

Abstract
Objective:
We aimed to compare the cytokine and chemokine profiles of patients with multifocal motor neuropathy (MMN) with those of patients with progressive muscular atrophy (PMA) and amyotrophic lateral sclerosis (ALS) to investigate immunologic differences in the CNS.
Methods:
CSF from 12 patients with MMN, 8 with PMA, 26 with sporadic ALS, and 10 with other noninflammatory neurologic disorders was analyzed for 27 cytokines and chemokines using the multiplex bead array assay. Cytokine titers of the 4 groups were compared, and correlations between the titers of relevant cytokines and clinical parameters were evaluated.
Results:
There were no obvious intrathecal changes except for interleukin (IL)-1 receptor antagonist in patients with MMN. In contrast, IL-4, IL-7, IL-17, eotaxin/CCL11, fibroblast growth factor-2 (FGF-2), granulocyte colony-stimulating factor (G-CSF), and platelet-derived growth factor BB titers were significantly elevated in patients with PMA and ALS; of these, FGF-2 and G-CSF titers were elevated compared with those in patients with MMN. IL-4 and IL-10 titers were high in patients with ALS, particularly patients with possible ALS presenting with a slowly progressive course or mild symptoms.
Conclusions:
The CSF cytokine profile of patients with MMN is distinct from that of patients with PMA and ALS. The similarity of the cytokine profiles between patients with PMA and ALS suggests that PMA shares common immunologic features with ALS in the CNS, even without clinical evidence of upper motor neuron involvement.
Multifocal motor neuropathy (MMN) is an immune-mediated neuropathy characterized by the lower motor neuron (LMN) syndrome, typically involving asymmetric muscle atrophy and weakness of the distal upper limbs.1–3 The early and accurate diagnosis of MMN is critical because immunotherapy, such as IV immunoglobulin, is often effective. The diagnostic features of MMN are conduction block (CB) in multiple peripheral nerves and anti-GM1 IgM antibodies.4–6 In cases lacking those features, however, MMN is often underdiagnosed7–9 or misdiagnosed as amyotrophic lateral sclerosis (ALS) or progressive muscular atrophy (PMA), a pure LMN variant of motor neuron disease.10 Conversely, ALS and PMA may be misdiagnosed as MMN because ALS may lack apparent upper motor neuron (UMN) signs11,12 and PMA by definition does not present any UMN signs during the whole course, even though it is suspected to be a form of ALS.13,14 It is important to distinguish MMN from PMA or ALS. We recently found that proinflammatory cytokines are elevated in the sera of patients with MMN, whereas few cytokine abnormalities are observed in the sera of patients with ALS.15 Instead, cytokine abnormalities have been reported in the CSF of patients with ALS, suggesting that CNS inflammation plays a crucial role.16–18 However, it is unclear whether CSF cytokine profiles differ between patients with MMN and patients with motor neuron disease (PMA and ALS) because of the paucity of data for MMN and PMA. To address this issue, we evaluated multiple cytokine/chemokine levels in the CSF of patients with MMN, PMA, and ALS.
METHODS
Patients.
We conducted a retrospective case-control study. The diagnosis of MMN was based on the diagnostic categories proposed by the European Federation of Neurological Societies and the Peripheral Nerve Society.19 We defined PMA according to the criteria described previously13: (1) diagnosed within 5 years; (2) clinical and electrophysiologic evidence of LMN involvement in 2 or more of 4 regions (bulbar, cervical, thoracic, and lumbosacral); (3) no CB in nerve conduction studies; and (4) no clinical UMN signs and symptoms. The diagnosis of ALS was made using the revised El Escorial criteria; patients fulfilling the “clinically definite,” “clinically probable,” “clinically probable—laboratory-supported,” or “possible” criteria were diagnosed with ALS.20 CSF samples were obtained from 10 patients with other noninflammatory neurologic disorders (ONDs), 12 with MMN (untreated), 8 with PMA, and 26 with ALS. All samples were immediately stored at −80°C until analysis. We determined sex, age, disease duration (time from symptom onset to CSF sampling), revised ALS Functional Rating Scale (ALSFRS-R) score,21 and disease progression rate (ΔALSFRS-R). ΔALSFRS-R was defined as [(ALSFRS-R full score – ALSFRS-R score at sampling)/disease duration expressed in months].18 Electrophysiologic studies were performed with commercially available EMG machines. Unilateral median, ulnar, tibial, and additional nerves when involvement was clinically suspected (e.g., radial, deep peroneal nerves) for motor studies, F-waves, and median, ulnar, and sural nerves for sensory studies were tested. CB was defined as a reduction in compound muscle action potential amplitude/area of >50% from distal to proximal stimulation in the absence of abnormal temporal dispersion.19 The presence of IgM and IgG antibodies against GM1, GM2, GD1a, GD1b, GM1b, GT1a, GT1b, GQ1b, GalNac-GD1a, and sulfate-3-glucuronyl paragloboside was tested in patients with MMN by conventional ELISA at Kinki University.22
Standard protocol approvals, registrations, and patient consents.
The procedures followed were in accordance with the Helsinki Declaration of 1975, as revised in 1983. This study was approved by the Ethics Committee of the Tokushima University Hospital. All participants gave written informed consent.
Cytokine and chemokine assays.
We performed multiplex bead array assay of serum and CSF samples using the Bio-Plex Pro Human Cytokine 27-plex Assay (Bio-Plex, Hercules, CA), as described previously.23 The panel was comprised of interleukin (IL)-1β; IL-1 receptor antagonist (IL-1ra); IL-2; IL-4; IL-5; IL-6; IL-7; IL-8/CXCL8; IL-9; IL-10; IL-12 (p70); IL-13; IL-15; IL-17; eotaxin/CCL11; fibroblast growth factor-2 (FGF-2); granulocyte colony-stimulating factor (G-CSF); granulocyte-macrophage colony-stimulating factor (GM-CSF); interferon (IFN)-γ; IFN-γ–induced protein 10 (IP-10)/CXCL10; monocyte chemotactic protein-1 (MCP-1)/CCL2; macrophage inflammatory protein (MIP) 1α/CCL3; MIP-1β/CCL4, platelet-derived growth factor BB (PDGF-BB); regulated on activation, normal T cell expressed and secreted/CCL5; tumor necrosis factor α (TNF-α); and vascular endothelial growth factor (VEGF). Soluble TNF receptor (TNFR1) was determined with an ELISA kit (BMS03; Cosmo Bio, Tokyo, Japan).
Statistical analysis.
Differences in cytokine levels among the 4 groups (ONDs, MMN, PMA, and ALS) were compared by the Kruskal-Wallis test followed by Dunn multiple comparison post hoc analysis. Subsequently, differences in cytokine levels among the subgroups (MMN, PMA, possible ALS, and ALS except possible) were also analyzed. Differences in clinical data were assessed with Kruskal-Wallis, χ2, Student t, and Mann-Whitney U tests. Correlations between cytokine levels and clinical parameters (age, disease duration, ALSFRS-R score, and ΔALSFRS-R) were assessed with Spearman rank correlation test. p values <0.05 were considered statistically significant. The data were analyzed using GraphPad Prism5 and SPSS20.
RESULTS
Clinical manifestations.
We enrolled 12 patients with MMN (12 male; age 41.1 ± 16.4 years), 8 with PMA (7 male; age 67.1 ± 9.4 years), and 26 with ALS (15 male; age 65.0 ± 8.9 years). The diagnosis of ALS was definite in 7, probable in 6, probable—laboratory-supported in 5, and possible in 8. Region of ALS onset was bulbar in 7, trunk in 3, upper limbs in 10, and lower limbs in 6. We enrolled 10 patients with ONDs (9 male; age 70.3 ± 11.4 years) as controls: 7 had idiopathic normal pressure hydrocephalus, 2 had myalgia/neuralgia, and 1 had tension headache. Of the 12 patients with MMN, 7 (58.3%) showed CB and 2 (16.7%) had anti-GM1 IgM antibodies. Other clinical and CSF findings are listed in table 1.
Table 1.
Clinical and CSF profiles
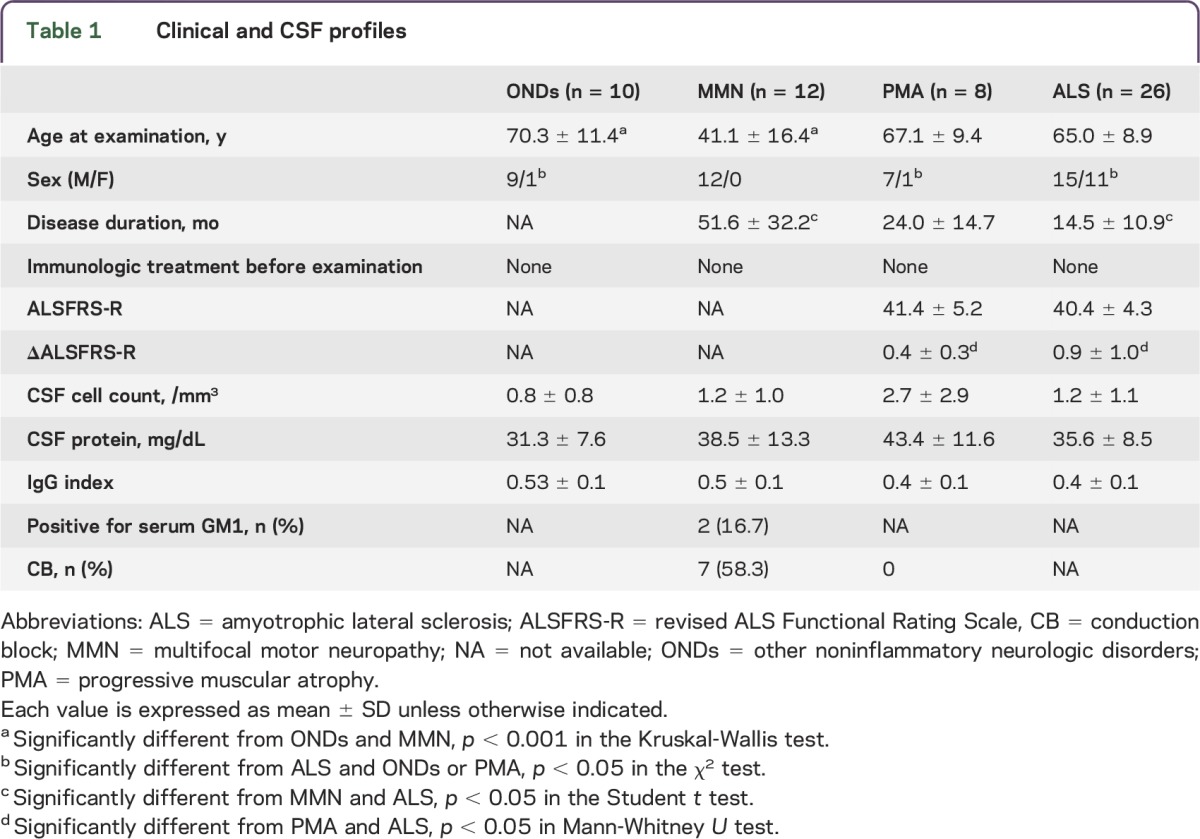
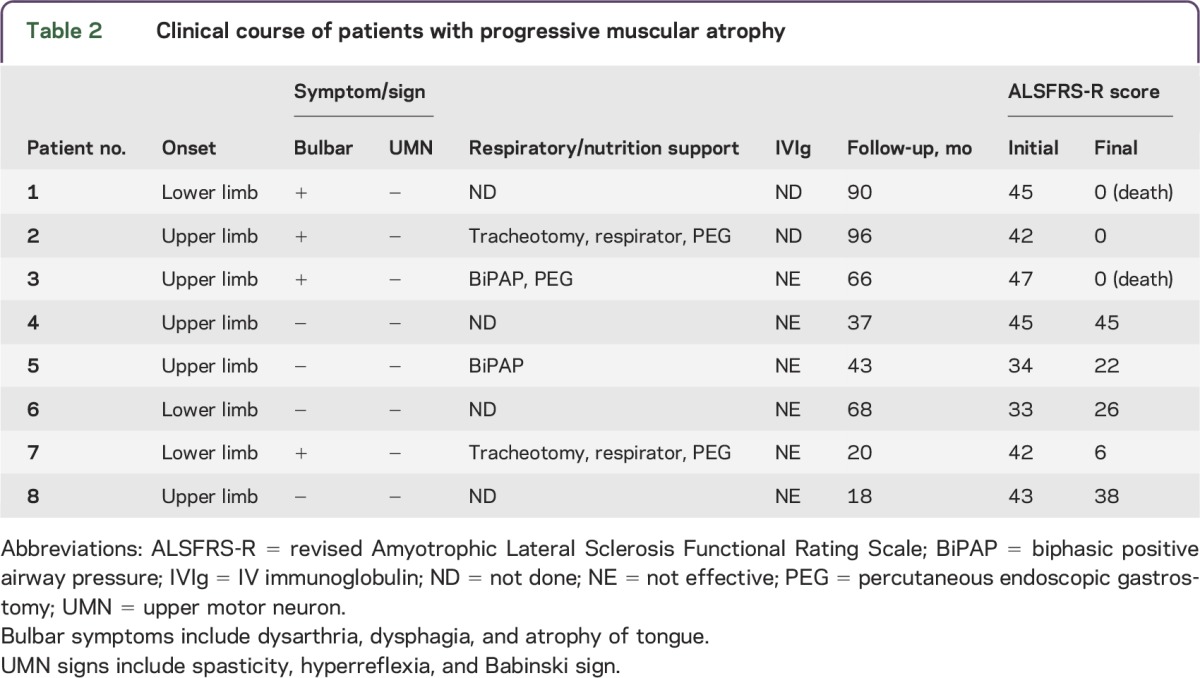
We compared the clinical profiles of patients with PMA with those of patients with ALS. The disease duration was longer in patients with PMA than in patients with ALS. Although ALSFRS-R scores did not differ (table 1), ΔALSFRS-R was lower in patients with PMA than in patients with ALS (p < 0.05). Region of PMA onset was upper limbs in 5 patients and lower limbs in 3. Four of the 8 patients with PMA required respiratory or nutrition support and 2 died of respiratory failure. Six patients with PMA received IV immunoglobulin (after CSF sampling) because the diagnosis of MMN without CB could not be excluded in the early stages; however, none responded to the treatment. None of the patients presented with UMN signs in the follow-up period, and all of them presented with progressive courses (observation time 54.8 ± 29.9 months). Overall, although none of the patients with PMA met the diagnostic criteria for ALS, the clinical features suggested that they presented with a variant phenotype of ALS (table 2). We also assessed the clinical profiles of patients with possible ALS and patients with probable/definite ALS. Patients with possible ALS showed slightly higher ALSFRS-R scores (43.6 ± 2.9) and lower ΔALSFRS-R (0.45 ± 0.22) than patients with probable/definite ALS (ALSFRS-R 40.4 ± 4.3; ΔALSFRS-R 0.76 ± 0.84), but there were no significant differences between them.
Table 2.
Clinical course of patients with progressive muscular atrophy
CSF cytokine profiles.
IL-1ra was the only cytokine whose level was increased in patients with MMN compared with patients with ONDs. In contrast, as many as 10 cytokines/chemokines showed higher levels in patients with PMA than patients with ONDs: IL-1ra, IL-7, IL-10, IL-17, eotaxin/CCL11, FGF-2, G-CSF, PDGF-BB, VEGF, and TNFR1. Of these, IL-10, FGF-2, G-CSF, and VEGF were markedly elevated compared with patients with MMN as well (table 3; figure 1). IL-4, IL-7, IL-17, eotaxin/CCL11, FGF-2, G-CSF, and PDGF-BB levels were higher in patients with ALS than patients with ONDs; of these, IL-4, IL-17, FGF-2, and G-CSF levels were elevated compared with patients with MMN as well (table 3; figure 1). Both patients with PMA and patients with ALS had elevated levels of IL-7, IL-17, eotaxin/CCL11, FGF-2, G-CSF, and PDGF-BB. CSF cytokine profiles were different between patients with MMN and patients with motor neuron disease but were similar between patients with the 2 motor neuron diseases (PMA and ALS).
Table 3.
Cytokine/chemokine profiles in CSF
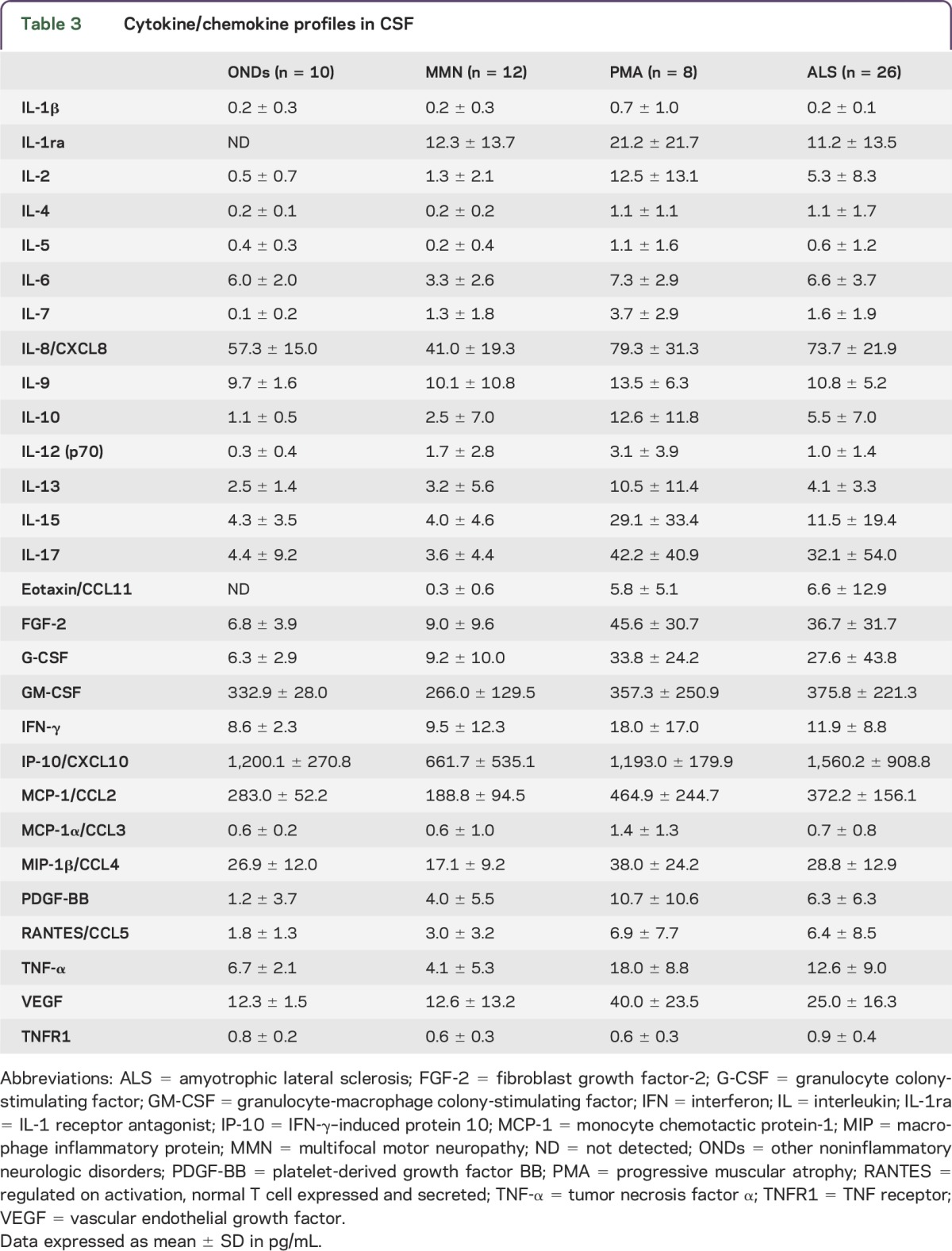
Figure 1. Cytokine titers determined by the multiplex bead assay.

Plotted are cytokine titers (pg/mL) in CSF of patients with other noninflammatory neurologic diseases (ONDs, n = 10), multifocal motor neuropathy (MMN, n = 12), progressive muscular atrophy (PMA, n = 8), and amyotrophic lateral sclerosis (ALS, n = 26).*p < 0.05, **p < 0.01. FGF-2 = fibroblast growth factor-2; G-CSF = granulocyte colony-stimulating factor; IL = interleukin; IL-1ra = IL-1 receptor antagonist; IP-10 = interferon-γ–induced protein 10; MCP-1 = monocyte chemotactic protein-1; PDGF-BB = platelet-derived growth factor BB; TNF-α = tumor necrosis factor α; VEGF = vascular endothelial growth factor.
IL-10 level was elevated in patients with ALS with high ALSFRS-R scores (r = 0.415, p = 0.035), indicating that elevation of IL-10 is associated with milder symptoms of the disease. IL-4 and eotaxin/CCL11 levels were higher in patients with ALS with lower ΔALSFRS-R (r = −0.454, p = 0.026 and r = −0.579, p = 0.003, respectively), indicating that those cytokines are found at higher levels in patients with slower disease progression (figure 2). In the subgroup analysis, cytokine patterns were not significantly different between patients with PMA and patients with possible ALS, but IL-4 and IL-10 levels tended to be higher in patients with PMA and patients with possible ALS than in patients with probable/definite ALS; however, this was not statistically significant (figure 2).
Figure 2. Correlation analysis and representative cytokine titers.

Left column shows correlations between cytokine levels in CSF and clinical parameters. Right column shows CSF cytokine titers (pg/mL) in multifocal motor neuropathy (MMN, n = 12), progressive muscular atrophy (PMA, n = 8), possible amyotrophic lateral sclerosis (ALS, n = 8), and ALS except possible ALS (n = 18). ALSFRS-R = revised Amyotrophic Lateral Sclerosis Functional Rating Scale; IL = interleukin.
DISCUSSION
Our results indicate that CSF inflammatory features are different between patients with MMN and those with motor neuron disease (PMA and ALS) but are similar between patients with motor neuron disease (PMA and ALS). In particular, FGF-2 and G-CSF levels were elevated in both patients with PMA and patients with ALS compared with patients with MMN and ONDs. Our epidemiologic study suggests that MMN is underdiagnosed because it is often misdiagnosed as “ALS without overt UMN signs.”7 Thus, it is critical to distinguish MMN from motor neuron diseases. This study included some patients with MMN who did not present with CB or with anti-GM1 IgM antibodies. However, the number of patients with MMN lacking CB and anti-GM1 IgM antibodies was too low to evaluate whether those cases showed a different cytokine profile from PMA and ALS. We have reported that proinflammatory cytokines, such as IL-1ra, IL-2, G-CSF, TNF-α, and TNFR1, are elevated in the sera of patients with MMN.15 In contrast, in the present study patients with MMN presented with abnormality only in CSF IL-1ra levels. Because IL-1ra is important for preventing ischemic or toxic brain injury in animal models,24 the elevated IL-1ra levels can be interpreted as a common regulatory mechanism to limit neuronal damage in MMN as well as ALS. In addition, CSF IL-1ra levels are elevated in patients with MMN with high serum IL-1ra levels (data not shown). Taken together, these findings indicate that cytokine abnormalities in MMN could be principally peripheral events. Although MMN may clinically resemble PMA and ALS, the CSF cytokine/chemokine profile in patients with MMN is quite distinct from that in patients with PMA and ALS. In our previous study,15 we also found that the levels of several proinflammatory cytokines were increased in the sera of patients with MMN, whereas no significant changes (except for IL-1ra level) were detected in the sera of patients with ALS. Therefore, the cytokine profiles in CSF complement rather than substitute for those in sera as useful biomarkers for MMN and motor neuron diseases.
IL-6, IL-8/CXCL8, IP-10/CXCL10, MCP-1/CCL2, and TNF-α levels were significantly higher in patients with PMA than patients with MMN, but not patients with ONDs. However, the levels of those cytokines (except IL-6) increased with age (data not shown), consistent with a previous study.25 Besides, aging itself might be associated with altered neuroinflammation.26 Therefore, the high levels of the 4 cytokines in patients with PMA could be associated with patient age, as patients in the MMN group were younger than those in the other groups.
In ALS mouse models with mutant Cu/Zn superoxide dismutase (SOD1), both beneficial (so-called M2 microglia, regulatory T cells, and Th2 cells) and deleterious (so-called M1 microglia and Th1 cells) immune responses can influence disease progression.27 We showed that IL-10 elevation was correlated with milder symptoms and that IL-4 and eotaxin/CCL11 elevation was correlated with slower disease progression in patients with ALS, suggesting that those cytokines may confer neuroprotection against ALS. Besides, subsequent analysis revealed that IL-4 and IL-10 tended to be higher in patients with PMA and possible ALS than in patients with probable/definite ALS, indicating slightly more severe symptoms in the latter. IL-10 has pleiotropic effects on immunoregulation.28 IL-4 is a Th2-associated cytokine.27 Eotaxin/CCL11 is a potent eosinophil chemoattractant cytokine associated with allergic airway inflammation.29 Thus, our results are consistent with the findings in SOD1 mouse models, in which regulatory T and Th2 cells can be beneficial.27 Furthermore, G-CSF was elevated in patients with motor neuron disease (PMA and ALS) compared with patients with MMN and disease controls. G-CSF can exert anti-apoptotic or anti-inflammatory effects as well as promote neurogenesis and angiogenesis in the CNS.30,31
Our results highlight the similarity in cytokine profiles between patients with PMA and patients with ALS. Both groups of patients have elevated levels of IL-7, IL-17, eotaxin/CCL11, FGF-2, G-CSF, and VEGF. In line with previous studies, patients with ALS also revealed abnormalities in the levels of such cytokines/chemokines as IL-7, IL-17, eotaxin/CCL11, FGF-2, G-CSF, and VEGF.16–18 IL-7 is a growth and differentiation factor for precursor B cells and plays a role in T cell activation.32 It is proposed that IL-17 contributes to several neurologic diseases, such as multiple sclerosis and neuromyelitis optica.33,34 FGF-2 deficiency prolonged survival and improved motor performance in the ALS mouse model,35 whereas FGF-2 was elevated in the present patients with PMA. The elevation of IL-7, IL-17, and FGF-2 may contribute to the CNS inflammatory process in patients with PMA and patients with ALS. Abnormalities in cytokine signaling have also been reported in other motor neuron diseases, such as spinal and bulbar muscular atrophy. In that disease, signaling of transforming growth factor β, a Th1-associated anti-inflammatory cytokine, is disrupted due to transcriptional dysregulation of its receptor, which is associated with polyglutamine-induced motor neuron damage.36 However, cytokines relevant to transforming growth factor β, such as IL-12 or IFN-γ, did not show abnormal levels in patients with PMA and ALS in the present study.
There is ongoing debate about whether PMA is a distinct disease entity or one spectrum of ALS. The current criteria for ALS diagnosis are based on the distribution of both UMN and LMN signs in various parts of the body, and thus “clinically pure LMN disease,” or PMA, is not classified as ALS. In fact, besides the UMN signs, PMA and ALS have somewhat different clinical characteristics. In the present study, the patients with PMA were mostly male and rarely had onset in the bulbar region, as previously reported.13,14 Nonetheless, several studies of clinical PMA cases revealed UMN abnormalities at autopsy.37–39 Moreover, a recent study reported that TAR DNA-binding protein 43 kDa pathology in the motor cortices or the hippocampus was common in both clinical ALS and PMA cases, suggesting that PMA is pathologically linked to ALS.40 The present study provides immunologic evidence that PMA and ALS are linked to each other.
ACKNOWLEDGMENT
This manuscript was completed with support from Dr. Mitsuyo Ishida (Itsuki Hospital, Tokushima, Japan).
GLOSSARY
- ALS
amyotrophic lateral sclerosis
- ALSFRS-R
revised ALS Functional Rating Scale
- CB
conduction block
- FGF-2
fibroblast growth factor-2
- G-CSF
granulocyte colony-stimulating factor
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- IFN
interferon
- IL
interleukin
- IL-1ra
IL-1 receptor antagonist
- IP-10
IFN-γ–induced protein 10
- LMN
lower motor neuron
- MCP-1
monocyte chemotactic protein-1
- MIP
macrophage inflammatory protein
- MMN
multifocal motor neuropathy
- OND
other noninflammatory neurologic disorder
- PDGF-BB
platelet-derived growth factor BB
- PMA
progressive muscular atrophy
- SOD1
Cu/Zn superoxide dismutase
- TNF-α
tumor necrosis factor α
- TNFR1
TNF receptor
- UMN
upper motor neuron
- VEGF
vascular endothelial growth factor
AUTHOR CONTRIBUTIONS
Dr. Furukawa: analysis/interpretation of data, drafting/revising the manuscript. Dr. Matsui: design/conceptualization of the study, acquisition of data, statistical analysis, analysis/interpretation of data, drafting/revising the manuscript. Dr. Fujita: acquisition of data, analysis/interpretation of data, drafting/revising the manuscript. Dr. Nodera: acquisition of data, revising the manuscript. Dr. Shimizu: acquisition of data, revising the manuscript. Dr. Takahashi: acquisition of data, revising the manuscript. Dr. Kanda: acquisition of data, revising the manuscript. Dr. Kusunoki: acquisition of data, revising the manuscript. Dr. Izumi: acquisition of data, analysis/interpretation of data, drafting/revising the manuscript. Dr. Kaji: acquisition of data, drafting/revising the manuscript. All authors gave their final approval of the version to be published.
STUDY FUNDING
Supported by JSPS KAKENHI Grant-in-Aid for Young Scientists (B) 25870477; Grants-in-Aid from the Research Committee of CNS Degenerative Diseases, the Ministry of Health, Labor and Welfare of Japan; and Health and Labor Sciences Research Grant on Rare and Intractable Diseases (Evidence-based Early Diagnosis and Treatment Strategies for Neuroimmunological Diseases) from the Ministry of Health, Labor and Welfare of Japan.
DISCLOSURE
T. Furukawa reports no disclosures. N. Matsui receives research support from JSPS KAKENHI, Neuroimmunological Disease Research Committee, the Ministry of Health, Labor and Welfare of Japan, and Research Committee of CNS Degenerative Diseases. K. Fujita reports no disclosures. H. Nodera is on the editorial board for Clinical Neurophysiology. F. Shimizu is employed by Biogen. K. Miyamoto reports no disclosures. Y. Takahashi received speaker honoraria from Ohtsuka, Benesis, Kyowa Hakko Kirin, GSK, Asteras, Eisai, Ono, Kaketsuken, MDS, Dianippon Sumitomo, and Tanabe Mitsubishi Pharmaceutical; is on the editorial board for Epileptologia; and received research support from Ministry of Education, Culture, Sports, Science and Technology, Health, Labour and Welfare Ministry and Japan Epilepsy Research Foundation. T. Kanda is the Editor-in-Chief for Clinical and Experimental Neuroimmunology, is an editorial advisory board member for Neuropathology, and received research support from Japan Society for the Promotion of Science. S. Kusunoki received travel funding and/or speaker honoraria from Teijin Pharma, Nihon Pharmaceuticals, Japan Blood Products Organization, Novartis, Dianippon Sumitomo Pharma, Kyowa Kirin, Asahi Kaesei, Bayer, Sanofi, and GlaxoSmithKline; is on the editorial board for Journal of Neuroimmunology and Clinical and Experimental Neurology; is an associate editor for Neurology and Clinical Neuroscience; and received research support from Novartis, GlaxoSmithKline, Dainippon Sumitomo Pharma, Teijin Pharma, Astellas, Sanofi, Japan Blood Products Organization, Nihon Pharmaceuticals, Ministry of Education, Culture, Sports, Science and Technology of Japan, and Ministry of Health, Labor and Welfare of Japan. Y. Izumi and R. Kaji report no disclosures. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Van Asseldonk JT, Franssen H, Van den Berg-Vos RM, Wokke JH, Van den Berg LH. Multifocal motor neuropathy. Lancet Neurol 2005;4:309–319. [DOI] [PubMed] [Google Scholar]
- 2.Katz JS, Wolfe GI, Bryan WW, Jackson CE, Amato AA, Barohn RJ. Electrophysiologic findings in multifocal motor neuropathy. Neurology 1997;48:700–707. [DOI] [PubMed] [Google Scholar]
- 3.Slee M, Selvan A, Donaghy M. Multifocal motor neuropathy: the diagnostic spectrum and response to treatment. Neurology 2007;69:1680–1687. [DOI] [PubMed] [Google Scholar]
- 4.Kaji R, Shibasaki H, Kimura J. Multifocal demyelinating motor neuropathy: cranial nerve involvement and immunoglobulin therapy. Neurology 1992;42:506–509. [DOI] [PubMed] [Google Scholar]
- 5.Dalakas MC, Stein DP, Otero C, Sekul E, Cupler EJ, McCrosky S. Effect of high-dose intravenous immunoglobulin on amyotrophic lateral sclerosis and multifocal motor neuropathy. Arch Neurol 1994;51:861–864. [DOI] [PubMed] [Google Scholar]
- 6.Pestronk A, Cornblath DR, Ilyas AA, et al. A treatable multifocal motor neuropathy with antibodies to GM1 ganglioside. Ann Neurol 1988;24:73–78. [DOI] [PubMed] [Google Scholar]
- 7.Miyashiro A, Matsui N, Shimatani Y, et al. ; Japanese Multifocal Motor Neuropathy Study Group. Are multifocal motor neuropathy patients underdiagnosed? An epidemiological survey in Japan. Muscle Nerve 2014;49:357–361. [DOI] [PubMed] [Google Scholar]
- 8.Katz JS, Barohn RJ, Kojan S, et al. Axonal multifocal motor neuropathy without conduction block or other features of demyelination. Neurology 2002;58:615–620. [DOI] [PubMed] [Google Scholar]
- 9.van Schaik IN, Bossuyt PM, Brand A, Vermeulen M. Diagnostic value of GM1 antibodies in motor neuron disorders and neuropathies: a meta-analysis. Neurology 1995;45:1570–1577. [DOI] [PubMed] [Google Scholar]
- 10.Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardiman O. Amyotrophic lateral sclerosis mimic syndromes: a population-based study. Arch Neurol 2000;57:109–113. [DOI] [PubMed] [Google Scholar]
- 11.Kiernan MC, Vucic S, Cheah BC, et al. Amyotrophic lateral sclerosis. Lancet 2011;377:942–955. [DOI] [PubMed] [Google Scholar]
- 12.Norris F, Shepherd R, Denys E, et al. Onset, natural history and outcome in idiopathic adult motor neuron disease. J Neurol Sci 1993;118:48–55. [DOI] [PubMed] [Google Scholar]
- 13.Raaphorst J, de Visser M, van Tol MJ, et al. Cognitive dysfunction in lower motor neuron disease: executive and memory deficits in progressive muscular atrophy. J Neurol Neurosurg Psychiatry 2011;82:170–175. [DOI] [PubMed] [Google Scholar]
- 14.Kim WK, Liu X, Sandner J, et al. Study of 962 patients indicates progressive muscular atrophy is a form of ALS. Neurology 2009;73:1686–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furukawa T, Matsui N, Fujita K, et al. Increased proinflammatory cytokines in sera of patients with multifocal motor neuropathy. J Neurol Sci 2014;346:75–79. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka M, Kikuchi H, Ishizu T, et al. Intrathecal upregulation of granulocyte colony stimulating factor and its neuroprotective actions on motor neurons in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 2006;65:816–825. [DOI] [PubMed] [Google Scholar]
- 17.Mitchell RM, Freeman WM, Randazzo WT, et al. A CSF biomarker panel for identification of patients with amyotrophic lateral sclerosis. Neurology 2009;72:14–19. [DOI] [PubMed] [Google Scholar]
- 18.Tateishi T, Yamasaki R, Tanaka M, et al. CSF chemokine alterations related to the clinical course of amyotrophic lateral sclerosis. J Neuroimmunol 2010;222:76–81. [DOI] [PubMed] [Google Scholar]
- 19.Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of multifocal motor neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society—first revision. J Peripher Nerv Syst 2010;15:295–301. [DOI] [PubMed] [Google Scholar]
- 20.Brooks BR, Miller RG, Swash M, Munsat TL; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293–299. [DOI] [PubMed] [Google Scholar]
- 21.Cedarbaum JM, Stambler N, Malta E, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS study Group (Phase III). J Neurol Sci 1999;169:13–21. [DOI] [PubMed] [Google Scholar]
- 22.Kusunoki S, Chiba A, Tai T, Kanazawa I. Localization of GM1 and GD1b antigens in the human peripheral nervous system. Muscle Nerve 1993;16:752–756. [DOI] [PubMed] [Google Scholar]
- 23.Fujita K, Matsui N, Takahashi Y, et al. Increased interleukin-17 in the cerebrospinal fluid in sporadic Creutzfeldt-Jakob disease: a case-control study of rapidly progressive dementia. J Neuroinflammation 2013;10:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arend WP, Palmer G, Gabay C. IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev 2008;223:20–38. [DOI] [PubMed] [Google Scholar]
- 25.Mitchell RM, Simmons Z, Beard JL, Stephens HE, Connor JR. Plasma biomarkers associated with ALS and their relationship to iron homeostasis. Muscle Nerve 2010;42:95–103. [DOI] [PubMed] [Google Scholar]
- 26.Keleshian VL, Modi HR, Rapoport SI, Rao JS. Aging is associated with altered inflammatory, arachidonic acid cascade, and synaptic markers, influenced by epigenetic modifications, in the human frontal cortex. J Neurochem 2013;125:63–73. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Zhao W, Beers DR, Appel SH. Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. J Neuroimmune Pharmacol 2013;8:888–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fitzgerald DC, Zhang GX, El-Behi M, et al. Suppression of autoimmune inflammation of the central nervous system by interleukin 10 secreted by interleukin 27-stimulated T cells. Nat Immunol 2007;8:1372–1379. [DOI] [PubMed] [Google Scholar]
- 29.Adzemovic MZ, Öckinger J, Zeitelhofer M, et al. Expression of Ccl11 associates with immune response modulation and protection against neuroinflammation in rats. PLoS One 2012;7:e39794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pitzer C, Krüger C, Plaas C, et al. Granulocyte-colony stimulating factor improves outcome in a mouse model of amyotrophic lateral sclerosis. Brain 2008;131:3335–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee ST, Park JE, Kim DH, et al. Granulocyte-colony stimulating factor attenuates striatal degeneration with activating survival pathways in 3-nitropropionic acid model of Huntington's disease. Brain Res 2008;1194:130–137. [DOI] [PubMed] [Google Scholar]
- 32.Jana M, Mondal S, Jana A, Pahan K. Interleukin-12 (IL-12), but not IL-23, induces the expression of IL-7 in microglia and macrophages: implications for multiple sclerosis. Immunology 2014;141:549–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ishizu T, Osoegawa M, Mei FJ, et al. Intrathecal activation of the IL-17/IL-8 axis in opticospinal multiple sclerosis. Brain 2005;128:988–1002. [DOI] [PubMed] [Google Scholar]
- 34.Hengstman GJ, Wesseling P, Frenken CW, Jongen PJ. Neuromyelitis optica with clinical and histopathological involvement of the brain. Mult Scler 2007;13:679–682. [DOI] [PubMed] [Google Scholar]
- 35.Thau N, Jungnickel J, Knippenberg S, et al. Prolonged survival and milder impairment of motor function in the SOD1 ALS mouse model devoid of fibroblast growth factor 2. Neurobiol Dis 2012;47:248–257. [DOI] [PubMed] [Google Scholar]
- 36.Katsuno M, Adachi H, Minamiyama M, et al. Disrupted transforming growth factor-β signaling in spinal and bulbar muscular atrophy. J Neurosci 2010;30:5702–5712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brownell B, Oppenheimer DR, Hughes JT. The central nervous system in motor neuron disease. J Neurol Neurosurg Psychiatry 1970;33:338–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van den Berg-Vos RM, Visser J, Kalmijn S, et al. A long-term prospective study of the natural course of sporadic adult-onset lower motor neuron syndromes. Arch Neurol 2009;66:751–757. [DOI] [PubMed] [Google Scholar]
- 39.Ince PG, Evans J, Knopp M, et al. Corticospinal tract degeneration in the progressive muscular atrophy variant of ALS. Neurology 2003;60:1252–1258. [DOI] [PubMed] [Google Scholar]
- 40.Riku Y, Atsuta N, Yoshida M, et al. Differential motor neuron involvement in progressive muscular atrophy: a comparative study with amyotrophic lateral sclerosis. BMJ Open 2014;4:e005213. [DOI] [PMC free article] [PubMed] [Google Scholar]