine. 2007;13:463–469. doi: 10.1038/nm1565. [DOI] [PubMed] [Google Scholar]
- 16.Ji P, Jayapal SR, Lodish HF. Enucleation of cultured mouse fetal erythroblasts requires Rac GTPases and mDia2. Nat Cell Biol. 2008;10:314–321. doi: 10.1038/ncb1693. [DOI] [PubMed] [Google Scholar]
- 17.Friedl P, Wolf K, Lammerding J. Nuclear mechanics during cell migration. Curr Opin Cell Biol. 2011;23:55–64. doi: 10.1016/j.ceb.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dahl KN, Booth-Gauthier EA, Ladoux B. In the middle of it all: mutual mechanical regulation between the nucleus and the cytoskeleton. J Biomech. 2010;43:2–8. doi: 10.1016/j.jbiomech.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184:205–213. doi: 10.1083/jcb.200806072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191:677–691. doi: 10.1083/jcb.201006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009;16:1438–1444. doi: 10.1038/cdd.2009.96. [DOI] [PubMed] [Google Scholar]
- 22.Yousefi S, et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med. 2008;14:949–953. doi: 10.1038/nm.1855. [DOI] [PubMed] [Google Scholar]
- 23.Hajishengallis G, Lambris JD. Crosstalk pathways between Toll-like receptors and the complement system. Trends Immunol. 2010;31:154–163. doi: 10.1016/j.it.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hakkim A, et al. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol. 2011;7:75–77. doi: 10.1038/nchembio.496. [DOI] [PubMed] [Google Scholar]
- 25.Yan J, et al. Glutathione Reductase Facilitates Host Defense by Sustaining Phagocytic Oxidative Burst and Promoting the Development of Neutrophil Extracellular Traps. J Immunol. 2012 doi: 10.4049/jimmunol.1102683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malawista SE, Van Blaricom G. Phagocytic capacity of cytokineplasts from human blood polymorphonuclear leukocytes. Blood Cells. 1986;12:167–177. [PubMed] [Google Scholar]
- 27.Malawista SE, Van Blaricom G. Cytoplasts made from human blood polymorphonuclear leukocytes with or without heat: preservation of both motile function and respiratory burst oxidase activity. Proc Natl Acad Sci U S A. 1987;84:454–458. doi: 10.1073/pnas.84.2.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malawista SE, Van Blaricom G, Breitenstein MG. Cryopreservable neutrophil surrogates. Stored cytoplasts from human polymorphonuclear leukocytes retain chemotactic, phagocytic, and microbicidal function. J Clin Invest. 1989;83:728–732. doi: 10.1172/JCI113939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malawista SE, Montgomery RR, van Blaricom G. Evidence for reactive nitrogen intermediates in killing of staphylococci by human neutrophil cytoplasts. A new microbicidal pathway for polymorphonuclear leukocytes. J Clin Invest. 1992;90:631–636. doi: 10.1172/JCI115903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Remijsen Q, et al. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. 2011;18:581–588. doi: 10.1038/cdd.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amulic B, Hayes G. Neutrophil extracellular traps. Curr Biol. 2011;21:R297–298. doi: 10.1016/j.cub.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 32.Berends ET, et al. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J Innate Immun. 2010;2:576–586. doi: 10.1159/000319909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sumby P, et al. Extracellular deoxyribonuclease made by group A Streptococcus assists pathogenesis by enhancing evasion of the innate immune response. Proc Natl Acad Sci U S A. 2005;102:1679–1684. doi: 10.1073/pnas.0406641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buchanan JT, et al. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol. 2006;16:396–400. doi: 10.1016/j.cub.2005.12.039. [DOI] [PubMed] [Google Scholar]
- 35.Walker MJ, et al. DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat Med. 2007;13:981–985. doi: 10.1038/nm1612. [DOI] [PubMed] [Google Scholar]
- 36.Ji P, Murata-Hori M, Lodish HF. Formation of mammalian erythrocytes: chromatin condensation and enucleation. Trends Cell Biol. 2011;21:409–415. doi: 10.1016/j.tcb.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malawista SE, De Boisfleury Chevance A. The cytokineplast: purified, stable, and functional motile machinery from human blood polymorphonuclear leukocytes. J Cell Biol. 1982;95:960–973. doi: 10.1083/jcb.95.3.960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kessenbrock K, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. 2009;15:623–625. doi: 10.1038/nm.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fuchs TA, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. 2010;107:15880–15885. doi: 10.1073/pnas.1005743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hakkim A, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A. 2010;107:9813–9818. doi: 10.1073/pnas.0909927107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manzenreiter R, et al. Ultrastructural characterization of cystic fibrosis sputum using atomic force and scanning electron microscopy. J Cyst Fibros. 2011 doi: 10.1016/j.jcf.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 42.Young RL, et al. Neutrophil extracellular trap (NET)-mediated killing of Pseudomonas aeruginosa: evidence of acquired resistance within the CF airway, independent of CFTR. PLoS One. 2011;6:e23637. doi: 10.1371/journal.pone.0023637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaltwasser M, Wiegert T, Schumann W. Construction and application of epitope- and green fluorescent protein-tagging integration vectors for Bacillus subtilis. Appl Environ Microbiol. 2002;68:2624–2628. doi: 10.1128/AEM.68.5.2624-2628.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bruckner R. Gene replacement in Staphylococcus carnosus and Staphylococcus xylosus. FEMS Microbiol Lett. 1997;151:1–8. doi: 10.1111/j.1574-6968.1997.tb10387.x. [DOI] [PubMed] [Google Scholar]
- 45.Diep BA, et al. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet. 2006;367:731–739. doi: 10.1016/S0140-6736(06)68231-7. [DOI] [PubMed] [Google Scholar]
- 46.Wu K, et al. Caenorhabditis elegans as a host model for community-associated methicillin-resistant Staphylococcus aureus. Clin Microbiol Infect. 2010;16:245–254. doi: 10.1111/j.1469-0691.2009.02765.x. [DOI] [PubMed] [Google Scholar]
- 47.Ho M, Hickey MJ, Murray AG, Andonegui G, Kubes P. Visualization of Plasmodium falciparum-endothelium interactions in human microvasculature: mimicry of leukocyte recruitment. The Journal of experimental medicine. 2000;192:1205–1211. doi: 10.1084/jem.192.8.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carvalho-Tavares J, et al. A role for platelets and endothelial selectins in tumor necrosis factor-alpha-induced leukocyte recruitment in the brain microvasculature. Circ Res. 2000;87:1141–1148. doi: 10.1161/01.res.87.12.1141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Live emigrated neutrophils rapidly form NETs in vivo within Gram-positive infected skin. Spinning disk confocal intravital microscopy was performed using SYTOX Orange to visualize extracellular DNA. Neutrophils (green) chemotax through tissue NETs (red) in response to S. aureus (Xen29). PMN do not take up the vital dye SYTOX. Neutrophils are hyperpolarized and form multiple pseudopods. White arrows demonstrate aberrant PMN crawling throughout NETs with hyperpolarization and multiple pseudopods.
Live emigrated neutrophils rapidly release extracellular histones as a component of NETs during a Gram-positive infection. PE-conjugated histone specific antibody demonstrates extracellular histone proteins (red) as a major component of NET structure. Live PMN chemotax throughout the tissue in response to S. aureus (Xen8.1).
Live emigrated PMN undergo NETosis while crawling during a Gram-positive infection. In vivo PMN nuclei were pre-labeled with the cell-permeable dye SYTO 60. The NETosing PMN continues to crawl toward the live GFP-staphylococcus. Released NETs surround the PMN. At the conclusion of the video we changed to z-stack imaging and this PMN is highlighted in Figure 2a. During the 3D imaging this PMN eventually engulfs the GFP-bacteria seen in the video.
Live PMN form NETs while crawling in vivo. Four-color spinning disk confocal intravital microscopy was used to visualize NET release from live PMN in response to Gram-positive skin infection. A PMN (yellow) that is crawling while releasing NETs is circled. Extracellular DNA NETs are visualized using the cell-impermeable DNA dye SYTOX Orange. GFP-staphylococcus can be seen within this crawling PMN. A PMN with retained intracellular nuclear dye (SYTO 60, blue) can be seen crawling to the right of the highlighted NETTing PMN. This cell is not releasing NETs.
In vivo intracellular imaging demonstrates PMN with normal, diffuse and absent nuclei during a Gram-positive skin infection. Normal nuclei are easily distinguished by their distinct multilobar morphology. Anuclear PMN are alive and continue to migrate. Examples are demonstrated of anuclear PMN engorged with GFP-staphylococcus as well as anuclear PMN devoid of bacteria. A PMN with an abnormal diffuse nucleus is highlighted for its multiple and erratic pseudopod formation and hyperpolarization. NETs are visualized within the area of anuclear PMN.